A standardized, extensible framework for optimizing classification improves marker-gene taxonomic assignments
Author and article information
Abstract
Background: Taxonomic classification of marker-gene (i.e., amplicon) sequences represents an important step for molecular identification of microorganisms.
Results: We present three advances in our ability to assign and interpret taxonomic classifications of short marker gene sequences: two new methods for taxonomy assignment, which reduce runtime up to two-fold and achieve high-precision genus-level assignments; an evaluation of classification methods that highlights differences in performance with different marker genes and at different levels of taxonomic resolution; and an extensible framework for evaluating and optimizing new classification methods, which we hope will serve as a model for standardized and reproducible bioinformatics methods evaluations.
Conclusions: Our new methods are accessible in QIIME 1.9.0, and our evaluation framework will support ongoing optimization of classification methods to complement rapidly evolving short-amplicon sequencing and bioinformatics technologies. Static versions of all of the analysis notebooks generated with this framework, which contain all code and analysis results, can be viewed at http://bit.ly/srta-012 .
Cite this as
2015. A standardized, extensible framework for optimizing classification improves marker-gene taxonomic assignments. PeerJ PrePrints 3:e934v2 https://doi.org/10.7287/peerj.preprints.934v2Author comment
This is a revision following our response to comments from reviewers at Microbiome.
Sections
Supplemental Information
Supplementary Figure 1. Mock community datasets analyzed in this study
Supplementary Figure 2. Mock community A composition
Supplementary Figure 3. Mock community B composition
Supplementary Figure 4. Mock community C composition
Supplementary Figure 5. Mock community D composition
Supplementary Figure 6
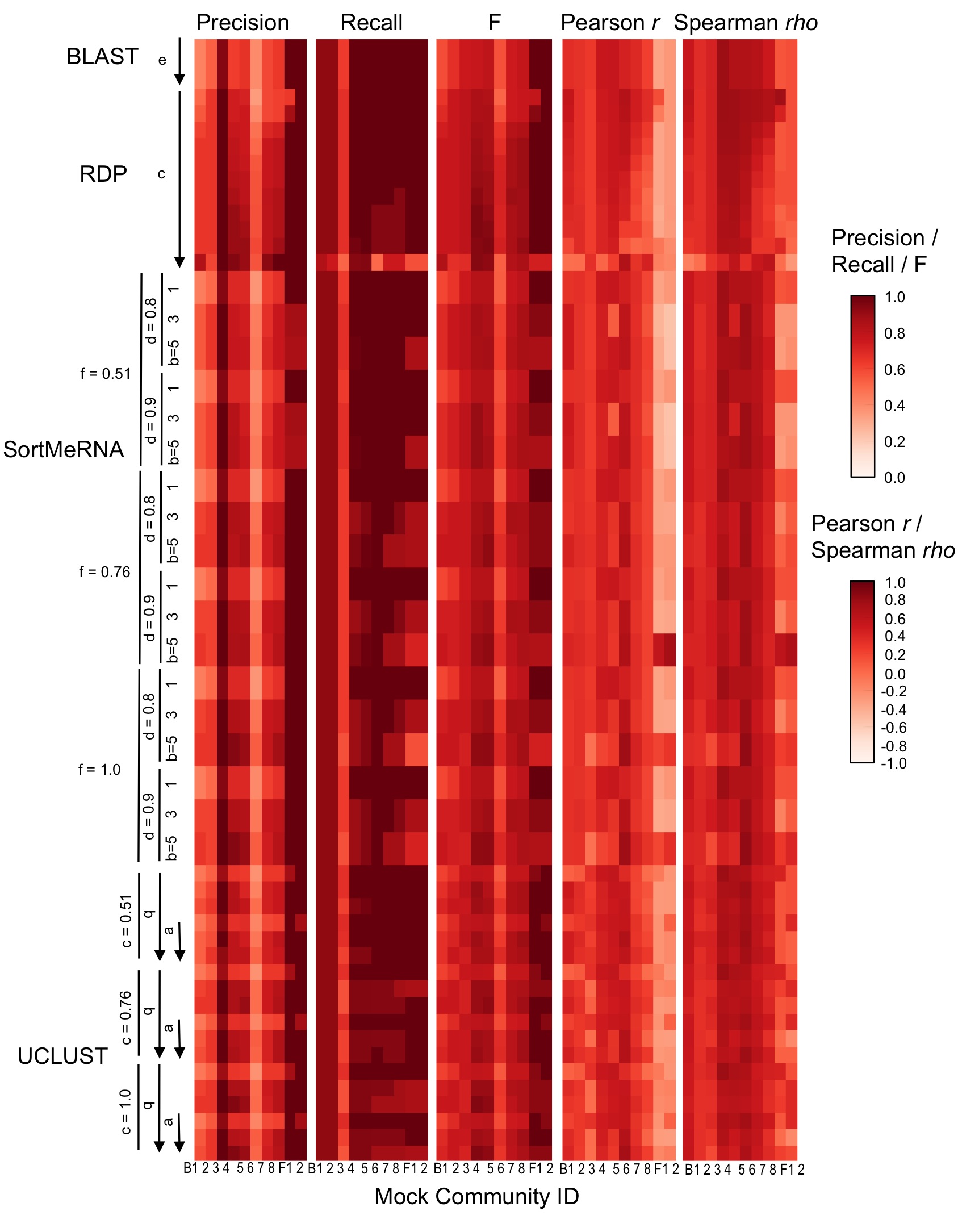
Taxonomy classifier configuration and mock community composition alter assignment accuracy at family-level.
Supplementary Figure 7
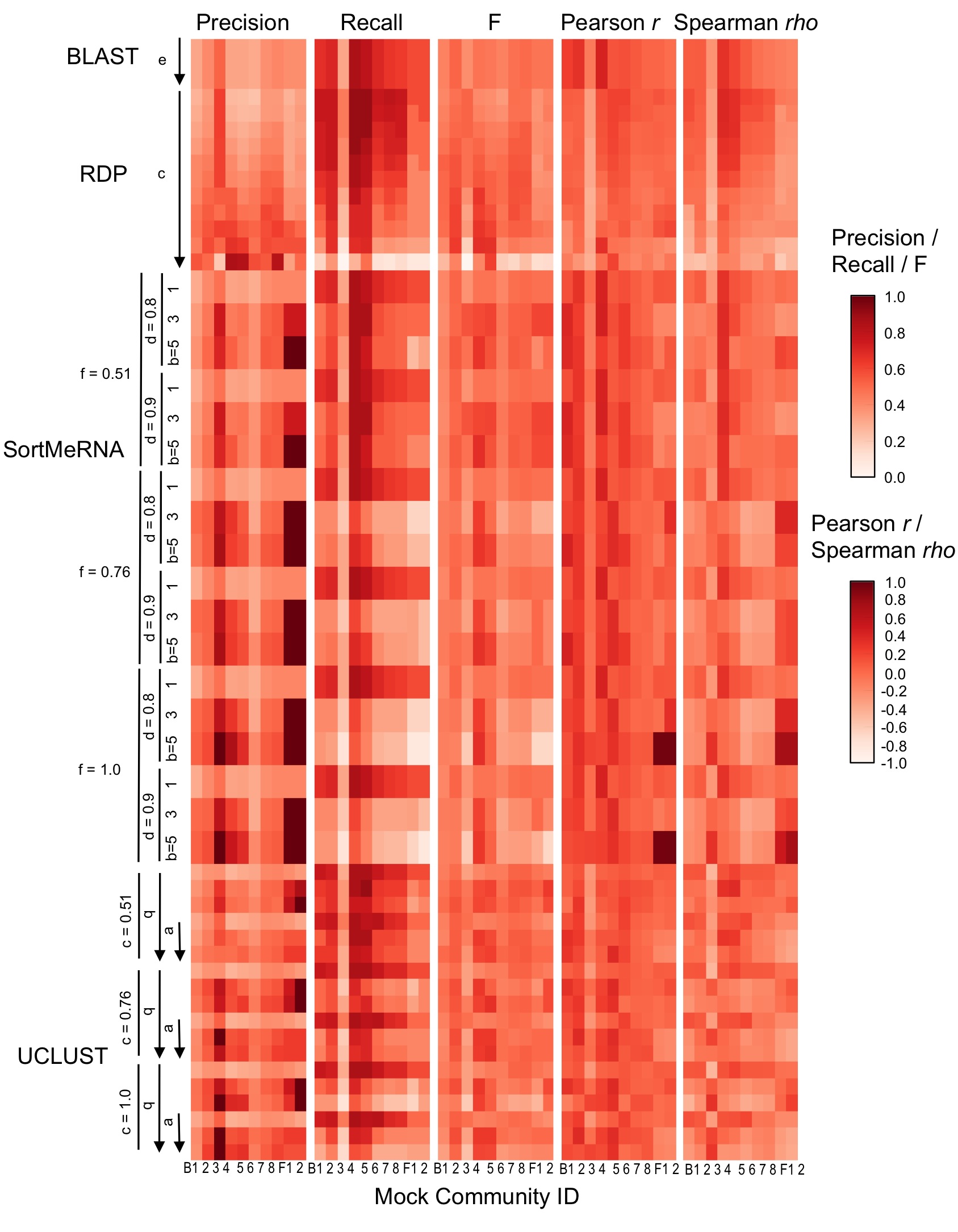
Taxonomy classifier configuration and mock community composition alter assignment accuracy at species-level.
