Transcriptome profiling by RNA-Seq reveals differentially expressed genes related to fruit development and ripening characteristics in strawberry (Fragaria × ananassa)
A peer-reviewed article of this Preprint also exists.
Author and article information
Abstract
Strawberry (Fragaria × ananassa) is an ideal plant for fruit development and ripening research due to the rapid substantial changes in fruit color, aroma, taste and softening. To gain deeper insights into the genes that play a central regulatory role in strawberry fruit development and ripening characteristics, transcriptome profiling was performed for the large green fruit, white fruit, turning fruit, and red fruit stages of strawberry. A total of 6,608 differentially expressed genes (DEGs) with 2,643 up-regulated and 3,965 down-regulated genes were identified in the fruit development and ripening process. The DEGs related to fruit flavonoid biosynthesis, starch and sucrose biosynthesis, the citrate cycle, and cell-wall modification enzymes played important roles in the fruit development and ripening process. Particularly, some candidate genes related to the ubiquitin mediated proteolysis pathway and MADS-box were confirmed to be involved in fruit development and ripening according to their possible regulatory functions. Five ubiquitin-conjugating enzymes and ten MADS-box transcription factors were differentially expressed between the four fruit ripening stages. The expression levels of DEGs relating to color, aroma, taste, and softening of fruit were confirmed by quantitative real-time polymerase chain reaction. Our study provides important insights into the complicated regulatory mechanism underlying the fruit ripening characteristics in Fragaria × ananassa.
Cite this as
2018. Transcriptome profiling by RNA-Seq reveals differentially expressed genes related to fruit development and ripening characteristics in strawberry (Fragaria × ananassa) PeerJ Preprints 6:e26870v1 https://doi.org/10.7287/peerj.preprints.26870v1Author comment
This is a submission to PeerJ for review.
Sections
Supplemental Information
URLs, annotation methods and parameters of seven databases
The comprehensive information of gene function comes from seven databases.
The information of software version and parameter
Each data indicate the information of software that produces all the transcriptome data.
The distribution of FPKM values of each library
FPKM: fragments per kilobaseof exon per million fragments mapped. FPKM is the most commonly used method of estimating gene expression level, which eliminates the expression level of technical deviation.
Primers used in this study
Each data indicates the detail information of candidate genes in quantitative real-time polymerase chain reaction.
The annotation results of KOG classification
Each data indicates the annotation result in seven databases.
The GO classification of unigenes
Each data indicates the classification of unigenes in GO database.
The KOG classification of unigenes
Each data indicates the classification of unigenes in KOG database.
The KEGG classification of unigenes
Each data indicates the classification of unigenes in KEGG database.
Differential analysis results of genes in different combinations
Each data is used to determine the differentially expressed genes (DEGs). The DEGs with padj < 0.05 and log2 (fold change) ≥ 1 are up-regulated, and those with padj < 0.05 and log2 (fold change) ≤ -1 are down-regulated. The other genes that do not meet the conditions of padj < 0.05 and log2 (fold change)| ≥ 1 are not DEGs.
Detailed information of genes in the flavonoid biosynthesis pathway
Each data indicates the average read_count of genes in each library.
Detailed information of genes in results
Each data indicates the corrected read_count value, differential analysis results and annotation information of genes in each library.
FPKM interval of all samples
FPKM: fragments per kilobaseof exon per million fragments mapped. The percentage of each sample's corresponding FPKM interval can be used to measure the difference in expression between samples.
