



Characterization of antimicrobial resistance genes in Haemophilus parasuis isolated from pigs in China
Author and article information
Abstract
Background
Haemophilus parasuis is a common porcine respiratory pathogen that causes high rates of morbidity and mortality in farmed swine. We performed a molecular characterization of antimicrobial resistance genes harbored by H. parasuis from pig farms in China.
Methods
We screened 143 H. parasuis isolates for antimicrobial susceptibility against six fluoroquinolone antibiotics testing by the broth microdilution method, and the presence of 64 antimicrobial resistance genes by PCR amplification and DNA sequence analysis. We determined quinolone resistance determining region mutations of DNA gyrase (gyrA and gyrB) and topoisomerase IV (parC and parE). The genetic relatedness among the strains was analyzed by pulsed-field gel electrophoresis.
Results
Susceptibility test showed that all isolates were low resistance to lomefloxacin (28.67%), levofloxacin (20.28%), norfloxacin (22.38%), ciprofloxacin (23.78%), however, high resistance levels were found to nalidixic acid (82.52%) and enrofloxacin (55.94%). In addition, we found 14 antimicrobial resistance genes were present in these isolates, including blaTEM-1, blaROB-1, ermB, ermA, flor, catl, tetB, tetC, rmtB, rmtD, aadA1, aac(3′)-llc, sul1, and sul2 genes. Interestingly, one isolate carried five antibiotic resistance genes (tetB, tetC, flor, rmtB, sul1). The genes tetB, rmtB, and flor were the most prevalent resistance genes in H. parasuis in China. Alterations in the gyrA gene (S83F/Y, D87Y/N/H/G) were detected in 81% of the strains and parC mutations were often accompanied by a gyrA mutation. Pulsed-field gel electrophoresis typing revealed 51 unique patterns in the isolates carrying high-level antibiotic resistance genes, indicating considerable genetic diversity and suggesting that the genes were spread horizontally.
Discussion
The current study demonstrated that the high antibiotic resistance of H. parasuis in piglets is a combination of transferable antibiotic resistance genes and multiple target gene mutations. These data provide novel insights for the better understanding of the prevalence and epidemiology of antimicrobial resistance in H. parasuis.
Cite this as
2018. Characterization of antimicrobial resistance genes in Haemophilus parasuis isolated from pigs in China. PeerJ 6:e4613 https://doi.org/10.7717/peerj.4613Main article text
Introduction
Haemophilus parasuis is the etiological agent of Glässer’s disease that causes significant morbidity and mortality as well as economic losses in the global pig industry (Oliveira & Pijoan, 2004). Antimicrobial therapy is used to prevent and control this infection even though antimicrobial agents are also used for growth promotion in pigs (Lancashire et al., 2005). However, extended agricultural use of antibiotics poses a risk for selecting antibiotic resistant pathogens, and antibiotic resistance in H. parasuis is increasing (Aarestrup, Seyfarth & Angen, 2004; De la Fuente et al., 2007; Markowska-Daniel et al., 2010; Walsh & Fanning, 2008; Wissing, Nicolet & Boerlin, 2001; Xu et al., 2018). In China, the resistance rate of H. parasuis to antimicrobials is also increasing, resulting in limited therapeutic choices (Zhou et al., 2010).
Increases in antibiotic resistance among bacteria is most often the result of antibiotic resistance gene (ARG) transfer mediated by mobile DNA elements such as plasmids, transposons and integrons in Gram-negative bacteria (Lancashire et al., 2005; San Millan et al., 2007). A long history of antibiotic use in the swine industry has generated a strong selective pressure for resistance transfer mediated by plasmids and transposons within and between bacterial species. Plasmids play a key role in this process by acting as vehicles for horizontal gene transfer (San Millan et al., 2016). The most prominent ARG types associated with resistance in H. parasuis include blaROB−1, tetB, tetL, qnrA1, qnrB6, aac (6′)-Ib-cr, lnu(C) and flor (Dayao et al., 2016; Guo et al., 2011; Kehrenberg et al., 2005; Lancashire et al., 2005; Li et al., 2015; San Millan et al., 2007). In China, blaROB−1, qnrA1, qnrB6, aac (6′)-Ib-cr, lnu(C) and flor have been identified in H. parasui s (Guo et al., 2012; Guo et al., 2011; Li et al., 2015). Horizontal gene transfer of ARG-carrying mobile elements and vertical gene transfer by the proliferation of ARG hosts facilitate resistance spread (Xu et al., 2018). Moreover, quinolone resistance determining region mutations (QRDR) of gyrA and parC were related to resistance. Therefore, studying ARG fates and their horizontal and vertical transfer-related elements and QRDRs can provide a comprehensive insight into resistance mechanisms.
