
Conservation of transcriptional elements in the obligate symbiont of the whitefly Bemisia tabaci
Author and article information
Abstract
Background
Bacterial symbiosis is widespread in arthropods, especially in insects. Some of the symbionts undergo a long-term co-evolution with the host, resulting in massive genome decay. One particular consequence of genome decay is thought to be the elimination of transcriptional elements within both the coding region and intergenic sequences. In the whitefly Bemisia tabaci species complex, the obligate symbiont Candidatus Portiera aleyrodidarum is of vital importance in nutrient provision, and yet little is known about the regulatory capacities of it.
Methods
Portiera genomes of two whitefly species in China were sequenced and assembled. Gene content of these two Portiera genomes was predicted, and then subjected to Kyoto Encyclopedia of Genes and Genomes pathway analysis. Together with two other Portiera genomes from whitefly species available previously, four Portiera genomes were utilized to investigate regulatory capacities of Portiera, focusing on transcriptional elements, including genes related with transcription and functional elements within the intergenic spacers.
Results
Comparative analyses of the four Portiera genomes of whitefly B. tabaci indicate that the obligate symbionts Portiera is similar in different species of whiteflies, in terms of general genome features and possible functions in the biosynthesis of essential amino acids. The screening of transcriptional factors suggests compromised ability of Portiera to regulate the essential amino acid biosynthesis pathways. Meanwhile, thermal tolerance ability of Portiera is indicated with the detection of a σ32 factor, as well as two predicted σ32 binding sites. Within intergenic spacers, functional elements are predicted, including 37 Shine-Dalgarno sequences and 34 putative small RNAs.
Cite this as
2019. Conservation of transcriptional elements in the obligate symbiont of the whitefly Bemisia tabaci. PeerJ 7:e7477 https://doi.org/10.7717/peerj.7477Main article text
Introduction
Over the last two decades, multiple functions of microbiota have been investigated, from nutrition provision to reproduction (Stouthamer, Breeuwer & Hurst, 1999; Moran, McCutcheon & Nakabachi, 2008; Akman & Douglas, 2009). During the long-term associations, microbes have gone through genome decay and gene loss, resulting in tiny genomes (Moran, McCutcheon & Nakabachi, 2008). On one hand, protein-coding regions of these tiny genomes, which are easily accessible, have received broad attention. Considerable efforts have been made to elucidate the mechanisms underpinning the symbiosis by investigating protein-coding regions, and have yielded insights into the dynamics and functions of symbionts within the host (Zientz, Dandekar & Gross, 2004). On the other hand, intergenic spacers (IGS) between protein-coding regions comprising of non-coding regions receive little attention. This situation has improved recently due to high-throughput sequencing method, which has allowed us to identify functional elements involved in gene regulation within IGSs (Degnan, Ochman & Moran, 2011; Hansen & Degnan, 2014). Questions concerning why microbiota choose to retain some regulatory elements in IGSs (Moran, Dunbar & Wilcox, 2005), are of particular interest.
The whitefly Bemisia tabaci is a genetically diverse group including multiple morphologically indistinguishable cryptic species (De Barro et al., 2011). Among this species complex, two invasive species, Middle East-Asia Minor 1 (MEAM1, also known as B whitefly) and Mediterranean (MED, also known as Q whitefly), can cause severe damages to crops and plants around the world (Liu et al., 2007). There are also indigenous whitefly species, for instance, Asia II 3 and China 1 in China, causing less harm (Hu et al., 2011). The symbionts in whitefly are of vital importance, contributing to nutrient provision (Calle-Espinosa et al., 2016; Santos-Garcia et al., 2018) and host fitness (Shan et al., 2017; Lv et al., 2018). Portiera, as the obligate symbiont, is uniformly harbored by all species of whiteflies (Thao & Baumann, 2004). Studies on Portiera from the two invasive species of whiteflies MEAM1 and MED, have indicated that this bacterium mainly retains essential amino acid (EAA) biosynthesis genes (Santos-Garcia et al., 2012; Sloan & Moran, 2012; Jiang et al., 2013). Besides, sequenced Portiera genomes contain around 30% IGSs, which is much higher than that of other obligate endosymbionts from insects (Shigenobu et al., 2000; Nakabachi et al., 2006). With such a largely reduced gene set and a high proportion of IGSs, compelling questions focusing on the regulatory capacities of Portiera arise. However, research on this issue has been restrained by various factors such as: (i) IGSs contain functional elements such as sRNAs and nonfunctional regions like decaying pseudogenes fragment, which limits their recognizability; (ii) Portiera in bacteriocytes is clustered with other secondary symbionts (Rao et al., 2015) and is hard to be isolated from insect tissues; (iii) Portiera cannot be cultured in vitro.
