
Mitochondrial and Wolbachia phylogenetics of the introduced Jorō spider, Trichonephila clavata (Araneae: Araneidae) in North America
- Published
- Accepted
- Received
- Academic Editor
- Graham Wallis
- Subject Areas
- Ecology, Entomology, Genetics
- Keywords
- Trichonephila clavata, COI, Wolbachia, United States
- Copyright
- © 2025 Russell et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2025. Mitochondrial and Wolbachia phylogenetics of the introduced Jorō spider, Trichonephila clavata (Araneae: Araneidae) in North America. PeerJ 13:e19952 https://doi.org/10.7717/peerj.19952
Abstract
The introduction of Trichonephila clavata (L. Koch, 1878) (Araneae: Araneidae: subfamily Nephilinae) in the United States was first recorded in Georgia in 2014. Since its introduction, T. clavata has become a prominent feature of the arthropod fauna in several southeastern US states. Many questions regarding the introduction event(s) remain unanswered; for instance, was the introduction a single discrete event followed by rapid spread, or were there multiple introductions? The mitochondrial cytochrome c oxidase subunit one gene region (COI), which was used to characterize the initial T. clavata observation in the US, has also been used to characterize within- and between-population genetic variation. One confounding factor for COI as a population genetic molecular marker, though, is the presence of cytoplasmic agents of selection such as intracellular bacteria in the genus Wolbachia. Given that Wolbachia infections have been detected in potential source populations of T. clavata, the present study sought to characterize mitochondrial genetic diversity and the status of Wolbachia infection in the North American population(s) closest to the originally proposed introduction site in Georgia. DNA sequencing revealed no mitochondrial genetic variation in the T. clavata population sampled in North America, and an exact sequence match to the previously reported T. clavata in Georgia and a sequence sample from Yunnan, China. Wolbachia was detected in the North American samples. However, phylogenetic analysis on a concatenated multi-locus type sequence suggested two distinct Wolbachia clades, one represented by samples collected in Georgia and another represented by a single sample collected in South Carolina. Sequence analyses of the multi-locus gene regions suggested that the Georgia T. clavata may be infected with two strains of Wolbachia (super-infection), and the South Carolina sample represented a separate single infection. The study’s results emphasize the need for further research, including expanded sampling in the introduced and potential source population regions, as well as a more detailed molecular characterization of the populations.
Introduction
Trichonephila clavata (L. Koch, 1878) (Araneae: Araneidae: subfamily Nephilinae) is an orbweaver spider native to eastern Asia, including Japan, Korea, and China (Hoebeke, Huffmaster & Freeman, 2015). The phylogenetic placement of the genus Trichonephila and other nephiline spiders relative to other Orbipurae orbweavers is a current topic of debate, with alternative placements in either their own family, Nephilidae (see Kuntner, 2006; Kuntner et al., 2019; Kuntner et al., 2023 and arguments within) or as a subfamily, Nephilinae, synonymized within the extremely large family of Araneidae (see Dimitrov & Hormiga, 2009; Dimitrov et al., 2017; Scharff et al., 2020; Kallal et al., 2020; Hormiga et al., 2023).
In late 2014, the first T. clavata were collected in Georgia, US (Hoebeke, Huffmaster & Freeman, 2015). This species, a congener of the native T. clavipes (Linnaeus, 1767; the golden silk orb weaver), quickly spread across the southern US in the years after its introduction, with one main population spanning Georgia, Tennessee, South Carolina, and North Carolina (Chuang et al., 2023), as well as recent observations which may represent new populations in Maryland, Pennsylvania, Virginia, and Massachusetts (https://john-deitsch.shinyapps.io/joroshiny/). Surveys on local orbweaver communities suggest there are fewer native orbweavers where T. clavata has been established the longest (Nelsen et al., 2023). As such, T. clavata requires further close observation and research in the North American habitats to determine its impacts. With overall research still in the early stages and a dearth of population genetic information despite the potential distribution of T. clavata across much of North America (Davis & Frick, 2022; Nelsen et al., 2023), questions remain about the introduction of this highly visible invasive species. Was T. clavata introduced to North America through a single introduction event? Could multiple introduction events be partially responsible for the species’ already large range in North America?
Population genetic tools can provide a better understanding of the past and future of T. clavata in North America. For example, when founded by a few closely related individuals, little or no genetic diversity is expected in a population, which restricts that population’s adaptive potential (Booy et al., 2000)—a condition that could be particularly pertinent for an introduced species in a novel environment. In 2014, the mitochondrial gene cytochrome c oxidase subunit I (COI) from these new North American samples was compared to the recorded literature and confirmed the species identification of T. clavata (Hoebeke, Huffmaster & Freeman, 2015). COI is useful as a “barcoding” gene, serving as a common identifier of species, and often has several distinct alleles in a population (Hebert et al., 2003), making this gene region particularly useful for both within- and between-population studies. Though COI has been used in many population-level studies of animals (Tavares et al., 2011; Karthika et al., 2017; Beebe, 2018), the use of any genetic marker is limited by the extent of prior study and the authenticity of previous sequences, often limiting its usefulness outside of already extensively-studied species (Dawnay et al., 2007). Additionally, mitochondrial population genetics can be confounded by many factors (Galtier et al., 2009), including highly oxidative environments, complex mutation processes, and other cytoplasmic agents of selection.
Bacteria in the genus Wolbachia (Alphaproteobacteria, Rickettsiales) can function as agents of selection. Wolbachia are cytoplasmic bacterial symbionts well represented across arthropod taxa, with one estimate suggesting that up to 66% of all insects may be infected (Hilgenboecker et al., 2008). In arthropods, Wolbachia exhibits several forms of genetic drive that selectively favor infected hosts through reproductive manipulations that either create reproductive barriers between infected and uninfected individuals or distort sex ratios in a manner that promotes the spread of Wolbachia within populations (Stouthamer, Breeuwer & Hurst, 1999). Given that cytoplasmic genetic elements are typically co-inherited, the result of Wolbachia infection in many arthropod populations is indirect selection on mtDNA (Turelli, Hoffmann & McKechnie, 1992; Jiggins, 2003; Narita et al., 2006). This can result in selective sweeps of Wolbachia and co-inherited mtDNA that reduce mitochondrial genetic diversity, as has been characterized in Drosophila simulans (Turelli, Hoffmann & McKechnie, 1992), Acraea encedon (Jiggins, 2003), and many other insect species (Cariou, Duret & Charlat, 2017). However, Baldo et al. (2008) found that strict cytoplasmic co-inheritance among Agelenopsis spiders was disrupted by apparent horizontal transfer among and within species, with some mitotypes associating with divergent Wolbachia strains, and individual Wolbachia strains associating with divergent mitotypes.