H. parasuis is one of the most important respiratory pathogens in pigs (Guo et al., 2012; Zhang et al., 2014; Zhou et al., 2010), so more information is needed on the characterization of resistance genes associated with the increase in antibiotic resistance for this bacterium. In the present study, we examined resistance determinants, QRDRs and genetic relatedness in H. parasuis strains from pig farms in China.
Materials and Methods
Bacterial strains
We isolated 143 H. parasuis strains from different diseased swine suffering polyserositis, pneumonia or meningitis between February 2014 and March 2017 in China. All strains were isolated from lung, brain, heart blood, pericardial effusion, pleural effusion, peritoneal effusion and joint fluid by aseptic inoculation ring, and cultured on tryptic soy agar (TSA) or tryptic soy broth (TSB) (Becton Dickinson, Owings Mills, MD, USA) containing 10 µg/ml nicotinamide adenine dinucleotide (NAD; Sigma, St. Louis, MO, USA) and 5% bovine serum (Gibco, Auckland, New Zealand). Plates were incubated at 37 °C for 24–48 h. All isolates were identified by PCR (Angen et al., 2007). The study was approved (No.2014-025).
Fluoroquinolone antimicrobial susceptibility testing
Nalidixic acid, ciprofloxacin, levofloxacin, enrofloxacin, norfloxacin and lomefloxacin were obtained from the National Institute for the Control of Pharmaceutical and Biological Products, Beijing, China. Minimal inhibitory concentrations (MIC) were determined in fastidious medium consisting of TSB with 5% bovine serum and 10 µg/mL NAD in 96-well microtiter plates. All plates were inoculated following the guidelines of the Clinical and Laboratory Standards Institute (CLSI) using Haemophilus influenzae and Haemophilus parainfluenzae M02 and M07(CLSI, 2015). The plates were incubated in an atmosphere containing 5% CO2 at 37 °C for 24 h. The MIC value was defined as the lowest concentration resulting in no visible bacterial growth. The reference strains H. influenzae ATCC 49247 and Escherichia coli ATCC 25922 served as quality controls for MIC determinations.
ARGs and integrons detection
DNA was extracted from whole organisms using the quick boiling method (Sambrook & Russell, 2001). PCR assays were used to screen for the presence of 64 ARG types including resistance to quinolones, β-lactams, macrolides, tetracycline, aminoglycosides, chloramphenicol, sulfonamides as well as for the integrase gene (Table 1). Purified PCR products were directly sequenced from both ends or cloned into plasmid vector pMD18-T, and then sequenced. DNA sequence similarity searches were performed against the GenBank database using BLAST software to confirm gene identity.
| Antibiotic | Resistance genes | Primers |
|---|---|---|
| quinolones | qepA, qnrA, qnrB, qnrC, qnrD, qnrS, oqxAB, aac(6′)-Ib-cr | Cavaco et al. (2009), Yang et al. (2017), Zhao et al. (2010) |
| β-lactams | blaTEM − 1, blaROB−1, SHV, CTX-M-1G, CTX-M-9G, CTX-M-2G, CTX-M-64, CTX-M-25 DHA, VIM-1, VIM-2, SPM-1, CMY-2, npmA, OXA, NDM, KPC, IMP, SPM, FOX | Grobner et al. (2009), Liu et al. (2007), San Millan et al. (2007), Weill et al. (2004) |
| tetracyclines | tetA, tetB, tetC, tetD, tetE, tetG, tetH, tetL-1, tetL-2 | De Gheldre et al. (2003), Matter et al. (2007), Miranda, Rodriguez & Galan-Vidal (2009) |
| aminoglycosides | rmtB, rmtC, armA, rmtA, rmtD, aadB[ant(2′)-la], aacC2 [aac(3)-Iic], aacC4 [aac(3)-Iva], aadA1,aac(6)-31 | Doi & Arakawa (2007), Matter et al. (2007) |
| macrolides | ermA, ermB, ermC, mefA/E | Hou et al. (2013), Matter et al. (2007), Sutcliffe et al. (1996) |
| chloramphenicol | catl, cmlA, flor, cfr | Maka & Popowska (2016), Wang et al. (2015) |
| sulfonamides | sul1, sul2, sul3, dfrA1, dfrB | Matter et al. (2007) |
| integrase gene | intl1, intl2, intl3 | Shibata et al. (2003) |
Detection of mutations in QRDRs of gyrA, gyrB, parC, and parE
Mutations in the quinolone resistance determining regions (QRDR) mutations in the gyrA, gyrB, parC and parE genes were identified after DNA sequencing of PCR products generated with the primers listed in Table 2.