In this study, in order to obtain information about overall regulatory capacities in Portiera genomes, we first sequenced the Portiera genomes from two indigenous whitefly species (Asia II 3 and China 1). We then analyzed the gene sets of these two Portiera genomes, together with two other Portiera genomes derived previously from two other species of whiteflies, MEAM1 and MED, of the B. tabaci complex. By comparing four Portiera genomes, we: (i) screened the transcription-related genes by using OrthoMCL and BLASTP; (ii) demonstrated two σ32 binding sites experimentally; (iii) identified the functional elements located in IGSs using a phylogenetic foot-printing approach.
Materials & Methods
Genome resources
Two Portiera genomes from two indigenous whitefly species (B. tabaci Asia II 3 and China 1) were sequenced and assembled according to a previously published protocol (Zhu et al., 2017). Genome sequences of Candidatus Portiera aleyrodidarum from B. tabaci Asia II 3 and China 1 were submitted to the GenBank databases under accession numbers CP016327 and CP016343. Another three Portiera genome sequences (Candidatus Portiera aleyrodidarum BT-B, Candidatus Portiera aleyrodidarum BT-Q and Candidatus Portiera aleyrodidarum TV) were downloaded from National Center Biotechnology Information (NCBI), and the accession numbers were listed in Table 1. In total, five Portiera genome sequences were analyzed and compared, with Portiera-B abbreviated for Candidatus Portiera aleyrodidarum from B. tabaci MEAM1 species, Portiera-Q for Candidatus Portiera aleyrodidarum of B. tabaci MED, Portiera-Z1 for Candidatus Portiera aleyrodidarum of B. tabaci Asia II 3, Portiera-Z3 for Candidatus Portiera aleyrodidarum of B. tabaci China 1 and Portiera-TV for Candidatus Portiera aleyrodidarum of greenhouse whitefly Trialeurodes vaporariorum.
| Portiera-Z1 | Portiera-Z3 | Portiera-B | Portiera-Q | Porteira-TV | |
|---|---|---|---|---|---|
| Genome size, bp | 354,523 | 359,359 | 358,242 | 357,472 | 280,663 |
| CDS number | 250 | 247 | 256 | 255 | 267 |
| Intergenic spacers, % | 32.9 | 39.4 | 33.5 | 33.5 | 7.7 |
| GC content, % (Protein-coding region) | 26.7 | 26.4 | 26.7 | 26.7 | 24.4 |
| GC content, % (Intergenic spacers) | 24.9 | 25.6 | 25.1 | 24.9 | 28.4 |
| Accession number | CP016327 | CP016343 | CP003708 | CP003835 | CP004358 |
Phylogenetic analysis
Whitefly species phylogeny was determined first by using whitefly mitochondrial cytochrome oxidase I (COI) genes downloaded from NCBI database. The tree was built using MrBayes (v.3.4.2) (Ronquist & Huelsenbeck, 2003), with the best-fitting model (TIM3 + G) generated by jModelTest (Darriba et al., 2012). For Portiera phylogenetic analysis, Candidatus Portiera aleyrodidarum TV served as an out-group member according its host taxonomic system. To obtain the homologous genes among the five Portiera genomes, protein-coding regions were extracted from each genome and subjected to OrthoMCL (Li, Stoeckert & Roos, 2003) with default parameters for orthologs identification. A total of 218 genes were defined as core-genes among the five genomes. The protein sequences of core-genes were concatenated, and aligned using MUSCLE (Edgar, 2004). The phylogeny was conducted by using Mrbayes, with the best-fitting model (cpREV + G + F) determined by ProtTest (Abascal, Zardoya & Posada, 2005).