Supplementary Figure 8
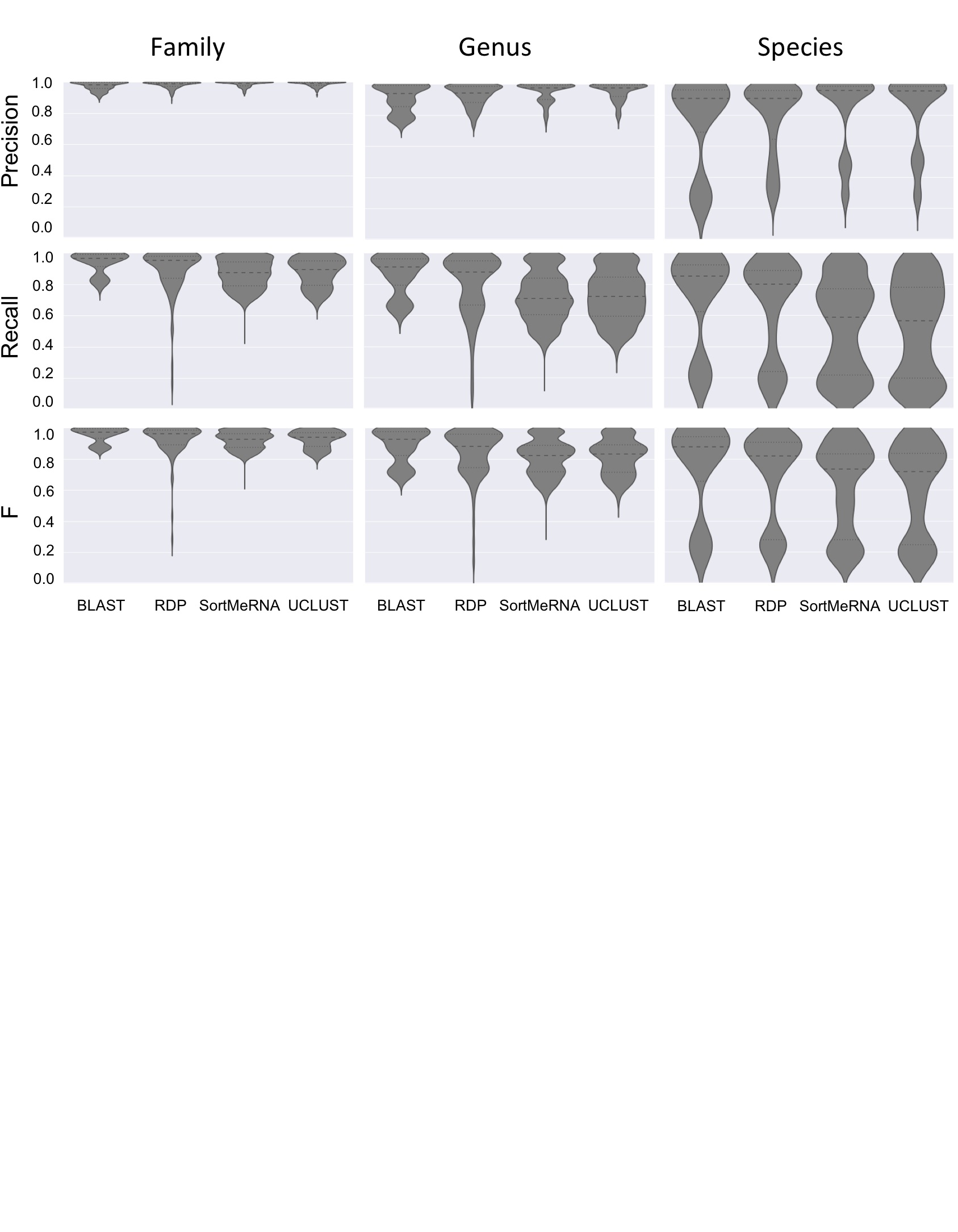
Taxonomy classifier selection critically shapes assignment accuracy of simulated communities. Violin plots illustrate the distribution of precision, recall, and F-measure values across all simulated communities and all parameter configurations for a given method for family-level (left), genus-level (middle), or species-level taxonomy assignments (right). Heavy dashed lines indicate median values, fine dashed lines indicate quartiles.
Supplementary Figure 9
Taxonomic lineages represented in reference databases
Supplementary Figure 10
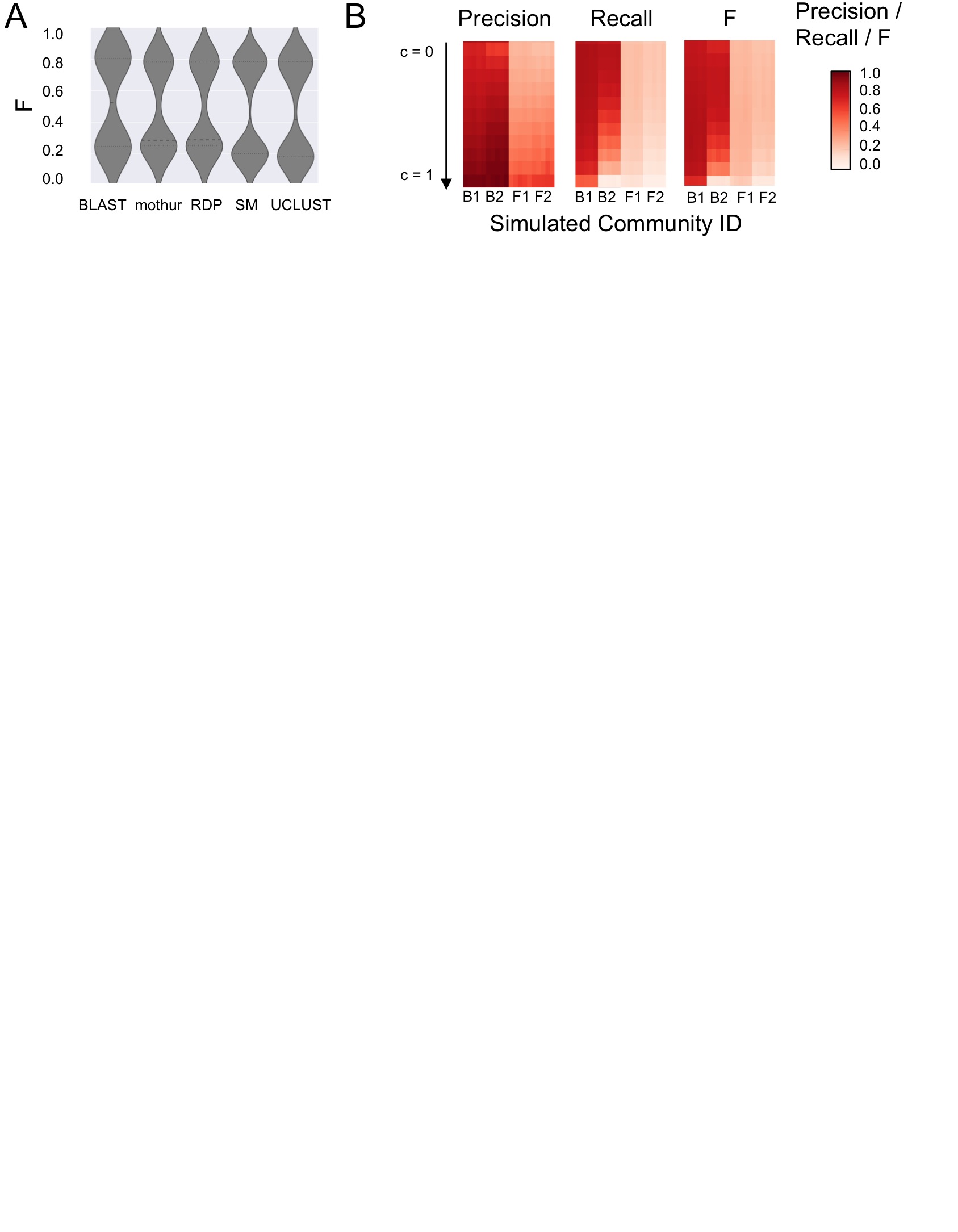
Evaluation of mothur taxonomy classifier. A, Distribution of F-measure scores across all partial-reference simulated communities and all parameter configurations for each method for species-level taxonomy assignments (right). Heavy dashed lines indicate median values, fine dashed lines indicate quartiles. SM = SortMeRNA. B, Confidence configuration and simulated community composition alter assignment accuracy at species-level. See figure 4 for full description of analysis and comparison to other classifiers and configurations.