Wolbachia have been detected in Chinese and Korean populations of T. clavata (Wang et al., 2010; Yang et al., 2021; Oh et al., 2000). Using strain-specific primers for the Wolbachia wsp gene region, Wang et al. (2010) discovered a double-infected T. clavata population in Wuhan, Hubei Province, China. Furthermore, Yang et al. (2021) characterized a single infection of T. clavata collected in Mangshan, Guangdong Province, China, using multi-locus sequence typing of gene regions (Baldo et al., 2006). These studies were part of large-scale Wolbachia prevalence investigations among spider species and did not sample T. clavata populations to any substantial degree (fewer than ten samples in each study), nor did they examine the potential impact of Wolbachia on host mitochondrial population genetics.
Our study took a phylogenetic approach, using mitochondrial and Wolbachia molecular markers, to investigate the founding population of T. clavata in the US. We addressed three questions: (1) What is the mitochondrial genetic diversity of the southeastern T. clavata population in North America? (2) Is the introduced population infected with Wolbachia? and (3) If the introduced population is infected with Wolbachia, to what extent is mitochondrial and Wolbachia genetic diversity linked? Given the recency of the introduction event, we hypothesized low levels of genetic diversity among T. clavata and associated low levels of Wolbachia diversity, assuming coinheritance between mitochondria and Wolbachia.
Materials & Methods
Sample collection
Mature female T. clavata were collected between August and December 2022 from various natural and managed locations (Table 1; Fig. 1) and mature female T. clavipes were collected in North Carolina before T. clavata invaded the area. Spiders were collected individually and stored separately by location and frozen at −20 °C. The collection of spider samples was approved by a Georgia Gwinnett College STEC 4500 research agreement.
| Abbreviation | City | State | Replicate # | Wolb. sample |
|---|---|---|---|---|
| AGA | Athens | GA | 3 | AGA1 |
| APGA | Buford | GA | 3 | APGA1 |
| APGB | Buford | GA | 1 | APGB |
| AUGA | Auburn | GA | 3 | AUGA3 |
| BGA | Braselton | GA | 4 | BGA2 |
| BUGA | Buford | GA | 3 | BUGA2 |
| CCGB | Jefferson | GA | 1 | CCGB |
| CLSC | Clemson | SC | 3 | CLSC1 |
| CPGB | Athens | GA | 1 | CPGB |
| DDGA | Decatur | GA | 5 | DDGA1 |
| RGA | Richland | GA | 3 | RGA3 |
| SGA | Suwannee | GA | 3 | SGA3 |
| TGA | Talmo | GA | 4 | TGA4 |
| WGA | Watkinsville | GA | 4 | WGA4 |
| WIGA | Winder | GA | 3 | WIGA1 |
| CTN | Collegedale | TN | 1 | N/A |
| CGA | Calhoun | GA | 3 | N/A |
| WBGA | Woodstock | GA | 4 | N/A |
| JGA | Jefferson | GA | 3 | JGA1 |
| GGA | Gainesville | GA | 3 | GGA3 |
| GGGB | Lawrenceville | GA | 1 | GGGB |
| FMGA | Lawrenceville | GA | 3 | FMGA1 |
| FMGB | Lawrenceville | GA | 1 | FMGB |
| HGA | Hoschton | GA | 3 | HGA2 |
| LGA | Loganville | GA | 3 | LGA2 |
| LMGB | Auburn | GA | 1 | LMGB |
| MGA | Morrow | GA | 5 | N/A |
| MGB | Morrow | GA | 4 | MGB1,2,4 |
| HEGA | Helen | GA | 3 | HEGA1 |
| HPGA | Dacula | GA | 4 | HPGA4 |
| SMGA | Hoschton | GA | 4 | N/A |
| SPGB | Hoschton | GA | 1 | SPGB |
| TMGB | Lawrenceville | GA | 1 | TMGB |
| FBGA | Flowery Branch | GA | 4 | N/A |
| MAGA | Marietta | GA | 2 | N/A |
| FNC | Fayetteville | NC | 2 | N/A |
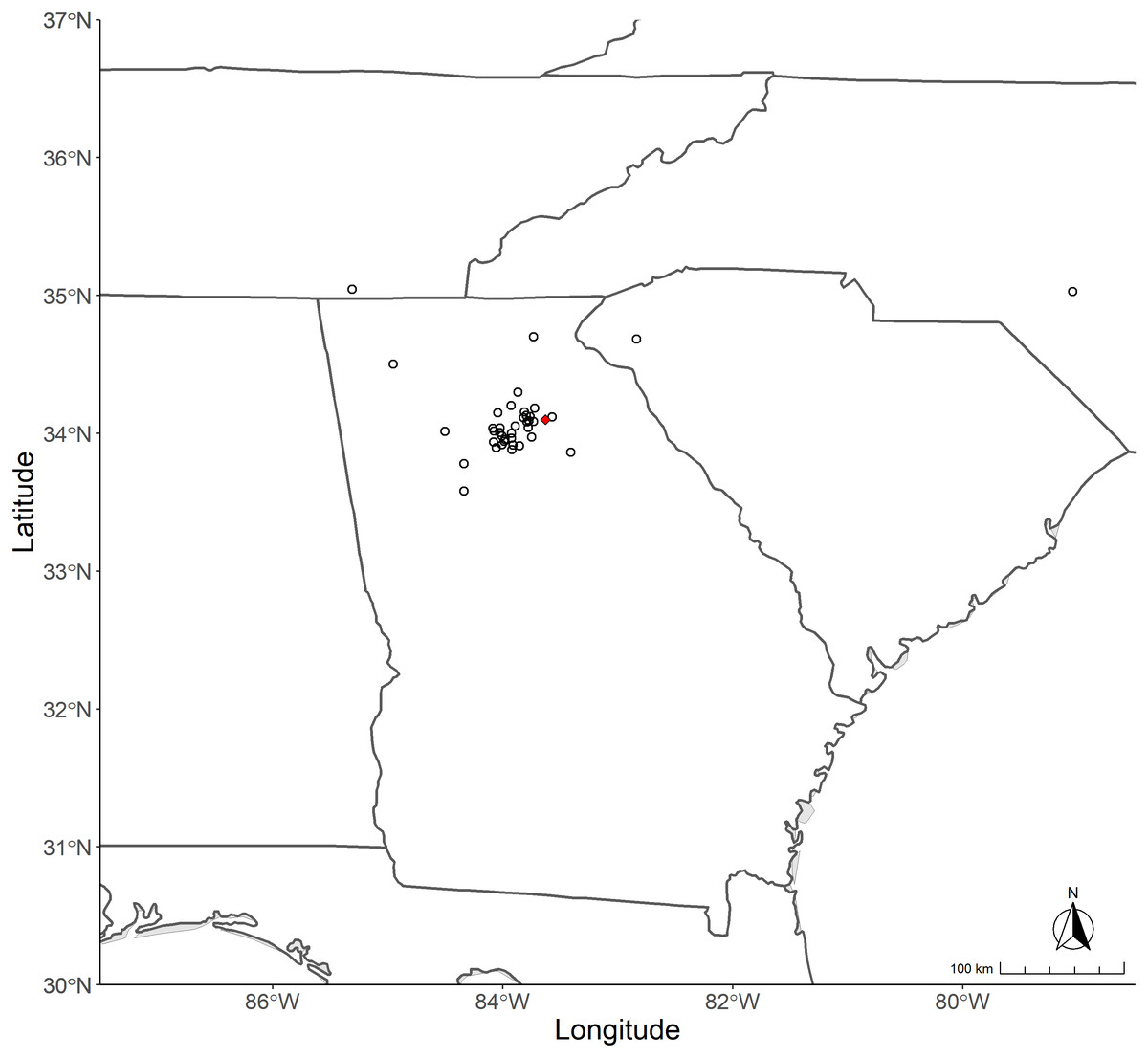
Figure 1: Map of collection sites for Trichonephila species.