Recognization of transcription-related genes
To focus on the Portiera from the whitefly B. tabaci only, 227 core-genes were determined by OrthoMCL among the four Portiera genomes from whitefly B. tabaci, and subjected to BLASTP (E-value <10−5) and the Kyoto Encyclopedia of Genes and Genome (KEGG) Automatic Annotation Server tool (Moriya et al., 2007) for functional prediction. Escherichia coli K12 (commonly used as a model organism) (Keseler et al., 2011) served as a reference to predict transcription-related genes (mainly the amino acid biosynthetic pathways) and heat regulon within the protein-coding genes of Portiera genomes. Ka/Ks ratios between different gene pairs of cspA were calculated using DnaSP (v 5.10.1) (Librado & Rozas, 2009). σ32 binding sites in the regulatory region of genes were determined based on consensus sequences of σ32 binding sites in E. coli (CTTGAAA [11–15] CCCCATnT) with the criteria previously described (Wilcox et al., 2003).
MED whitefly heat shock treatment
Insects are ectotherms and they are not able to control body temperatures. Hence, the elevation of environmental temperature will influence the bacterial symbionts within whitefly. In order to investigate possible function of predicted σ32 binding sites, MED whiteflies were subjected to a high temperature of 40 °C for 3 h, 25 °C as control. This high temperature of 40 °C has been shown to cause thermal stress of whiteflies (Wang et al., 2019). Temperature exposure experiments were conducted in climatic chambers (Sanyo, MLR-350H; Sanyo Electric Co., Ltd., Osaka, Japan) which offered precise control of temperatures within ± 0.5 °C of the set value. Newly emerged whiteflies were placed in tubes (2.5 cm × 0.5 cm diam) covered with gauze, and then exposed to 40 °C and 25 °C (control) for 3 h.
DNA (lysis buffer method) (Frohlich et al., 1999) and RNA (TRIzol protocol) (Rio et al., 2010) were extracted from whitefly samples, four biological replicates for DNA and 4 for RNA, with each replicate containing five males and five females. RNA was then reverse transcribed into cDNA using PrimeScript RT reagent Kit (Takara, Japan) after RNase-free DNase I treatment. Both DNA and cDNA samples were then subjected to quantitative PCR (qPCR) according to manufacturer’s instructions. The primers utilized were listed in Table S1. The qPCR results were calculated using the comparative CT method (2−ΔΔCT). The 2−ΔΔCT method normalized the amount of target genes in each sample against whitefly nuclear gene encoding beta-Actin (DNA samples), and gene expression level of target genes against whitefly nuclear gene encoding the ribosomal protein L29 (RPL29) (cDNA samples) (Li et al., 2013). The significance level was determined by using Student’s t-test.
Functional elements searching in intergenic spacers
Orthologous IGSs were defined as sequences flanked by orthologous genes in genomes when comparing different genomes (Degnan, Ochman & Moran, 2011). IGSs from four Portiera genomes (from whitefly B. tabaci) were extracted and clustered in groups based on orthologous relationship. The lengths of paired orthologous IGSs were scattered to investigate the length variations between Portiera genomes from different species to examine their relationship. The orthologous IGSs were then aligned with MUSCLE. Identically aligned loci with more than 20 nucleotides in length were extracted and subjected to further analysis using a variety of means to investigate functional elements retained. Shine-Dalgarno sequences were predicted by RBSfinder (Suzek et al., 2001), with default parameters. Putative small RNAs were selected by using RNAFold (Lorenz et al., 2011), only if the minimum free energy was below 0 kcal/mole.
These transcriptional elements were investigated in another three Portiera genomes available, Portiera-TV and two other Portiera genomes (from Aleurodicus dispersus and Aleurodicus floccissimus, NCBI accession number LN649255 and LN734649). According to the orthologous IGSs containing the transcriptional elements in the Portiera genomes from Bemisia tabaci, sequences of the orthologous IGSs in the other three Portiera genomes (not from whitefly Bemisia tabaci) were extracted, and only sequences with a length over 20 nucleotides were kept. The investigation of transcriptional elements was performed as described previously.
Results and Discussion
The genome of Candidatus Portiera aleyrodidarum BT-Z1 from whitefly Asia II 3 was 354,523 base pairs (bp) and contained 288 protein coding genes. For Candidatus Portiera aleyrodidarum BT-Z3 from whitefly China 1, the genome size was 359,359 bp, with 291 protein-coding genes identified. Both Portiera genomes were AT-rich, with 33 tRNA-coding genes and three rRNA-coding genes (5S, 16S and 23S). Together with Portiera-B from the whitefly MEAM1 and Portiera-Q from the whitefly MED, Portiera genomes in these four species of whiteflies (Bemisia tabaci) showed higher (than Portiera genome from whitefly Trialeurodes vaporariorum) similarity in genome size, coding sequence number and GC content (Table 1).