Additional Information
Competing Interests
The authors declare they have no competing interests.
Author Contributions
Nicholas A Bokulich conceived and designed the experiments, performed the experiments, analyzed the data, wrote the paper, prepared figures and/or tables, reviewed drafts of the paper.
Jai Ram Rideout conceived and designed the experiments, performed the experiments, analyzed the data, wrote the paper, prepared figures and/or tables, reviewed drafts of the paper.
Evguenia Kopylova performed the experiments, analyzed the data, contributed reagents/materials/analysis tools, reviewed drafts of the paper.
Evan Bolyen performed the experiments, analyzed the data, prepared figures and/or tables, reviewed drafts of the paper.
Jessica Patnode performed the experiments, analyzed the data, reviewed drafts of the paper.
Zach Ellett performed the experiments, analyzed the data, reviewed drafts of the paper.
Daniel McDonald performed the experiments, analyzed the data, reviewed drafts of the paper.
Benjamin Wolfe contributed reagents/materials/analysis tools, reviewed drafts of the paper.
Corinne F Maurice contributed reagents/materials/analysis tools, reviewed drafts of the paper.
Rachel J Dutton contributed reagents/materials/analysis tools, reviewed drafts of the paper.
Peter J Turnbaugh conceived and designed the experiments, contributed reagents/materials/analysis tools, reviewed drafts of the paper.
Rob Knight conceived and designed the experiments, reviewed drafts of the paper.
J Gregory Caporaso conceived and designed the experiments, performed the experiments, analyzed the data, wrote the paper, prepared figures and/or tables, reviewed drafts of the paper.
Data Deposition
The following information was supplied regarding the deposition of related data:
http://github.com/gregcaporaso/short-read-tax-assignment
Funding
This work was supported in part by the 2014 Kinsella Memorial Award (NAB), NIH P50 GM068763 (PJT and CFM), NSF IGERT grant number 1144807 (DM), the NIH and the Howard Hughes Medical Institute (RK), and the Arizona Board of Regents TRIF (JGC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}