The states from which Trichonephila spp. were sampled is shown above. State outlines containing polygons indicating collection sites are clockwise from top left: Tennessee, North Carolina, South Carolina, and Georgia. Red dot indicates the approximate location where T. clavata were initially found in 2014.{kind=link}
Molecular methods
Single-leg tissue samples were used for DNA extraction using 5% Chelex (Bio-Rad Laboratories, Hercules, CA, US) according to modified methods described in Walsh, Metzger & Higuchi (1991). PCR reactions contained one microliter of template DNA, 12.5 microliters of Promega GoTaq® master mix, one microliter of each 10 µM primer, and 9.5 µl of molecular-grade water. A portion of the cytochrome oxidase subunit one (COI) gene region was amplified using primers LCO (5′-GGTCAACAAATCATAAAGAT ATTGG-3′) and HCO (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′) (Folmer et al., 1994). Thermocycle conditions consisted of an initial denaturation at 95 °C for four minutes, followed by 34 cycles of 94 °C for forty-five seconds, 55 °C for thirty seconds, 72 °C for one minute and thirty seconds, followed by a final extension at 72 °C for ten minutes.
Wolbachia multi-locus sequence typing (MLST) primers for coxA, fbpA, gatB, and hcpA identified by Baldo et al. (2006) were used to amplify the respective gene regions using the previously established protocols. Wolbachia A and B supergroup-specific wsp primers (Zhou, Rousset & O’Neill, 1998) were used to identify the spider’s superinfection status. All PCR products were analyzed with 2% agarose gel electrophoresis, and successful amplicons were prepared for sequence analysis with ExoSAP-IT® (Applied Biosystems, Waltham, MA, US). Of the 94 T. clavata samples successfully amplified for COI, a subset of 31 were used to amplify Wolbachia-specific gene regions, including the MLST gene regions.
Additional COI and MLST sequences were acquired from accession numbers provided from Hoebeke, Huffmaster & Freeman (2015), Yang et al. (2021), and the Wolbachia MLST database (http://www.pubmlst.org/wolbachia/) (Tables 2A–2B).
| Sample name | Location | Source |
|---|---|---|
| (A) Cytochrome oxidase subunit one (COI) | ||
| Georgia 1 | Georgia, USA | Hoebeke, Huffmaster & Freeman (2015) |
| Georgia 2 | Georgia, USA | Hoebeke, Huffmaster & Freeman (2015) |
| Guangdong 1 | Guangdong, China | Yang et al. (2021) |
| Yunnan 1 | Yunnan, China | Hoebeke, Huffmaster & Freeman (2015) |
| Yunnan 2 | Yunnan, China | Hoebeke, Huffmaster & Freeman (2015) |
| Zhejiang 1 | Zhejiang, China | Hoebeke, Huffmaster & Freeman (2015) |
| Zhejiang 2 | Zhejiang, China | Hoebeke, Huffmaster & Freeman (2015) |
| South Korea 1 | South Korea | Hoebeke, Huffmaster & Freeman (2015) |
| Taiwan 1 | Taiwan | Hoebeke, Huffmaster & Freeman (2015) |
| (B) Wolbachia Multiple Locus Sequence Types (MLST) | ||
| Araneus ventricosus | Guangdong, China | Yang et al. (2021) |
| Mesida yini | Guangdong, China | Yang et al. (2021) |
| Pardosa mionebulosa | Guangdong, China | Yang et al. (2021) |
| Trichonephila clavata | Guangdong, China | Yang et al. (2021) |
| Agelenopsis aperta | Oklahoma, USA | MLST database |
| Acraea encedon | NA | MLST database |