The divergence and function of Portiera
In order to determine the phylogeny of Portiera in whitefly species, 218 orthologs among five Portiera genomes were generated, including four Portiera genomes from B. tabaci whitefly species and the Portiera genome from the greenhouse whitefly T. vaporariorum (as an out-group member). Sequence similarities between different Portiera pairs were calculated (Table S2). Interestingly, Portiera-B and -Q from invasive whitefly species showed the highest similarity (99.8%), and meanwhile, Portiera-Z1 and Z3 also present strong similarity (99.5%). However, the similarity of Portiera genomes between B. tabaci whitefly and greenhouse whitefly was only about 77%. The classification of Portiera was also evidenced by the phylogenetic tree, where four Portiera genomes from B. tabaci obviously formed two different clades, in coincidence with the classification of whitefly B. tabaci species (Fig. 1). Previous study on the divergence of whiteflies has implied that the divergence of the greenhouse whitefly T. vaporariorum and whitefly B. tabaci is dated back to 85.95 Ma, while the divergence of the species of whitefly B. tabaci is quite recent, approximately 17.73 Ma (Santos-Garcia et al., 2015). We believe that long-term co-existence within respective whitefly species following the divergence of whiteflies, probably contributes to the differences of Portiera genomes. Meanwhile, it is also revealed that although B. tabaci lineage (Portiera genomes from whitefly B. tabaci) is evolving faster than other lineages, the diversity among the B. tabaci lineage is smaller when compared to other lineages (Santos-Garcia et al., 2015). Our results on the similarity (Table S2) and phylogeny (Fig. 1) are in accordance with this finding.
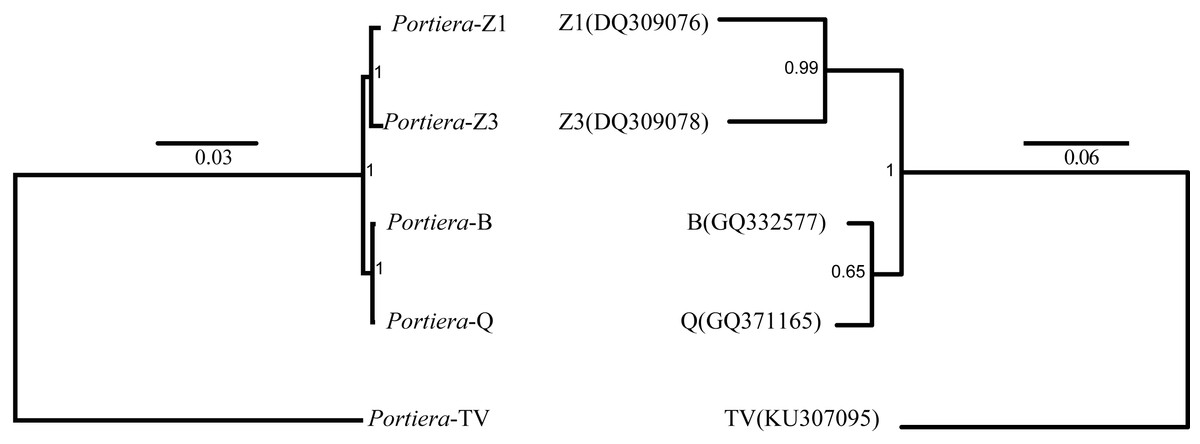
Figure 1: Bayes trees showing the phylogeny of Portiera and whitefly gene mitochondrial cytochrome oxidase I (COI) from five whitefly species.
Portiera analysis was based on 218 core-genes determined by OrthoMCL. Portiera genome and COI gene from Trialeurodes vaporariorum served as out-group members, respectively. Posterior probabilities are given on the nodes.Despite the differences, from the orthologs and KEGG pathway analysis, two Portiera genomes from indigenous whitefly species in this study kept an identical set of genes dedicated to EAA biosynthesis and metabolisms (Fig. 2), indicating that Portiera in indigenous whitefly species may also be responsible for EAA provision. Meanwhile, Portiera-TV and two other Portiera genomes (from Aleurodicus dispersus and Aleurodicus floccissimus, NCBI accession numbers LN649255 and LN734649) are all predicted to contain the ability in amino acid biosynthesis (Santos-Garcia et al., 2015). Our results provided further evidence that Portiera enters into whitefly before the divergence, and is conserved as an essential part of whitefly genome, contributing to EAA provision.