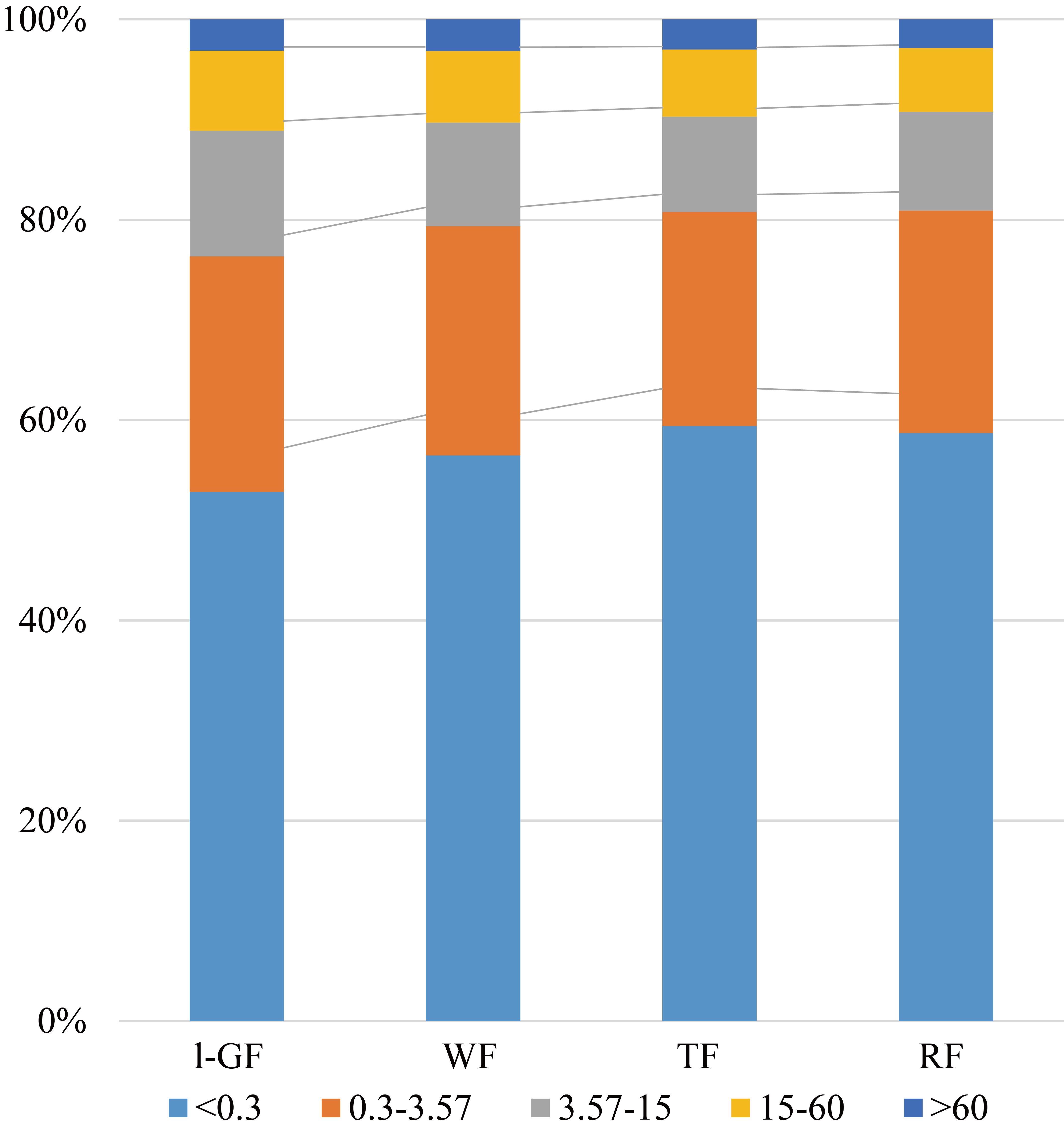{kind=link}
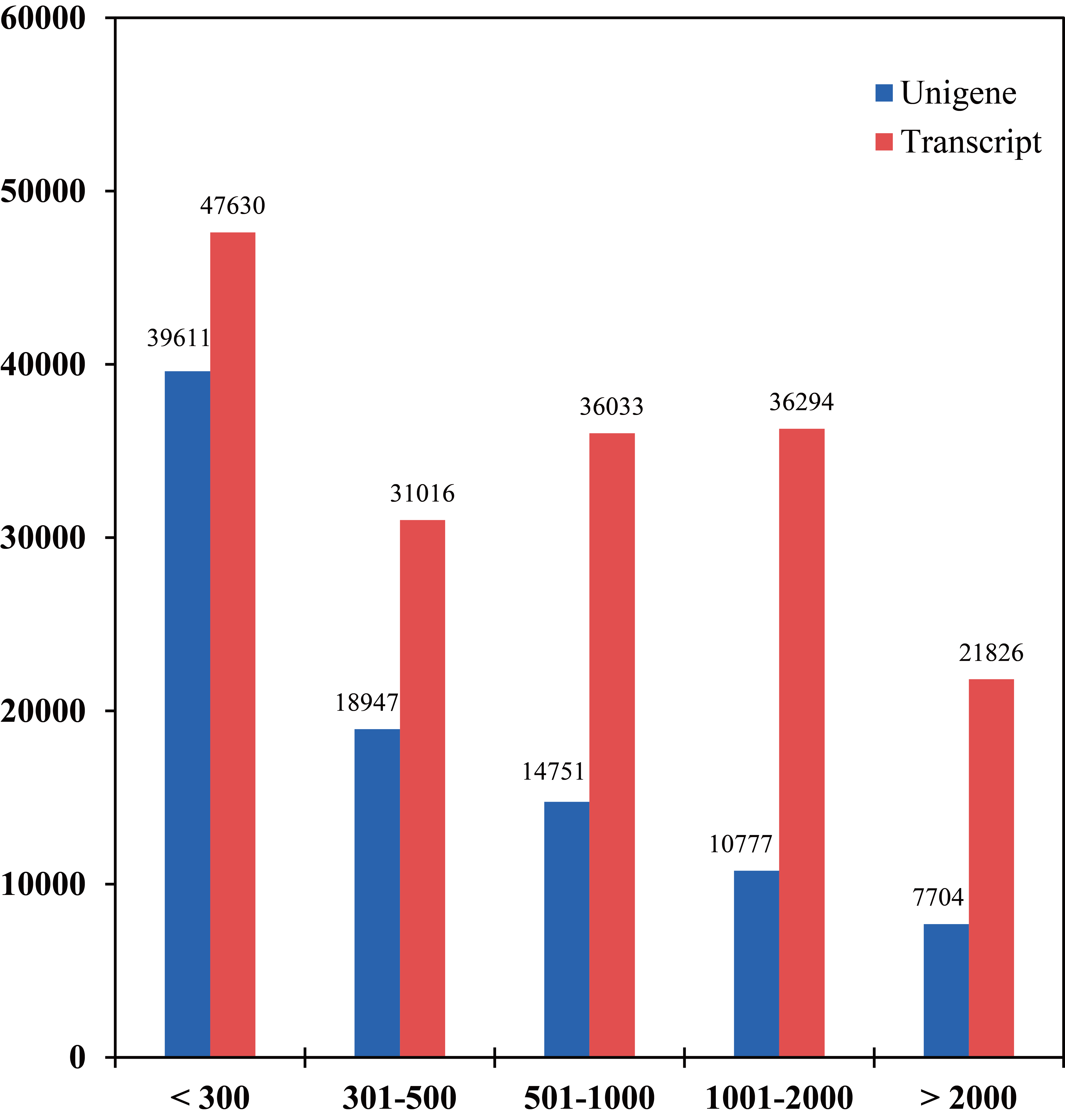
Length distribution of transcripts and unigenes
The x-axis represents the length interval of transcript/unigene, and the y-axis represents the number of times for each length of the transcript/unigene.