| Gene | Primers | Sequence (5′-3′) | Size (bp) | Reference |
|---|---|---|---|---|
| gyrA | GyrA-F GyrA-R |
AGCGTTACCAGATGTGCGAGATG TTGCCACGACCTGTACGATAAGC |
620 | This study |
| gyrB | GyrB-F GyrB-R |
TACATACGCTGTAGGTTCAAGGA CAAGATAATACGGAAATGGAGC |
500 | This study |
| parC | ParC-F ParC-R |
AACTTCAACATTACCACTTAGCCCTCG TACCTCACCAAGCCTCGCCATCT |
1,445 | This study |
| parE | ParE-F ParE-R |
CGATAATTCCCTTGAAGTCGTTG ATTGATCTGCTCGCCACCCTCTG |
609 | This study |
Pulsed-field gel electrophoresis
Genetic relatedness of H. parasuis strains carrying ARGs was determined by pulsed field electrophoresis (PFGE) of CpoI- (TaKaRa, Beijing, China) digested genomic DNA samples (Zhang et al., 2011). PFGE typing used a CHEF Mapper electrophoresis system (BioRad, Hercules, CA, USA) with 2.16–63.8 sfor 21 h. Salmonella enterica serovar Braenderup H9812 DNA digested with CpoI was used for a size standard. Interpretation of the PFGE patterns was accomplished using BioNumerics 6.6 software (Applied Maths, Sint-Martens-Latem, Belgium) (Tenover et al., 1995).
Results
Bacterial strains analysis
In the current study, 143 H. parasuis strains were isolated and 73 carried antibiotic resistance genes. Information on isolation site, isolation time and resistance gene content are listed in Table 3.
| Isolates | Separation site | Date | Organ | Resistance gene |
|---|---|---|---|---|
| HP001 | Fujian | 2016 | lung | rmtB |
| HP008 | Fujian | 2015 | nasal cavity | tetB |
| HP011 | Meizhou | 2014 | pericardial effusion | sul2+ blaROB−1 |
| HP012 | Jinan | 2017 | lung | blaTEM−1 |
| HP013 | Zengcheng | 2016 | nasal cavity | catl1+tetB+ blaROB−1 |
| HP016 | Laiyang | 2015 | brain | ermA |
| HP017 | Dongguan | 2015 | joint fluid | tetB+tetC |
| HP018 | Qingdao | 2016 | lung | ermB |
| HP019 | Hebei | 2017 | lung | sul2+tetB |
| HP020 | Jilin | 2016 | lung | tetB |
| HP022 | Huadou | 2015 | lung | blaTEM−1 |
| HP025 | Zengcheng | 2016 | joint fluid | catl1+tetB+aac(3′)-IIc |
| HP026 | Guangxi | 2014 | lung | tetB+flor |
| HP029 | Guangxi | 2015 | heart blood | tetB+flor+ aac(3′)-IIc |
| HP032 | Chengde | 2014 | joint fluid | aadA1 |
| HP035 | Guangxi | 2017 | heart blood | catl1 |
| HP037 | Fujian | 2015 | nasal cavity | rmtB |
| HP039 | Hebei | 2015 | pericardial effusion | aac(3′)-IIc |
| HP040 | Jiangmen | 2016 | lung | sul1+ aac(3′)-IIc |
| HP044 | Jiangsu | 2014 | lung | tetB |
| HP050 | Jiangsu | 2016 | pleural effusion | tetB |
| HP051 | Jiangsu | 2014 | heart blood | tetC |
| HP053 | Yunnan | 2016 | lung | tetB+flor |
| HP054 | Guangzhou | 2016 | lung | tetB |
| HP056 | Zhucheng | 2017 | lung | rmtB+sul1 |
| HP059 | Guangxi | 2016 | lung | catl1 +tetB |