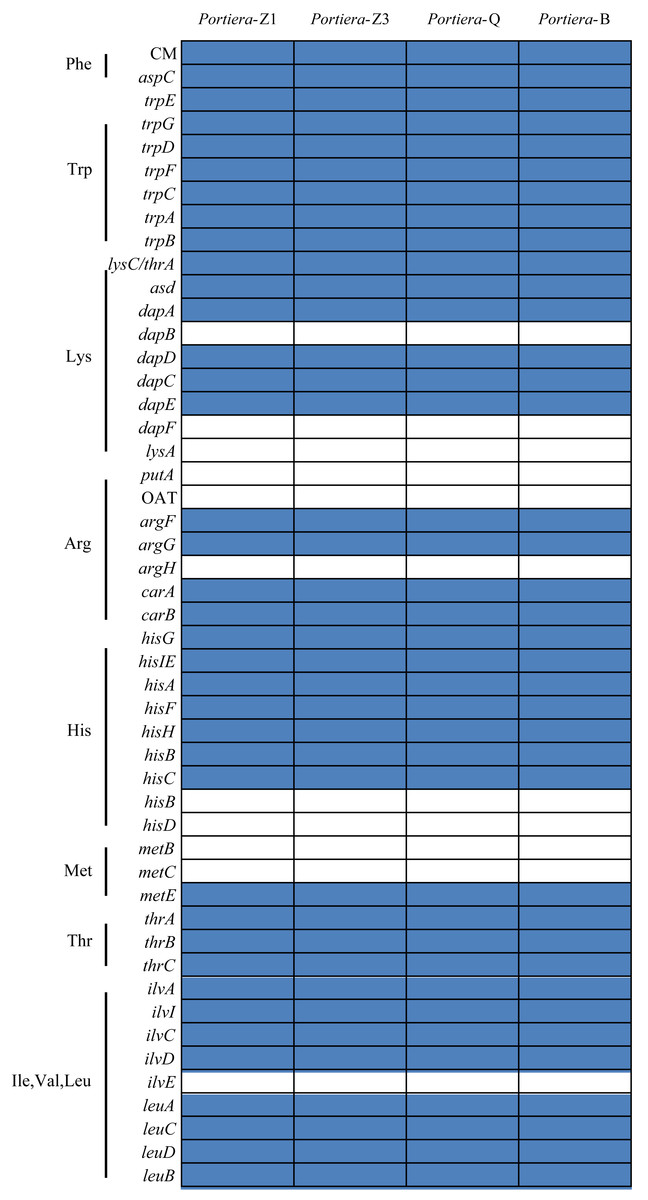
Figure 2: Genes involved in amino acids biosynthesis pathways present in four Portiera genomes.
The blue rectangle indicates that the gene is present in the genome.Genes related with transcription
To generate and compare a putative set of genes globally retained by Portiera from B. tabaci, 227 orthologous genes were determined from four Portiera genomes of whitefly B. tabaci. Regulatory genes related with amino acid biosynthetic pathways were all missing compared to E. coli (Table S3). The only transcription factor found in Portiera was gene cspA, which encodes a major cold-shock protein in E. coli (Schindelin et al., 1994). Besides, purifying selection was found acting on this gene (Table S4).
The lack of EAA regulation genes means a crippled ability of Portiera to regulate the biosynthesis of EAA. On the other hand, it has been pointed out that the genome of whitefly contains several genes that are within EAAs biosynthetic pathways (Luan et al., 2015). Hence, those genes from whitefly genome, other than genes from Portiera genomes, are supposed to be possible candidates that regulate biosynthesis pathways of EAAs.
