Culture-independent detection and characterisation of Mycobacterium tuberculosis and M. africanum in sputum samples using shotgun metagenomics on a benchtop sequencer
A peer-reviewed article of this Preprint also exists.
Author and article information
Abstract
Tuberculosis remains a major global health problem. Laboratory diagnostic methods that allow effective, early detection of cases are central to management of tuberculosis in the individual patient and in the community. Since the 1880s, laboratory diagnosis of tuberculosis has relied primarily on microscopy and culture. However, microscopy fails to provide species- or lineage-level identification and culture-based workflows for diagnosis of tuberculosis remain complex, expensive, slow, technically demanding and poorly able to handle mixed infections. We therefore explored the potential of shotgun metagenomics, sequencing of DNA from samples without culture or target-specific amplification or capture, to detect and characterise strains from the Mycobacterium tuberculosis complex in smear-positive sputum samples obtained from The Gambia in West Africa. Eight smear- and culture-positive sputum samples were investigated using a differential-lysis protocol followed by a kit-based DNA extraction method, with sequencing performed on a benchtop sequencing instrument, the Illumina MiSeq. The number of sequence reads in each sputum-derived metagenome ranged from 989,442 to 2,818,238. The proportion of reads in each metagenome mapping against the human genome ranged from 20% to 99%. We were able to detect sequences from the M. tuberculosis complex in all eight samples, with coverage of the H37Rv reference genome ranging from 0.002X to 0.7X. By analysing the distribution of large sequence polymorphisms (deletions and the locations of the insertion element IS6110) and single nucleotide polymorphisms (SNPs), we were able to assign seven of eight metagenome-derived genomes to a species and lineage within the M. tuberculosis complex. Two metagenome-derived mycobacterial genomes were assigned to M. africanum, a species largely confined to West Africa; the others that could be assigned belonged to lineages T, H or LAM within the clade of “modern” M. tuberculosis strains. We have provided proof of principle that shotgun metagenomics can be used to detect and characterise M. tuberculosis sequences from sputum samples without culture or target-specific amplification or capture, using an accessible benchtop-sequencing platform, the Illumina MiSeq, and relatively simple DNA extraction, sequencing and bioinformatics protocols. In our hands, sputum metagenomics does not yet deliver sufficient depth of coverage to allow sequence-based sensitivity testing; it remains to be determined whether improvements in DNA extraction protocols alone can deliver this or whether culture, capture or amplification steps will be required. Nonetheless, we can foresee a tipping point when a unified automated metagenomics-based workflow might start to compete with the plethora of methods currently in use in the diagnostic microbiology laboratory.