| HP060 | Guangxi | 2015 | heart blood | tetC+flor |
| HP061 | Qingdao | 2016 | lung | rmtB |
| HP063 | Hebei | 2015 | lung | blaTEM−1 |
| HP065 | Qingyuan | 2015 | lung | rmtB+ blaTEM−1 |
| HP066 | Guangxi | 2016 | lung | sul1+ aac(3′)-IIc |
| HP067 | Qingdao | 2015 | lung | tetB+aadA1 |
| HP068 | Qingyuan | 2015 | heart blood | tetB |
| HP069 | Hunan | 2017 | heart blood | flor |
| HP071 | Hebei | 2015 | lung | tetB |
| HP072 | Zhucheng | 2017 | nasal cavity | tetB |
| HP073 | Zhuhai | 2016 | joint fluid | blaROB−1 |
| HP075 | Fujian | 2014 | pericardial effusion | sul1 |
| HP076 | Henan | 2017 | heart blood | tetB |
| HP078 | Henan | 2015 | lung | rmtB+ blaTEM−1 |
| HP079 | Jining | 2016 | lung | rmtB |
| HP080 | Jinan | 2014 | joint fluid | rmtB+sul1 |
| HP082 | Qingdao | 2016 | lung | tetB |
| HP085 | Liaoning | 2016 | lung | tetB |
| HP091 | Shaoguan | 2017 | pericardial effusion | catl1+tetB |
| HP094 | Hebei | 2015 | lung | catl1+tetB+ blaROB−1 |
| HP095 | Huadou | 2017 | lung | catl1+tetB |
| HP096 | Zhengzhou | 2014 | heart blood | rmtB |
| HP097 | Hunan | 2016 | pericardial effusion | tetB |
| HP098 | Hebei | 2014 | lung | tetB |
| HP102 | Anyang | 2015 | lung | blaROB−1+aadA1 |
| HP103 | Hunan | 2017 | lung | catl1+tetB+flor |
| HP104 | Jiangsu | 2016 | joint fluid | flor+aadA1 |
| HP108 | Guangxi | 2016 | lung | tetB+flor+rmtB |
| HP109 | Zhaoqing | 2017 | heart blood | tetB |
| HP111 | Jiangxi | 2015 | lung | rmtB |
| HP112 | Sihui | 2016 | heart blood | tetB+blaROB−1 |
| HP113 | Henan | 2015 | lung | tetB+tetC+flor |
| HP116 | Boluo | 2014 | lung | blaTEM−1 |
| HP117 | Hebei | 2017 | heart blood | rmtD+rmtB+blaTEM−1 |
| HP118 | Huizhou | 2016 | lung | rmtB+aac(3′)-IIC |
| HP120 | Hebei | 2016 | lung | tetB |
| HP121 | Hebei | 2016 | heart blood | flor |
| HP123 | Anhui | 2015 | lung | flor |
| HP127 | Jilin | 2015 | joint fluid | flor |
| HP131 | Yunnan | 2014 | heart blood | sul1 |
| HP133 | Huizhou | 2016 | lung | catl1+rob-1 |
| HP134 | Zhucheng | 2014 | lung | rmtB+sul1 |
| HP135 | Shaoguan | 2016 | pleural effusion | tetB |
| HP137 | Yangzhou | 2015 | lung | catl1+tetB+ blaTEM−1 |
| HP140 | Conghua | 2014 | heart blood | rmtB+ blaTEM−1 |
| HP141 | Yangzhou | 2017 | lung | rmtB+sul1 |
| HP142 | Henan | 2016 | lung | tetB+tetC+flor+rmtB+sul1 |
Fluoroquinolone antimicrobial susceptibility testing
The results of the fluoroquinolone antimicrobial susceptibility of 143 H. parasuis isolates are listed in Supplemental Information 1. It showed that 82.52% and 55.94% of all isolates were resistant to nalidixic acid and enrofloxacin, respectively. Resistance of lomefloxacin, levofloxacin, norfloxacin, ciprofloxacin were 28.67%, 20.28%, 22.38%, 23.78%, respectively.