In addition, some other common regulatory elements were detected. Three genes (nusA, nusG and rho) involved in transcriptional termination were present, indicating that the termination of transcription in Portiera may fall into both intrinsic termination and rho-dependent termination (Nudler & Gottesman, 2002). Core RNA polymerase (α2ββ′) was supposed to be complete, with the presence of rpoA, rpoB, and rpoC, which encodes α, β, and β′ subunit. RNA polymerase (RNAP) is of transcriptional importance in bacteria. Two σ factors functioning in the RNAP recognition of promoter sequences, were also suggested by the Portiera genomes. Along with primary σ factor σ70 (encoded by rpoD), σ32 (encoded by rpoH) was also detected, which is similar to E. coli (Helmann & Chamberlin, 1988) and Buchnera in aphids (Wilcox et al., 2003). Generally, this σ32 factor can compete with σ70, when the bacterium is confronted with heat stress, to redirect RNAP to promoters which contain σ32 binding sites.
Heat shock treatment
The detection of σ32 factor led to a set of 14 genes retained in Portiera as heat shock regulon, according to heat shock regulon of E. coli (Nonaka et al., 2006) (Table 2). These genes are predicted to encode relevant proteins responsible for cellular homeostasis, which indicates that heat shock response is probably partially conserved in Portiera. Meanwhile, promoter loci of the 14 genes were screened, with two possible σ32 binding sites detected (Fig. 3). Intriguingly, the σ32 binding sites were conserved among 4 Portiera genomes in whitefly B. tabaci, with identical −35 and −10 loci. Additionally, in E. coli and Buchnera genomes from aphids, σ32 binding sites are also found before genes grpE and dnaKJ (respectively), suggesting a possible universal mechanism of conserving σ32 recognition sequences before these three genes (Wilcox et al., 2003).
| Genes | Log2FC | FC (MEAN ± S.E.M.) | p-value |
|---|---|---|---|
| groESb | 2.96 | 7.79 ± 1.949 | * |
| dnaKa | 2.66 | 6.31 ± 0.868 | *** |
| ftsH | 2.64 | 6.24 ± 0.809 | *** |
| dnaJa | 2.42 | 5.34 ± 1.174 | * |
| grpEa | 2.26 | 4.79 ± 1.606 | * |
| hslV | 2.04 | 4.12 ± 0.642 | ** |
| lon | 1.74 | 3.33 ± 0.523 | ** |
| ybeY | 1.67 | 3.18 ± 0.773 | * |
| groELb | 1.65 | 3.14 ± 0.347 | ** |
| ileS | 1.55 | 2.92 ± 0.383 | ** |
| rpoH (σ32 factor) | 1.54 | 2.91 ± 0.325 | ** |
| valS | 1.46 | 2.75 ± 0.237 | *** |
| hslU | 1.39 | 2.61 ± 0.393 | ** |
| rpoD | 1.22 | 2.33 ± 0.300 | ** |
| glnS | 0.94 | 1.92 ± 0.331 | * |

Figure 3: σ32 binding sites conserved in seven Portiera genomes.
Whitefly species indicates the origin of the Portiera. ‘Bemisia tabaci’ contains four species of whitefly Bemisia tabaci. The σ32 binding sites were determined based on the consensus sequence of that in Escherichia coli with certain criteria. The grey shaded loci mean the same nucleotides as that in consensus sequence.To identify possible function and efficiency of σ32 binding sites, MED whiteflies were treated at two temperatures, 40 °C and 25 °C (as control) for 3 h, and collected immediately. qPCR was conducted on Portiera 16S rDNA to quantify the density of Portiera, and 14 predicted heat-related genes in MED whitefly (using cDNA) to determine the possible differential expression of them between the heat shock treatment samples and control samples. The density of Portiera didn’t significantly increase after heat shock (control vs. heat shock treatment, 1.00 ± 0.114 vs. 1.27 ± 0.187, Mean ± Stand Error Mean, t-test, p = 0.259). However, all the 15 genes (including rpoH encoding σ32factor) tested present significant variations (Table 2).
Our results of Portiera are quite similar to that of Buchnera (Wilcox et al., 2003), that only modest dynamics of gene expression are observed compared to free-living E. coli. This is probably due to the weakness of the σ32 promoters (Wilcox et al., 2003). On the other hand, we focused on six genes dnaK, groEL, groES, ftsH, dnaJ and hslV, with log2FoldChange >2. The results suggested that genes with predicted σ32 binding sites showed higher upregulation under heat shock. One σ32 binding site was proposed in the promoter of grpE, dnaK and dnaJ. These three genes were conjunct to each other, and supposed to be in the same operon according to genomic data. This σ32 binding site containing promoter may control the transcription of the three genes. Meanwhile, genes groEL and groES may also be in the same operon and be regulated together, with the σ32 binding sites in the upstream of groES. However, ftsH and hslV did not have recognized σ32 binding sites in immediate upstream spacer regions. How the transcription of these two genes were induced remains unclear.