| Aedes albopictus | Koh Samui, Thailand | MLST database |
| Armadillidium vulgare | NA | MLST database |
| Brugia malayi | NA | MLST database |
| Drosophila melanogaster | NA | MLST database |
| Nasonia giraulti | NA | MLST database |
| Rhagoletis cerasi | Czech Republic | MLST database |
Sequence analysis
All successfully cleaned amplicons were sent to Eurofins Genomics (Louisville, KY, US) for Sanger sequencing. Received sequence reads were aligned in MUSCLE using default parameters. Chromatograms (https://technelysium.com.au/wp/chromas/) were used to inspect sequence reads by eye and verify polymorphisms. Cytochrome oxidase subunit one (COI), all MLST Wolbachia loci, and concatenation of MLST sequences were phylogenetically analyzed in BEAST (Suchard et al., 2018) using Bayesian inference methods and in MEGA 11 (Tamura, Stecher & Kumar, 2021) using maximum likelihood methods with 1000 bootstrap replications (see Supplementary Materials). Appropriate evolutionary models were determined using the model test function in MEGA 11 and the Akaike information criterion scores. BEAST parameters for the COI analysis included the Tamura-Nei substitution model (Tamura & Nei, 1993), a chain length of 10,000,000 generations with log parameters saved every 1,000 generations and a burn-in of 3,000 samples. All T. clavata sequences were recovered in a monophyletic clade with a posterior probability of 1.0. Parameters for the concatenated Wolbachia MLST analysis were identical to the COI analysis with the exception of a Hasegawa-Kishino-Yano substitution model (Hasegawa, Kishino & Yano, 1985). BEAST generated trees were visualized in FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Results
COI
Spider samples from one collection location, Tennessee, failed to successfully amplify and were excluded from subsequent analyses. The North Carolina collection location produced two T. clavipes samples, the only congener to T. clavata in North America. Three of the 94 T. clavata samples sent for sequencing were not readable and were excluded from analysis, resulting in a final n = 91. No polymorphisms were observed among the 91 T. clavata COI sequence samples from Georgia and South Carolina. The haplotype was identical across the 620 sites analyzed to samples collected in Georgia (Hoebeke, Huffmaster & Freeman, 2015) and to Yunnan 1 (HQ441928.1). The COI tree (Fig. 2) resolved two main T. clavata clades with a posterior probability of 1.0, one of which included all US samples, Yunnan 1, Guangdong, and Zhejiang 1, and a sister clade that included South Korea as an outgroup to Yunnan 2, Zhejiang 2, and Taiwan. Maximum likelihood analysis returned a similar topology with a T. clavata polytomy that included support for a US + Yunnan 1 clade and a clade that included South Korea, Yunnan 2, Zhejiang 2, and Taiwan. Guangdong and Zhejiang 1 were unassigned to either clade in the polytomy (see Material S1). Given the limited sample size, taxonomic breadth, and estimated recent evolutionary time frame for the taxa used in the COI analyses, low levels of support for some of these clades are not unexpected.
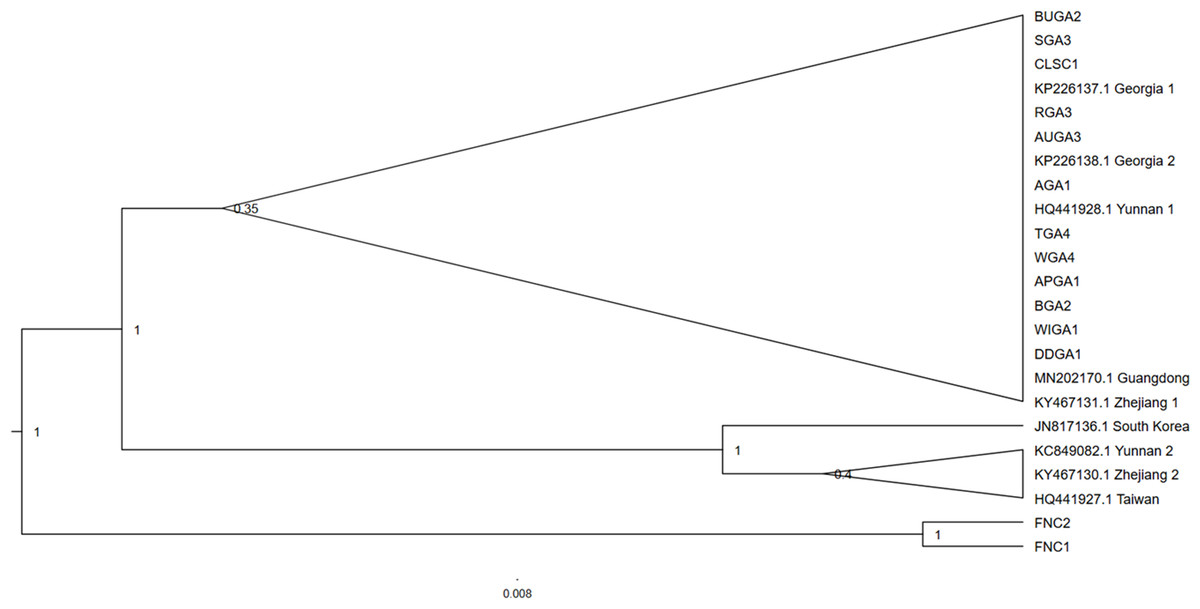
Figure 2: Cladogram of the cytochrome oxidase subunit one (COI) gene region of Trichonephila clavata.