ARG and integron prevalence and detection
We examined 143 H. parasuis strains and 16 (11.2%) carried β-lactamases including blaTEM−1 and blaROB−1. Tetracycline resistant strains carried tetB and tetC. There were two isolates (1.40%) also yielded the erythromycin resistance genes: 1 for ermA, and 1 for ermB. A higher proportion (16.1%) carried chloramphenicol resistance genes including 10 catl and 13 flor. Aminoglycoside resistance was also high (11.9%) and included the genes rmtB, rmtD, aadA1 and aac(3′)- llc. The sulfonamide resistance genes were represented by sul1 and sul2 and were found in 9 (6.3%) and 2 (1.4%) of the isolates, respectively (Table 4).
| Gene | Number identified | Prevalence (%) |
|---|---|---|
| ermA | 1 | 0.70 |
| ermB | 1 | 0.70 |
| catl | 10 | 6.99 |
| flor | 13 | 9.09 |
| tetB | 34 | 23.78 |
| tetC | 5 | 3.50 |
| rmtD | 1 | 0.70 |
| rmtB | 17 | 11.89 |
| aadA1 | 4 | 2.80 |
| aac(3′)- IIc | 6 | 4.20 |
| sul1 | 9 | 6.29 |
| sul2 | 2 | 1.40 |
| blaTEM−1 | 9 | 6.29 |
| blaROB−1 | 7 | 4.90 |
The resistance gene patterns were diverse and 39 isolates carried one gene, 24 carried two and nine isolates carried three genes. Interestingly, strain HP142 carried five genes tetB, tetC, flor, rmtB and sul1. Overall, tetB, rmtB and flor were the most prevalent resistance genes in these H. parasuis isolates from Chinese pig farms (Table 5). Other genes were not detected in this study.
| Pattern | No. of isolates | Pattern | No. of isolates |
|---|---|---|---|
| ermA | 1 | flor+aadA1 | 1 |
| ermB | 1 | catl1+tetB | 3 |
| tetB | 16 | rmtB+ aac(3′)-IIc | 1 |
| catl1 | 1 | rmtB+ blaTEM−1 | 3 |
| tetC | 1 | rmtB+sul1 | 4 |
| rmtB | 6 | sul1+ aac(3′)-IIc | 2 |
| flor | 4 | sul2+ blaROB−1 | 1 |
| sul1 | 2 | sul2+tetB | 1 |
| aadA1 | 1 | blaROB−1+aadA1 | 1 |
| aac(3′)-IIc | 1 | catl1+tetB+ blaTEM−1 | 1 |
| blaTEM−1 | 4 | catl1+tetB+ blaROB−1 | 2 |
| blaROB−1 | 1 | catl1+tetB+flor | 1 |
| catl1+blaROB−1 | 1 | catl1+tetB+aac(3′)-IIc | 1 |
| tetB+flor | 2 | tetB+flor+rmtB | 1 |
| tetB+aadA1 | 1 | tetB+flor+ aac(3′)-IIc | 1 |
| tetB+ blaROB−1 | 1 | tetB+tetC+flor | 1 |
| tetB+tetC | 1 | rmtD+rmtB+ blaTEM−1 | 1 |
| tetC+flor | 1 | tetB+tetC+flor+rmtB+sul1 | 1 |
Mutations in QRDRs of gyrA, gyrB, parC, and parE
We also identified several QRDR mutations among the resistant H. parasuis strains. Mutations in gyrA (S83F/Y, D87Y/N/H/G) were detected in 116 (81%) of the strains. In addition, 79 strains had parC mutations (L379I/ Y557C/ V648I/E678D) and most of these were accompanied by gyrA mutations. Only nine strains had single parC mutations that were either L379I, Y557C, E678D, L379I or Y557C. The strains with gyrA mutations at either codon 83 or 87 showed higher MIC values compared with the 18 strains lacking mutations. The MIC values of the strains with single parC mutations were not significantly different from controls. No mutations were found in gyrB and parE (Table 6).