Furthermore, we investigated the σ32 binding sites in another three available Portiera genomes, which are from different whitefly species (Fig. 3). The σ32 binding site upstream of grpE in 7 Portiera genomes showed weak similarities, while σ32 binding site close to groES presented an almost identical sequence. It is indicated that the regulation of groES and groEL might be universally kept in Portiera of whitefly (including whitefly Bemisia tabaci), and thermal tolerance ability may conserved among Portiera in different species of whiteflies.
Putative functional elements in intergenic spacers of Portiera genomes
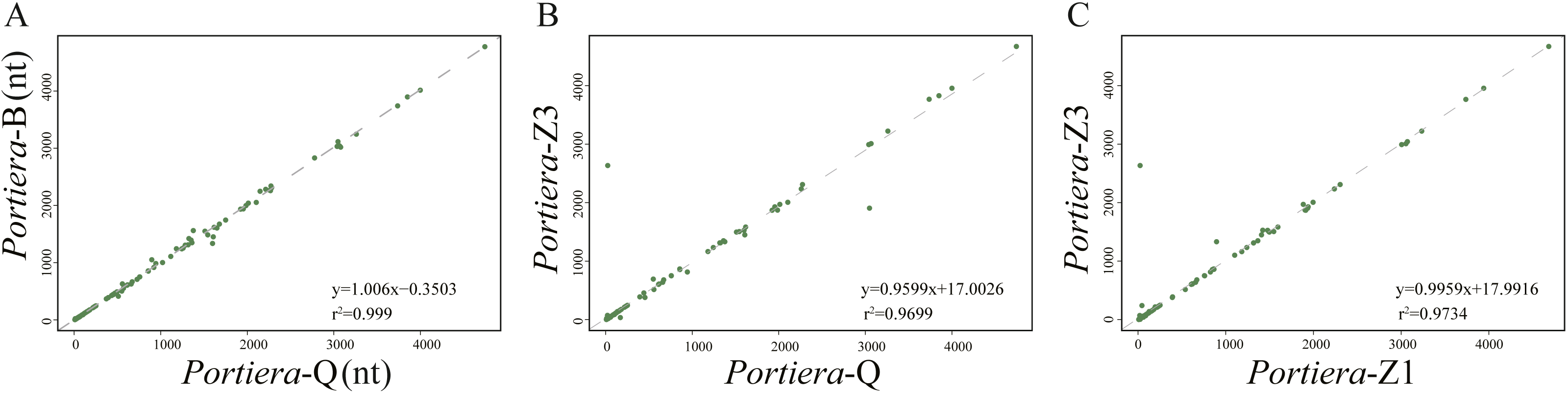
For intergenic spacers, we focused on Portiera genomes from whitefly B. tabaci species only. Intergenic spacers occupy over 30% of the Portiera genomes from whitefly B. tabaci, thus leading to the investigation for possible regulatory elements in intergenic spacers. Strong associations between lengths of orthologous IGSs were observed between four Portiera genomes (Fig. 4), which suggests that four Portiera genomes are coherently arranged even in the non-coding region with only subtle variations, indicating genome stasis (Tamas et al., 2002). Notably, there is a closer relationship between the two invasive species and between the two indigenous species, as evidenced by r2 values (Fig. 4), which is in agreement with phylogeny (Fig. 1).

Figure 4: Length associations of orthologous IGSs between different pairs of Portiera genomes.
(A) Comparison between Portiera-B and Portiera-Q; (B) comparison between Portiera-Z3 and Portiera-Q; and (C) comparison between Portiera-Z3 and Portiera-Z1.Those determined IGSs were then aligned among four Portiera from different whitefly species, which identified 95 IGSs (Data S1) orthologous (over 20 nucleotides in length) ranging from 20 to approximately 4700 nucleotides. The conserved loci are quite AT-rich with GC contents only around 23.5%. Furthermore, identically aligned loci among four Portiera occupied only a total of 62,438 nt, about 52% of all the orthologous IGSs. The stretch of identically aligned loci was probably interrupted by decayed pseudogenes at varying stages or random mutations in some preserved functional elements. Notably, 51 loci (Data S1) were over 20 nucleotides in length and were subjected to further analysis. In all, 37 perfectly conserved or nearly perfectly conserved Shine-Dalgarno sequences (Data S1) were detected (consensus sequence AGGAG), as well as 34 putative small RNAs predicted (Data S1) by using RNAFold. Since functional elements tend to evolve slower than nonfunctional elements (Ganley & Kobayashi, 2007), the predicted functional loci within IGSs may be involved in the process of transcriptional regulation. The predicted loci need further study for a better understanding of transcriptional regulation within the non-coding region.