Bayesian inference phylogenetic tree for the cytochrome oxidase subunit one (COI) gene region of Trichonephila clavata obtained in Georgia (GA) and South Carolina (SC). Reference samples obtained in Genbank are included with accession numbers. Trichonephila clavipes (FNC) was used to root the tree. Posterior probabilities are indicated at nodes. T. clavata samples, within the collapsed clade with posterior probability of 0.35, represent a subset of 91 samples sequenced that were identical.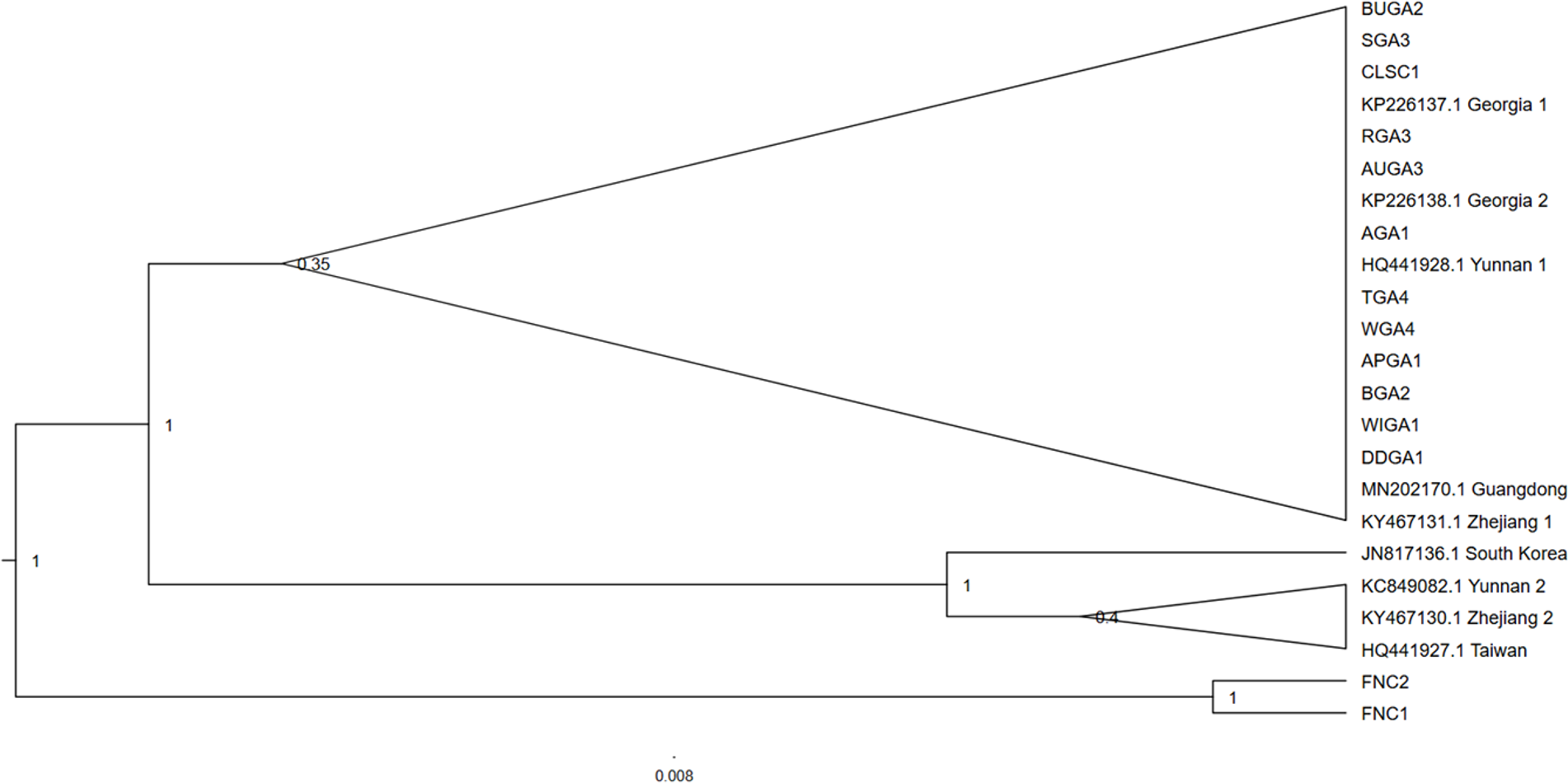{kind=link}
Trichonephila clavipes samples FNC1 and FNC2 served as an outgroup for the T. clavata clade and were used to root the tree. Due to the genetic monomorphism of sampled T. clavata, only those samples that were also used in the Wolbachia analysis were reported for COI phylogenetic analysis (see Material S2 table for accession numbers).
Wolbachia
Wolbachia supergroup A and B-specific wsp primers (Zhou, Rousset & O’Neill, 1998) were used to detect superinfection among collected samples. Although the amplification success of the two primer regions was limited, both primers successfully amplified some of the Georgia samples, suggesting superinfection of A and B supergroup Wolbachia in those samples (8/8 or 100% A-specific amplification, 22/31 or 71% B-specific amplification, with all A-specific amplifications also amplifying the B-specific gene region). One sample used in phylogenetic analysis, CLSC1, failed to amplify either wsp gene region.
Due to limited success at amplifying all MLST loci, a subset of T. clavata samples and MLST loci (wsp and ftsZ sequences were omitted) were used for analysis. Wolbachia supergroup A and B specific primers (Zhou, Rousset & O’Neill, 1998) successfully amplified a further subset of T. clavata samples but were not used for sequencing. Concatenation of MLST loci coxA, fbpA, gatB, and hcpA among 12 collected T. clavata samples was used for phylogenetic analysis (see Material S2 table for accession numbers). The gene regions chosen for concatenation were selected based on visual inspection of the chromatograms.
All observed polymorphisms in T. clavata were bimodal. Upon visual inspection of polymorphisms, double peaks corresponding to the alternative allele were found for all loci, a result that could be indicative of double infection. CLSC, the one sample from South Carolina, was unique among the T. clavata samples with no observed double peaks at polymorphic sites and an identical genotype to T. clavata from China (Yang et al., 2021) for three of the four MLST loci (coxA, gatB, hcpA) used in this study (Fig. 3). The nearest match allele profiles, based on the Wolbachia MLST database, for CLSC were unique among the sampled T. clavata for all loci except hcpA. All other samples shared nearest match allele profiles with other sampled T. clavata for three out of the four MLST loci.
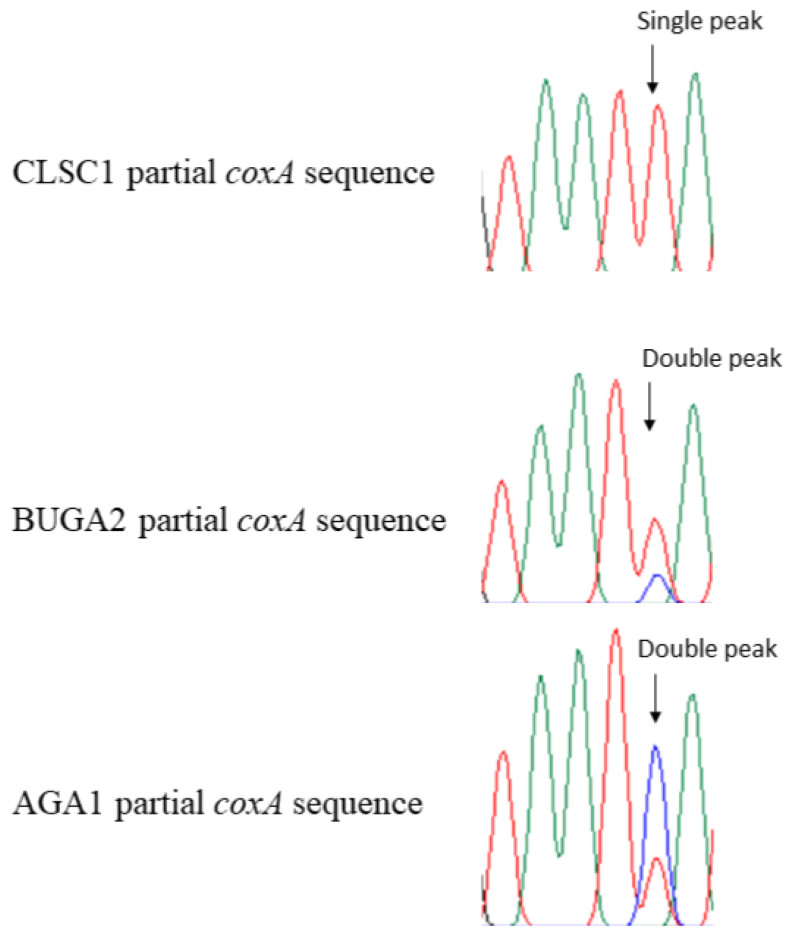
Figure 3: Partial chromatogram results from the Wolbachia coxA gene region of Trichonephila clavata.