| QRDR mutation | Number of strains | MICs (µg/mL) | ||||||
|---|---|---|---|---|---|---|---|---|
| gyrA | parC | Nalidixic acid | Levofloxacin | Ciprofloxacin | Enrofloxacin | Norfloxacin | Lomefloxacin | |
| – | – | 18 | 0.25–128 | <0.25–2 | <0.25–4 | <0.25–2 | <0.25–4 | <0.25–1 |
| S83F/Y | – | 8 | 1–>512 | 0.25–16 | 0.25–16 | <0.25–8 | 0.25–256 | <0.25–4 |
| S83F/Y, D87Y/N/H/G | – | 38 | 4–>512 | 0.25–32 | 1–>512 | 0.25–32 | 0.25–>512 | 0.25–64 |
| S83F/Y | aL379I/Y557C/ V648I | 20 | 32->512 | 0.25–64 | 0.25–32 | <0.25–32 | 0.25–16 | <0.25–128 |
| D87Y/H | bL379I/Y557C | 2 | 4, 16 | 0.25, 0.5 | 0.25, 0.5 | 2 | 1, 4 | 0.25, 0.5 |
| S83F/Y, D87Y/N/Y/G/H | cL379I/Y557C/ V648I/E678D | 48 | 1–>512 | 2–128 | 2–64 | 0.25–32 | 0.25–>512 | <0.25–64 |
| – | dL379I/Y557C/ L379I, Y557C, E678D/L379I, Y557C | 9 | 0.5–>512 | 0.25–8 | 0.25–16 | 0.5-16 | 0.25–>512 | 0.5–64 |
PFGE
The 73 H. parasuis strains carrying resistance determinants were typed by PFGE and were genomically heterogenic. We identified 51 unique CpoI patterns but no evidence of clonality (Fig. 1).
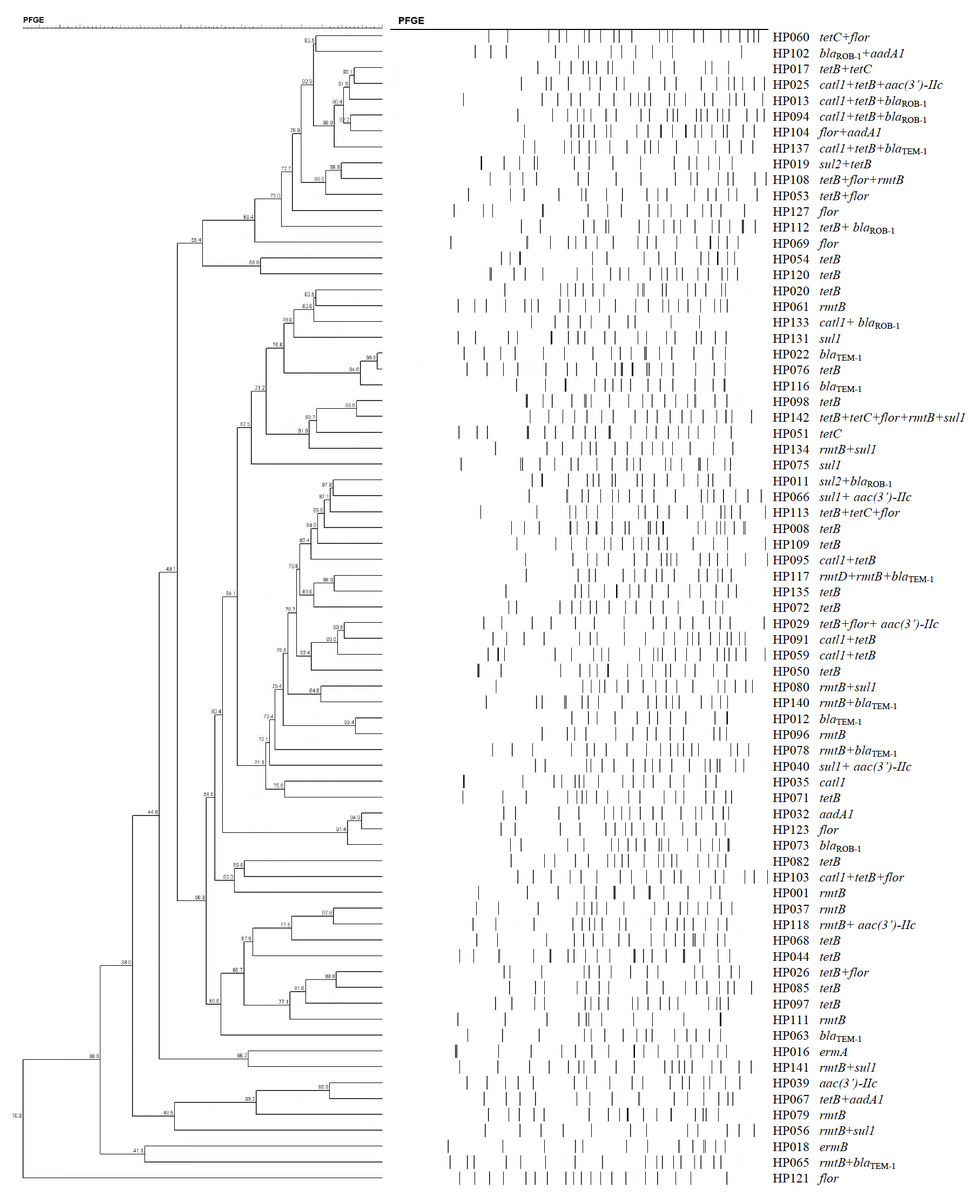
Figure 1: Dendrogram of patterns generated by PFGE of 73 ARG-containing H. parasuis isolates
Discussion
In the current study, we observed high-level resistance to nalidixic acid and enrofloxacin. Similar results have been reported such as 84.8% to nalidixic acid (Xu et al., 2011) and 60.1% and 45.5% to enrofloxacin (Xu et al., 2011; Zhang et al., 2013). These differed from results in the United Kingdom and Spain (0 and 20%) (De la Fuente et al., 2007). We described the fluoroquinolone antimicrobial resistance profiles for H. parasuis strains isolated between 2014–2017. When compared with 2002–2009 and 2008-2010, our data indicated that fluoroquinolone antimicrobial resistance in H. parasuis was very serious in China during the last 15 years.