{kind=link}
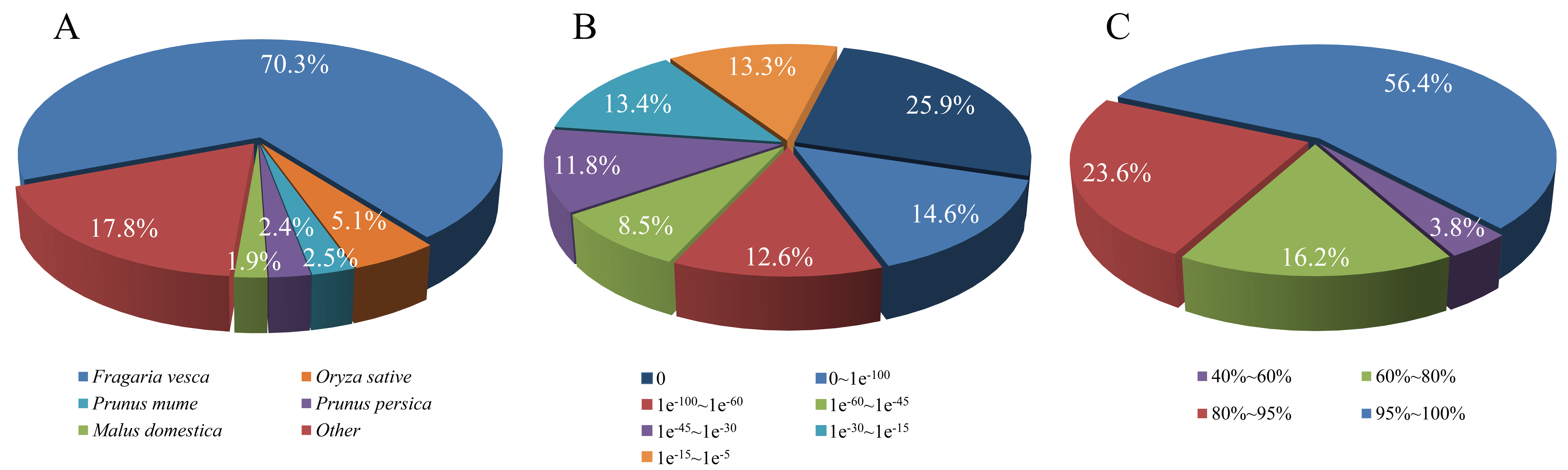
Characteristics of homology search of Illumina sequences against the Nr database
(A) Percentage of the total homologous sequences of 5 top species against the Nr database; (B) E-value distribution of the top BLASTx hits against the Nr database; (C) Similarity distribution of the top BLASTx hits for each sequence.
{kind=link}
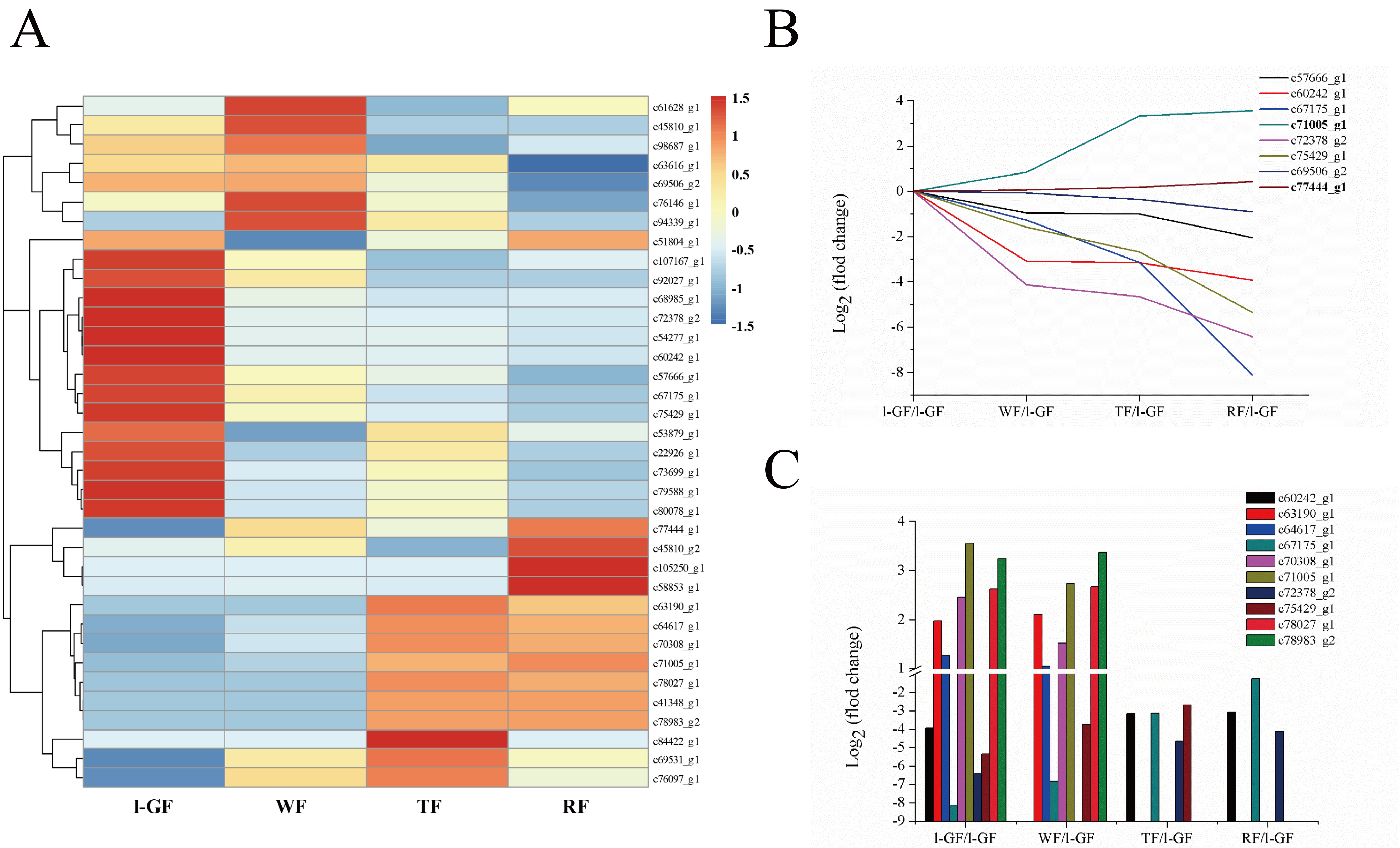
Expression pattern of genes in the flavonoid biosynthetic pathway
(A) Cluster analysis of genes in flavonoid biosynthetic pathway. Expression level was showed by different colors, the redder the higher expression and the bluer the lower. The values of red to blue is log10 (read_count). (B) The relative expression of up- and down-regulated genes in flavonoid biosynthetic pathway. (C) The expression pattern of DEGs in flavonoid biosynthetic pathway.
{kind=link}
Expression pattern of MYB and bHLH transcription factors
(A/C) The relative expression of up- and down-regulated MYB and bHLH transcription factors. Black Fonts indicate the up-regulated gene ID. (B/D) The expression pattern of DEGs of MYB and bHLH transcription factors.
{kind=link}
The expression level of candidate genes in transcriptome data
Each data indicates the expression pattern of candidate genes with strawberry ripening in transcriptome data.