Examples of distinguishing features for South Carolina (CLSC1) and Georgia (BUGA2 and AGA1) Wolbachia MLST sequence results are shown in partial coxA chromatograms above. Polymorphic Wolbachia MLST sequence sites were represented by single peaks in the South Carolina sample, and double peaks in Georgia samples.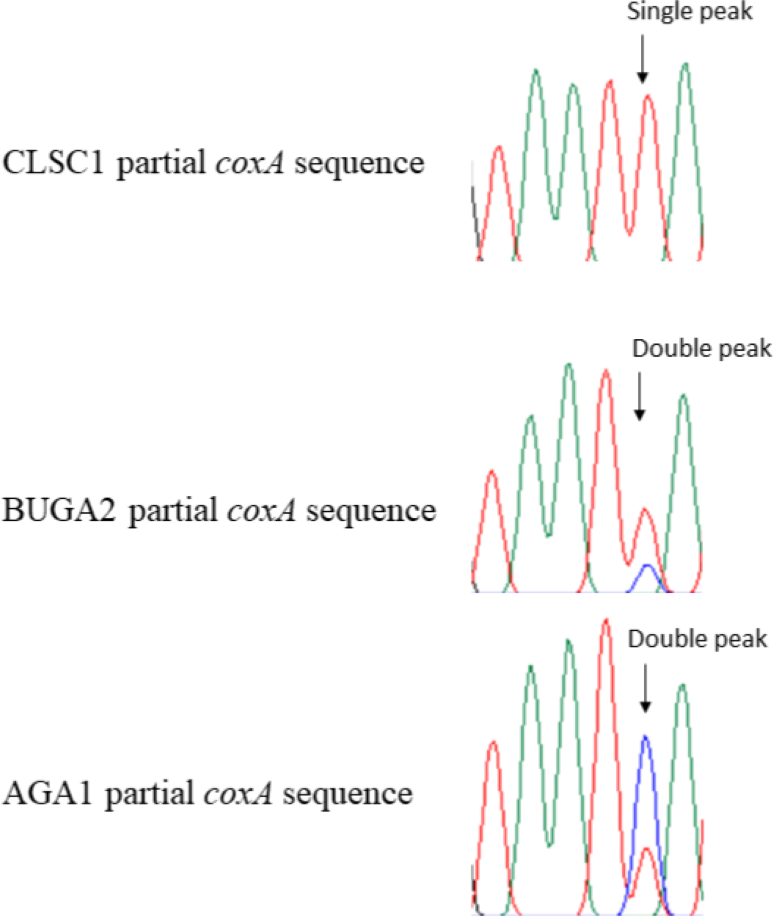{kind=link}
Phylogenetic analysis of the concatenated Wolbachia MLST loci, with Brugia mayali serving as an outgroup, returned two deeply branched clades, which can be attributed to Wolbachia supergroups A and B (Fig. 4). All sampled T. clavata are recovered in the Wolbachia A supergroup. The monophyletic Georgia T. clavata Wolbachia clade with Wolbachia from the spider species Agelenopsis aperta as sister, was phylogenetically distinct from the clade containing the South Carolina sample, CLSC (posterior probability 1.0). The South Carolina MLST profile was sister to T. clavata from Yang et al. (2021) and shared a clade with Wolbachia MLST sequences derived from the parasitic wasp Nasonia giraulti. Maximum likelihood analysis returned generally similar results regarding the disjunct topology of the US T. clavata Wolbachia with a polytomy that included a Georgia clade (75% bootstrap support) and a poorly supported clade (59%) that included South Carolina and T. clavata from Yang et al. (2021) (Material S3).
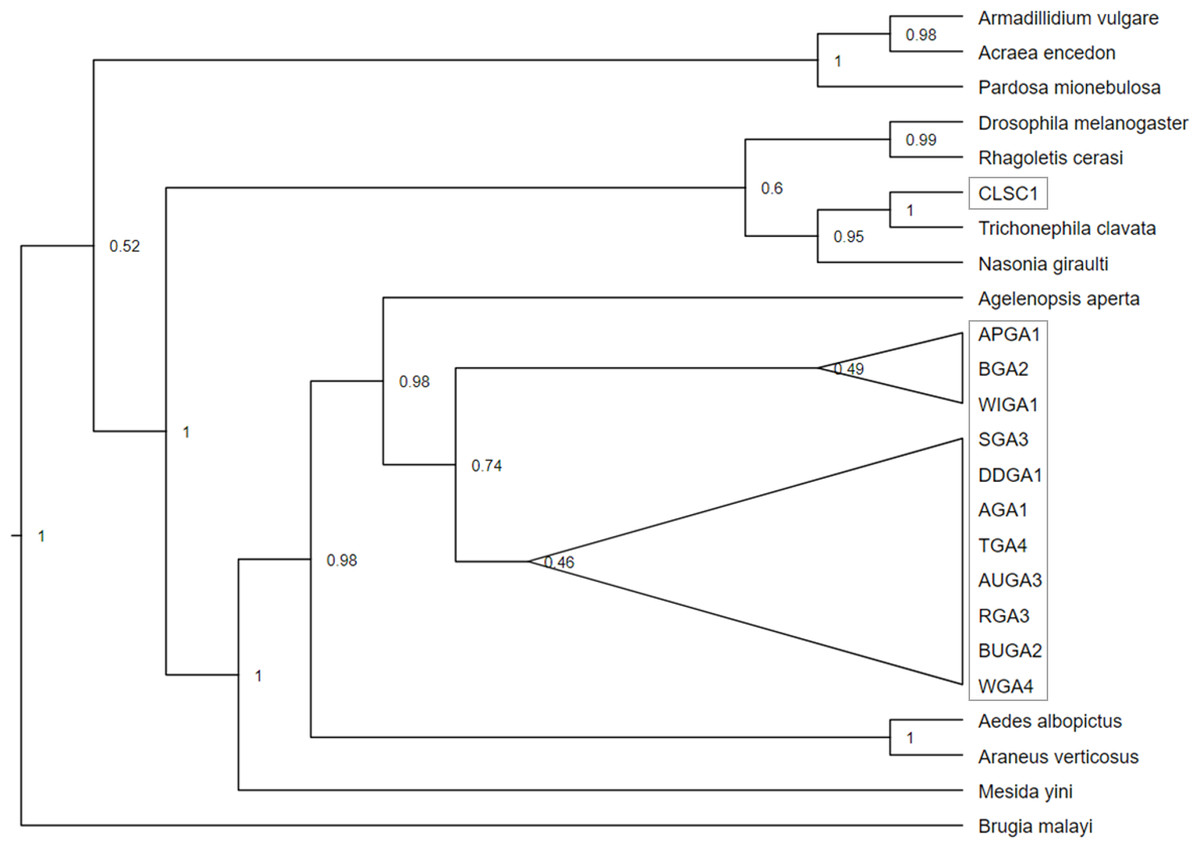
Figure 4: Cladogram of a concatenation of Trichonephila clavata Wolbachia MLST gene regions.