There have been few complete and systematic molecular studies of antimicrobial resistance in H. parasuis. The genes blaROB−1, tetB, tetL, qnrA1, qnrB6, aac (6′)-Ib-cr, lnu(C) and flor were the only that were previously identified and that correlated with resistance (Dayao et al., 2016; Guo et al., 2011; Kehrenberg et al., 2005; Lancashire et al., 2005; Li et al., 2015; San Millan et al., 2007). Cephalosporinases, which are naturally present in some enterobacterial species, can be mobilized by transposons and migrate via plasmids into other species. Moreover, the abuse of antimicrobial agents increases the number of carbapenem-resistant strains generating a public health concern (Yang et al., 2017). In the Enterobacteriaceae, the blaTEM−1 β-lactamase is the predominant genotype (Yang et al., 2017). In our study, we identified both blaTEM−1 and blaROB−1 β-lactamase genes which are widespread among H. parasuis and Pasteurella spp (Guo et al., 2012; San Millan et al., 2007). blaTEM−1 and blaROB−1 are usually present in H. influenzae and have particular geographic distributions in different countries (Farrell et al., 2005). These geographic differences may also be present in H. parasuis. The first reports of blaTEM−1 and blaROB−1 were in China and Spain, respectively(Guo et al., 2012; San Millan et al., 2007). blaROB−1 was located on plasmid pB1000 and recently a novel 2,661 bp plasmid (pJMA-1) bearing blaROB−1 has been identified. This plasmid possessed a backbone found in small Pasteurellaceae plasmids and was 100% stable with a lower biological cost than pB1000 (Moleres et al., 2015).