Meanwhile, these predicted transcriptional elements were searched against another three Portiera genomes available, Portiera-TV, and Portiera genomes from A. dispersus and A. floccissimus. Among the 37 Shine-Dalgarno sequences detected in Portiera genomes of whitefly, eight of them were also detected in the orthologous IGSs from the additional three Portiera genomes, with another three Shine-Dalgarno sequences detected only in one or two additional Portiera genomes. Moreover, according to the 34 orthologous IGSs containing putative small RNAs in Portiera genomes of whitefly B. tabaci, corresponding regions in the extra three Portiera genomes were extracted, resulting in 22 orthologous IGS groups (Data S1). Each group contains at least one orthologous IGSs sequence from another three Portiera genomes. Interestingly, there were 11 groups, in which seven sequences were predicted to contain a putative small RNA, meaning these sRNAs were universally kept by Portiera genomes investigated, and within the orthologous IGS regions. Previous prediction on the divergence of whitefly species, wherein the separation between Portiera strains from A. dispersus and A. floccissimus, and T. vaporariorum and B. tabaci, was around 129 Ma, implies that the conserved elements found here might have been conserved at least 129 million years. However, the functions of these sRNAs need further investigation.
Conclusions
Taken as a whole, our study indicates similar function of Portiera in whitefly B. tabaci and the first to investigate putative transcriptional regulation processes in Portiera of whitefly B. tabaci. Several regulatory elements in both coding and non-coding regions are revealed, by the combination of orthologous and phylogenetic footprinting approaches. For the coding regions, four Portiera genomes retained a similar set of genes that are responsible for transcriptional regulation. In particular, the possible thermal tolerance ability of Portiera was revealed genetically and experimentally. Although non-coding regions contain decayed pseudogenes, Portiera genomes of whitefly B. tabaci still retain functional elements including putative sRNAs. This finding demonstrates regulation within symbiont genomes and gives insights into symbiosis mechanisms.
Supplemental Information
Sequence similarities (%) between different Portiera species pairs
No retention of regulatory genes for essential amino acid biosynthesis pathways in Portiera inferred from Escherichia coli
Calculation of Ka/Ks ratio of genes cspA between orthologous pairs from different Portiera species
List of 95 intergenic spacers (IGS) orthologs, 51 perfectly aligned IGS (over 20 nucleotides), 37 Shine-Dalgarno sequences and 34 putative small RNAs
Additional Information and Declarations
Competing Interests
The authors declare there are no competing interests.
Author Contributions
Dan-Tong Zhu conceived and designed the experiments, performed the experiments, analyzed the data, prepared figures and/or tables, authored or reviewed drafts of the paper.
Chi Zou and Fei-Xue Ban performed the experiments.
Hua-Ling Wang prepared figures and/or tables, authored or reviewed drafts of the paper.
Xiao-Wei Wang conceived and designed the experiments, contributed reagents/materials/analysis tools.
Yin-Quan Liu conceived and designed the experiments, contributed reagents/materials/analysis tools, authored or reviewed drafts of the paper, approved the final draft.
Data Availability
The following information was supplied regarding data availability:
The sequences of Portiera from whitefly B. tabaci Asia II 3 (previously referred to as ZHJ1 ‘biotype’) (Portiera BT-Z1) and Portiera from whitefly B. tabaci China 1 (previously referred to as ZHJ3 ‘biotype’) (Portiera BT-Z3) are available in GenBank under accession numbers CP016327 and CP016343.
Funding
Financial support for this study was provided by the National Key Research and Development Program (2016YFC1200601), the China Agriculture Research System (CARS-23-D07), National Natural Science Foundation of China (31390421), and the China Postdoctoral Science Foundation (Grant Number 517000-X91502). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}