Bayesian inference phylogenetic tree for concatenated Wolbachia gene regions coxA, gatG, fbpA, and hcpA of Trichonephila clavata obtained in Georgia (GA) and South Carolina (SC), indicated with grey outline. Reference samples indicated by genus species were obtained from the Wolbachia MLST database. Brugia mayali was used as an outgroup to root the tree.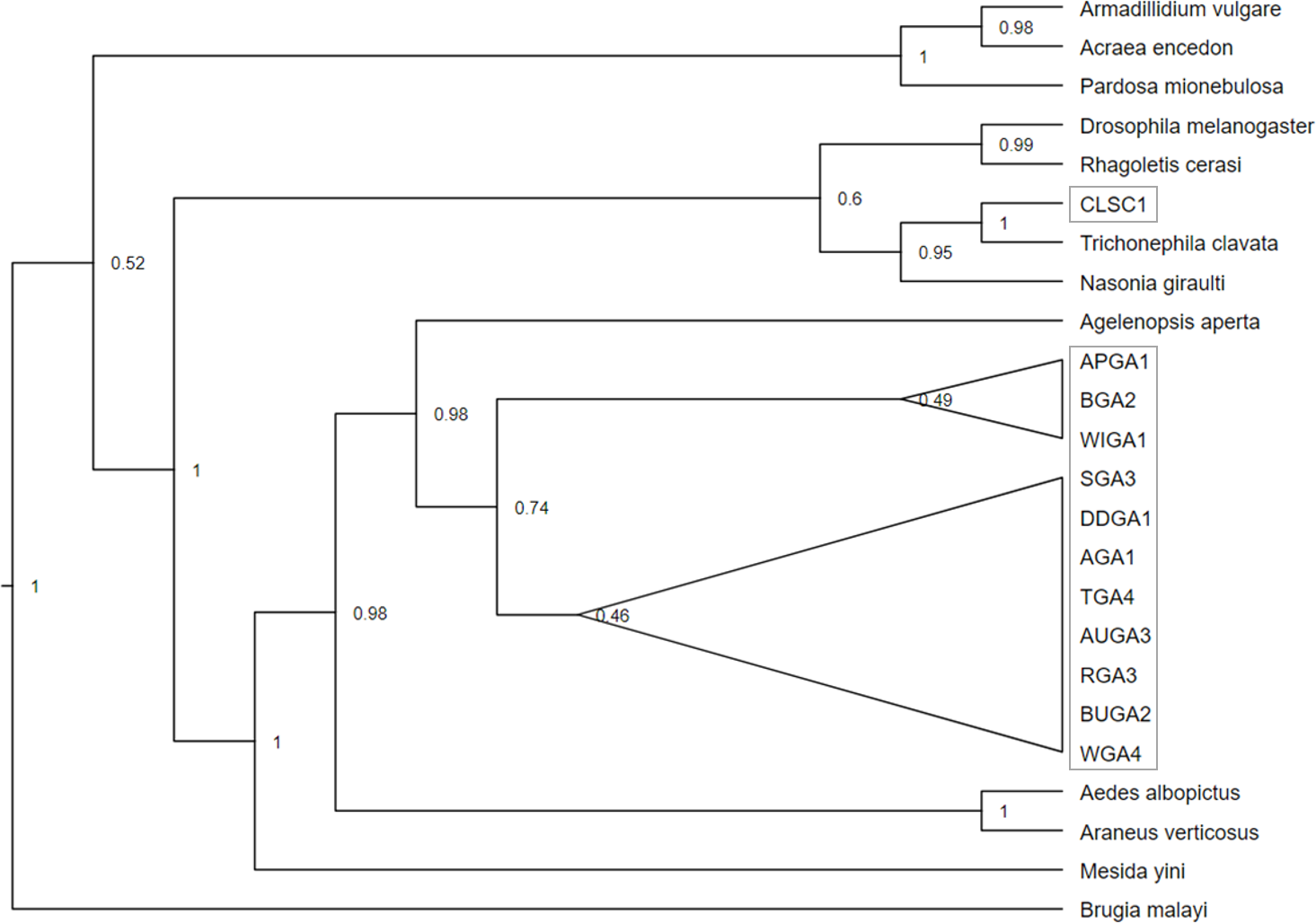{kind=link}
Discussion
Phylogenetic analysis of T. clavata from the US (focused on Georgia and South Carolina) reveals contrasting patterns of cytoplasmic diversity for the mitochondrial (COI) and Wolbachia MLST loci (coxA, fbpA, gatB, and hcpA) (Figs. 2 and 4). Like the previous limited analysis by Hoebeke, Huffmaster & Freeman (2015), which was primarily used to confirm the species status of the recently discovered spider in the US, no mitochondrial genetic diversity was observed in the larger sample size of the present study. These findings suggest a population bottleneck, likely due to founder effects associated with the introduction of T. clavata in the US. Subsequent introduction events in the same sample collection range would appear to have come from the same mitochondrial population of the originally-introduced T. clavata.