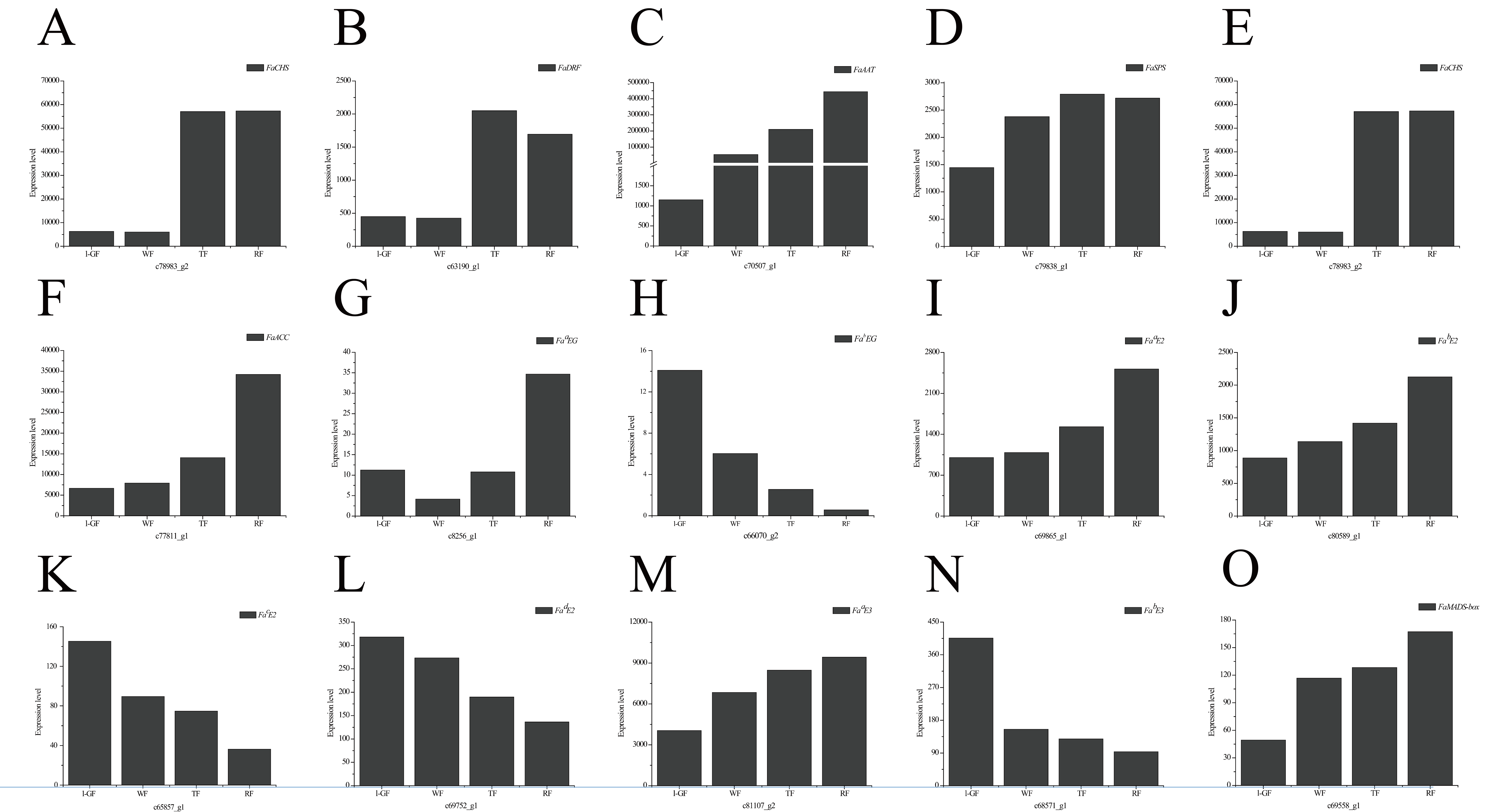{kind=link}
Expression pattern of genes in starch and sucrose biosynthesis and citrate cycle
(A/B) The relative expression of up- and down-regulated genes in starch and sucrose biosynthesis. (C-E) The expression pattern of DEGs in starch and sucrose biosynthesis. (F) The relative expression of up- and down-regulated genes in citrate cycle. (G) The expression pattern of DEGs in citrate cycle.
