Biases in genome reconstruction from metagenomic data
- Published
- Accepted
- Subject Areas
- Bioinformatics, Genomics, Microbiology
- Keywords
- genome reconstruction, binning, genomes from metagenomes, metagenomics
- Copyright
- © 2017 Nelson et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Preprints) and either DOI or URL of the article must be cited.
- Cite this article
- 2017. Biases in genome reconstruction from metagenomic data. PeerJ Preprints 5:e2953v1 https://doi.org/10.7287/peerj.preprints.2953v1
Abstract
Background: Technological advances in sequencing, assembly and segregation of resulting contigs into species-specific bins has enabled the reconstruction of individual genomes from environmental metagenomic data sets. Though a powerful technique, it is shadowed by an inability to truly determine whether assembly and binning techniques are accurate, specific, and sensitive due to a lack of complete reference genome sequences against which to check the data. Errors in genome reconstruction, such as missing or mis-attributed activities, can have a detrimental effect on downstream metabolic and ecological modeling, and thus it is important to assess the accuracy of the process.
Methods: We compared genomes reconstructed from metagenomic data to complete genome sequences of 10 organisms isolated from the same community to identify regions not captured by typical binning techniques. The nucleotide content, as %G+C and tetranucleotide frequencies, and sequence redundancy within both the genome and across the metagenome were determined for both the captured and uncaptured regions. This direct comparison allowed us to evaluate the efficacy of nucleotide composition and coverage profiles as elements of binning protocols and look for biases in sequence characteristics and gene content in regions missing from the reconstructions.
Results: We found that repeated sequences were frequently missed in the reconstruction process as were short sequences with variant nucleotide composition. Genes encoded on the missing regions were strongly biased towards ribosomal RNAs, transfer RNAs, mobile element functions and genes of unknown function.
Conclusions: Our observation of increased mis-binning of short regions, especially those with variant nucleotide content, and repeated regions implies that factors which affect assembly efficiency also impact binning accuracy. To a large extent, mis-binned regions appear to derive from mobile elements. Our results support genome reconstruction as a robust process, and suggest that reconstructions determined to be >90% complete are likely to effectively represent organismal function.
Author Comment
This is a submission to PeerJ for review.
Supplemental Information
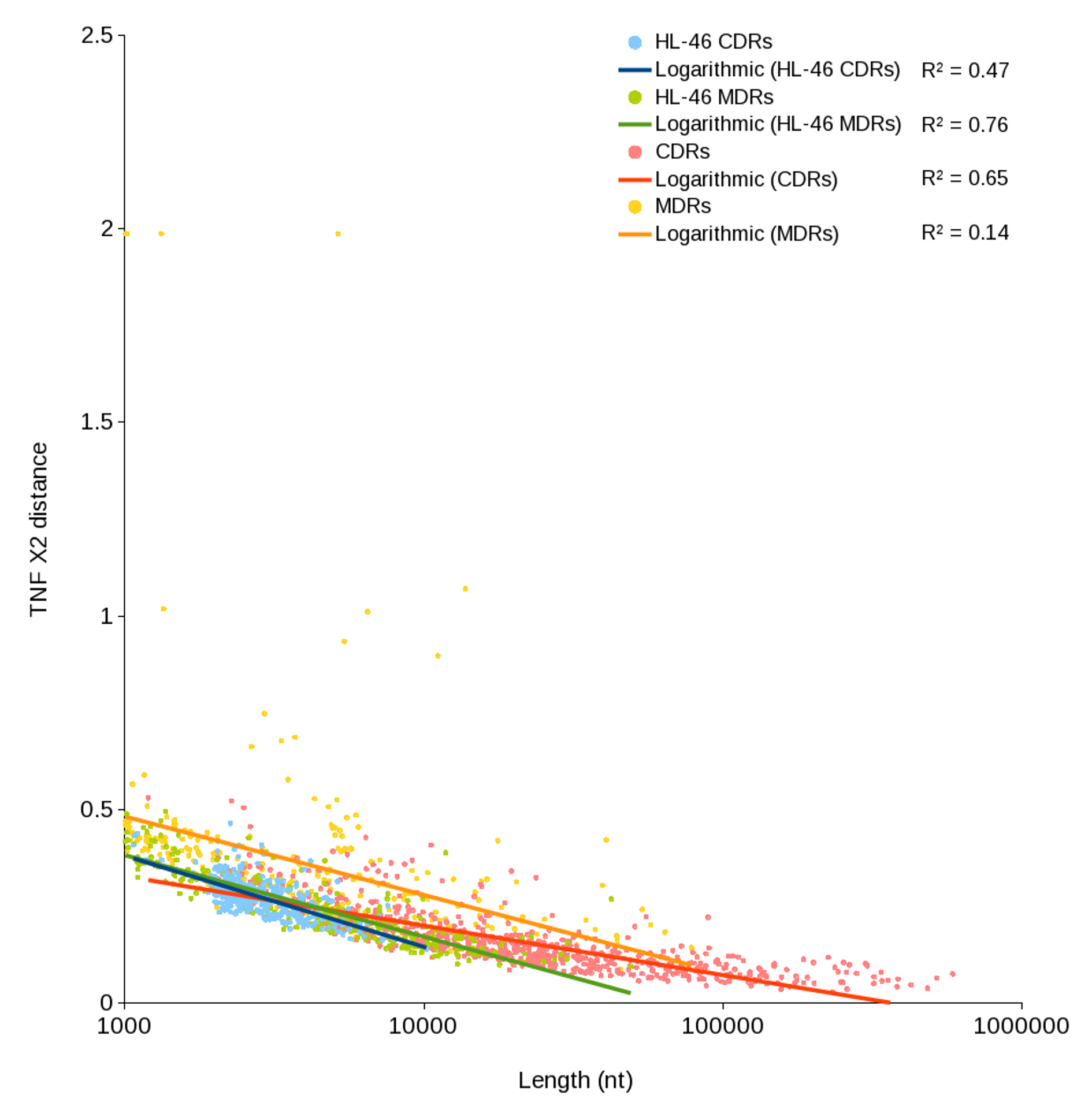
Compositional variance as a function of CDR/MDR length
Tetranucleotide frequency χ2 values were calculated for each CDR and MDR as described in the Methods and plotted against the length of each region. HL-46 serves as a control; having less than half of its genome represented in the reconstruction, CDRs (blue) and MDRs (green) should be indistinguishable. A general negative relationship is observed, however, experimental CDRs (red) show a strong relationship, whereas MDRs (yellow) are poorly explained by this model, suggesting variant sequence comprises a greater proportion of MDRs.
{kind=link}
Analysis summaries
Representation of summarized data for all genomes. Data depiction is as described for Figure 2.
{kind=link}