Yunnan 1 T. clavata represented the only identical non-US COI sequence that grouped with the USA samples. The closely related Guangdong sample shared sequence similarity with one nucleotide polymorphism (A/G) at position 527 in the alignment. In addition to the polymorphisms mentioned in Hoebeke, Huffmaster & Freeman (2015), an additional polymorphic site among the COI sequences was observed that distinguished the Yunnan 1/US clade from the other Asian sequences: a C/T polymorphism 339 bp further along the alignment from the previously cited polymorphisms in which the US/Yunnan 1/Guangdong T. clavata share a cytosine at that position. Though our results are limited by sample size, the observed mitochondrial haplotype match suggests the founding US population is more closely related to T. clavata previously sampled from Yunnan than other samples from Asia used in the current dataset (Fig. 2). It should be noted that the clade representing the T. clavata outgroup contains sample Yunnan 2. Yunnan is the most southwest province in China, bordering Myanmar, Laos, and Vietnam, and represents one of the westernmost extents of the native range of T. clavata, excluding the Himalayas. The observed phylogenetic discordance of COI within this province, and that observed for COI samples from Zhejiang province, may simply result from limited sample size (only two samples from each region) or an indication of mtDNA diversity within the regions. Regional and population-level analyses of T. clavata in its native range are necessary to address questions of genetic diversity and the potential source population for the US founding of T. clavata.
The phylogenetic bifurcation observed for the concatenated Wolbachia MLST T. clavata sequences from the US was unexpected, given the uniform COI sequence identities of the samples (Fig. 4). The single South Carolina sample (CLSC1) grouped with T. clavata Wolbachia, which was collected from Guangdong Province (also represented in the COI tree as Guangdong) and was recovered in a clade that included Drosophila melanogaster, Rhagoletis cerasi, and Nasonia giraulti MLST sequences. All Georgia T. clavata MLST sequences formed a clade sister to Agelenopsis aperta and were recovered in a larger grouping of other spider species, Mesida yini and Araneus ventricosus, as well as the mosquito Aedes albopictus. The apparent phylogenetic discordance of the MLST data from the US suggests T. clavata was introduced in the US more than once, with South Carolina and Georgia representing distinct Wolbachia populations. The sequence similarities of the Guangdong MLST data to the South Carolina sample and the exact COI sequence match for Yunnan 1 and all US samples may be the result of a source population in southern China, with a relatively uniform mitochondrial background (one COI nucleotide polymorphism) and distinctly different Wolbachia populations. Previous empirical and theoretical analyses have established a selective sweep effect by Wolbachia on mitochondrial genetic variation, whereby single or repeated waves of Wolbachia infection reduce mitochondrial genetic variation (Turelli, Hoffmann & McKechnie, 1992; Kriesner et al., 2013). Wolbachia selective sweeps have also been associated with accelerated rates of molecular evolution for both mitochondria and Wolbachia (Baldo et al., 2010; Russell, Saum & Williams, 2022; Schulenburg et al., 2000), potentially complicating conclusions drawn from phylogenetic analyses (Hurst & Jiggins, 2005).
Mitochondrial-Wolbachia phylogenetic discordance is common among spiders and other arthropods (Yang et al., 2021; Baldo et al., 2008; Wendt et al., 2022; Russell, Saum & Williams, 2022). Previous studies of Wolbachia infection among spiders in China have yielded conflicting phylogenetic placement of T. clavata Wolbachia, with some studies placing T. clavata Wolbachia in the B supergroup (Wang et al., 2010) and others in the A supergroup (Yang et al., 2021). Incongruent phylogenies among species have been explained as a result of horizontal transfer between species (Baldo et al., 2008; Rowley, Raven & McGraw, 2004), which appears to be a common inference for incongruent Wolbachia phylogenies (Boyle et al., 1993; Heath et al., 1999; Vavre et al., 1999; Baldo et al., 2008). Discordant mitochondrial-Wolbachia phylogenies, associated with a common mitochondrial genetic background and divergent Wolbachia infections, were observed in the spider Agelenopsis aperta (Baldo et al., 2008). Whether the divergent South Carolina Wolbachia strain observed is a subset of a double-infected Georgia strain or is a unique strain itself, horizontal transmission of Wolbachia within distinct source populations of T. clavata could explain the phylogenetic discordance observed in the present study.
Amplification of the South Carolina sample and analysis of the chromatogram results showed no double peaks characteristic of double infection. In contrast, the Georgia samples all showed double peaks at specific locations corresponding to A and B supergroup reference samples (Fig. 3). We cannot discount the possibility that the MLST alignments used, particularly the Georgia samples, may be compromised by a mix of A and B supergroup sequences. Regardless, the available data suggests a double infection for the Georgia samples and a single infection for the South Carolina sample. Double infection status is also supported by the observation of successful amplification of A- and B-specific wsp sequences. Therefore, without molecular cloning, it was impossible to eliminate the possibility of double-infected T. clavata samples from Georgia returning MLST sequences that are not some mosaic of A- and B-supergroup Wolbachia. Further, we cannot assume that the South Carolina A-supergroup Wolbachia sequence is not identical to an A-supergroup Wolbachia sequence in a double-infected Georgia population. However, the status of a single-infected South Carolina population and a double-infected Georgia population appears to distinguish the two populations.
Conclusions
The introduction of T. clavata to the US, first reported in the state of Georgia (Hoebeke, Huffmaster & Freeman, 2015), has resulted in a spreading, established population. Our investigation found the mitochondrial genetic structure of the since-established population in Georgia to be monomorphic for the COI gene region, the same gene region sequence used to first identify the species in the US. Like T. clavata in native potential source populations in Asia, the Georgia population was found to be infected with Wolbachia. Phylogenetic constructions using COI and Wolbachia MLST gene regions found mitochondrial-Wolbachia discordance associated with a divergent Wolbachia strain from a single sample collected in South Carolina. The presence of evidence for super-infection among Georgia T. clavata and a single infection in the South Carolina sample supports the contention that distinct Wolbachia populations are present within a uniform mitochondrial background in the area sampled. Future research should expand the sample area within the US and beyond to include the native ranges in Asia. A more comprehensive sampling protocol would provide valuable population genetic information that could help identify potential source populations for the introduced US population(s) and provide context for the cytoplasmic population structure observed in the introduced population.
Supplemental Information
Maximum likelihood phylogenetic tree of T. clavata COI gene region
Acquired COI and Wolbachia accession numbers
Wolbachia multi-locus sequence typing (MLST) loci and the associated accession numbers are shown below.