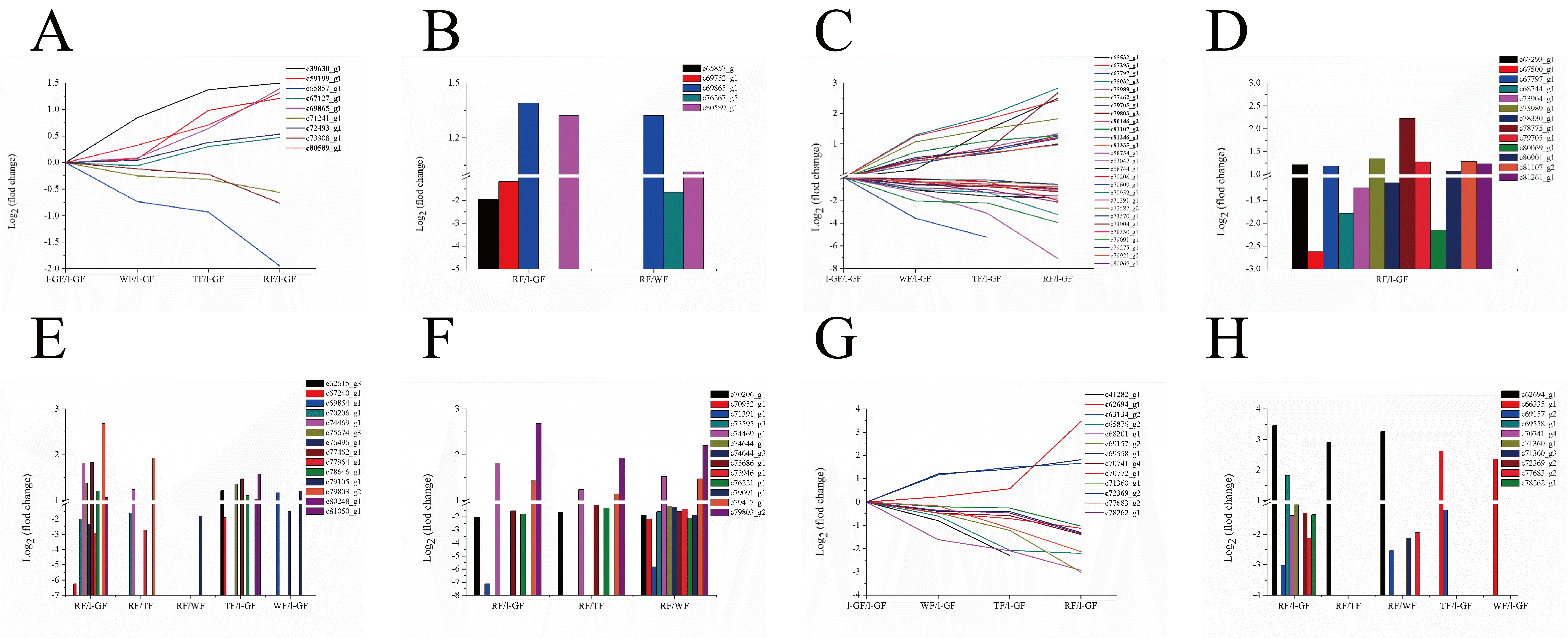{kind=link}
The assembled unigene transcriptome data of unigene sequences
This file showed the sequences of unigenes in this text.
Additional Information
Competing Interests
The authors declare that they have no competing interests.
Author Contributions
Panpan Hu performed the experiments, analyzed the data, prepared figures and/or tables, approved the final draft.
Gang Li contributed reagents/materials/analysis tools, approved the final draft.
Xia Zhao contributed reagents/materials/analysis tools, approved the final draft.
Fengli Zhao approved the final draft, manage and provide the materials.
Liangjie Li approved the final draft, manage and provide the materials.
Houcheng Zhou conceived and designed the experiments, authored or reviewed drafts of the paper, approved the final draft.
Data Deposition
The following information was supplied regarding data availability:
Funding
The research was supported by the Central Public-interest Scientific Institution Basal Research Fund (1610192016608, 1612382017204), the Natural Science Foundation of Henan Province (162300410329), and the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2017-ZFRI). There was no additional external funding received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.