Cite this as
2014. Culture-independent detection and characterisation of Mycobacterium tuberculosis and M. africanum in sputum samples using shotgun metagenomics on a benchtop sequencer. PeerJ PrePrints 2:e482v1 https://doi.org/10.7287/peerj.preprints.482v1Author comment
This submission has been accepted for publication at PeerJ.
Sections
Supplemental Information
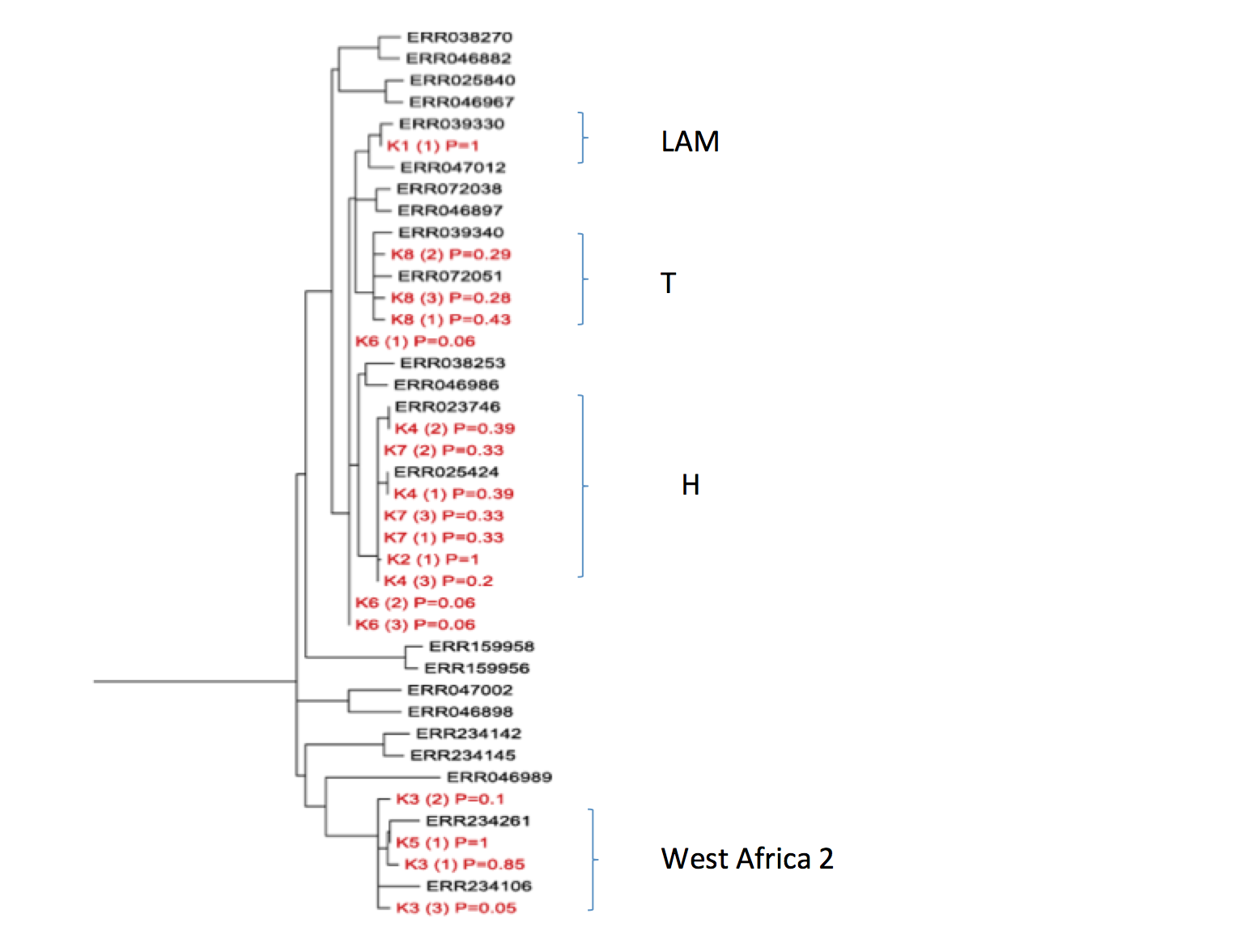
Detection and characterisation of Mycobacterium tuberculosis in sputum samples using shotgun metagenomics Figure S1 Detailed phylogenetic placement of metagenome-derived genomes
For each sample, the majority bases at each reference SNP position (or gaps if there was no coverage at that position) were concatenated and the sequence was placed in the reference tree using pplacer (see Methods). The output from pplacer (jplace file) was parsed and a new file produced, such that each alternative placement for a sample could be displayed in the tree along with the posterior probability of that placement. Trees were generated from the modified place file, using guppy from the pplacer suite of programs. Only the top 3 placements for each sample are shown. The combined pp values for each alternative placement of sample in a clade were used to ascertain the likelihood of that sample belonging to that clade.
SNP matrix used to generate tree shown in Figure 1
Additional Information
Competing Interests
There are no competing interests.
Author Contributions
Emma L Doughty conceived and designed the experiments, performed the experiments, analyzed the data, wrote the paper, prepared figures and/or tables, reviewed drafts of the paper.
Martin J Sergeant conceived and designed the experiments, performed the experiments, analyzed the data, wrote the paper, prepared figures and/or tables, reviewed drafts of the paper.
Ifedayo M.O Adetifa conceived and designed the experiments, contributed reagents/materials/analysis tools.
Martin Antonio conceived and designed the experiments, reviewed drafts of the paper.
Mark J Pallen conceived and designed the experiments, wrote the paper, prepared figures and/or tables, reviewed drafts of the paper.
Human Ethics
The following information was supplied relating to ethical approvals (i.e., approving body and any reference numbers):
The joint Gambia Government/MRC Ethics Committee approved this investigation under reference SCC 1232 and informed written consent was obtained for all participants.
DNA Deposition
The following information was supplied regarding the deposition of DNA sequences:
Metagenomic sequence reads from this study (excluding those that mapped to the human genome) will be deposited in the European Nucleotide Archive (project accession number pending).
Funding
Support for Emma Doughty's PhD studentship and research costs was provided by Warwick Medical School and MRC Unit, The Gambia. Support for Martin Sergeant's salary was provided by Warwick Medical School. The Enhanced Case Finding project was funded and sponsored by the MRC Unit, The Gambia. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.

{kind=link}