We also identified genes encoding tetracycline efflux pumps (tetB and tetD) in this study. The first tetracycline resistant gene identified in H. parasuis was tetB and this gene is the most common tetracycline resistance gene in Actinobacillus pleuropneumoniae and Pasturella multocida (Dayao et al., 2016; Matter et al., 2007). The genes tetH and tetM are present in other members of the Pasteurellaceae (Roberts, 2012). Furthermore, the tetB-carrying plasmid pHS-Tet in H. parasuis was similar to a tetL-carrying plasmid in Pasteurella isolates (Kehrenberg et al., 2005; Lancashire et al., 2005). This is the first report of the tetD gene in H. parasuis isolates from China and needs further study. Tetracycline resistance genes are often associated with conjugative and mobile genetic elements enabling horizontal transfer (Dayao et al., 2016; Roberts, 2012). The presence of tetD suggests that tetracycline resistance in H. parasuis relies on efflux pumps.
In bacteria with animal origins, five florfenicol resistance genes (floR, fexA, fexB, cfr and optrA) have been reported (Schwarz et al., 2004; Wang et al., 2015). In Gram-negative bacteria, floR makes the greatest contribution to florfenicol resistance and this has been described for a number of bacterial species (He et al., 2015; Meunier et al., 2010; Schwarz et al., 2004; Wang et al., 2015). The emergence of florfenicol resistance in H. parasuis isolates was attributable to a novel small plasmid pHPSF1 bearing floR. This novel plasmid was similar to other Pasteurellaceae plasmids suggesting these species prefer to exchange genetic elements with each other.
High-level aminoglycoside resistance mediated by the production of the 16S rRNA methylases armA, rmtA to H and npmA, and resistance is increasing among Gram-negative pathogens (Du et al., 2009), being sometimes clonal spread of a single pulsotype (Hopkins et al., 2010). In our case, a clone bearing rmtB HP118 and HP037, was present in two different regions. However, until now, few studies have described the presence of the armA and rmtB genes in H. parasuis isolates, although they have been frequently reported on Enterobacteriaceae from food animals. The strains in our study also carried rmtB, rmtD, aadA1 and aac (3′) IIc and these warrants further investigation.
The macrolide-resistance genes erm A and erm B showed a low frequency in our H. parasuis isolates. These genes are responsible for ribosomal binding site modifications that are the most important macrolide resistance mechanisms(Takaya et al., 2010).
The sul1, sul2 and sul3 genes are dihydropteroate synthases involved in sulfonamide resistance of Gram-negative bacteria and are usually associated with an integron system and a conjugative plasmid (Vo et al., 2006). In the current study, we identified both sul1 and sul2, and these genes most likely accounted for the observed resistance to trimethoprim-sulfamethoxazole. These results are similar to others in Gram-negative bacteria (Koljalg et al., 2009; Matter et al., 2007).
This is the first report describing the presence of the tetC, sul1, sul2, ermA, ermB, catl, rmtB, rmtD, aadA1 and aac (3′)-IIc genes in H. parasuis, to the best of our knowledge. Nevertheless, we did find several isolates with reduced antibiotic susceptibility that did not harbor any of the tested resistance genes. This suggests that H. parasuis possesses other resistance mechanisms such as mutations, decreases in permeability and increases in efflux pump activity or yet unknown antibiotic resistance mechanisms. In addition, the widespread dissemination of resistance genes and integrons could potentially fuel the rapid development of antimicrobial resistance due to their high transfer capabilities (Hussein et al., 2009). Therefore, more study is needed on this subject.
There have been numerous studies demonstrating gyrA and parC mutations engendering fluoroquinolone resistance in Gram-negative bacteria and Gram-positive bacteria from pigs such as Salmonella spp., E. coli or Streptococcus suis (Cao et al., 2017; Escudero et al., 2007). In H. parasuis, the gyrA mutations S83Y, S83F, D87Y, D87N and D87G are correlated with fluoroquinolone resistance. In addition, the parC mutations Y577C, V648I, E678D, S669F, A464V and A466S and parE mutations S283G, A227T and G241S were also found in these strains(Guo et al., 2011). In another study, mutations of gyrA D87N, parC S73R and parE T551A were involved in fluoroquinolone resistance, but other mutations such as in gyrA (452D∧V/G, 627G∧E), gyrB (211V∧I, 254D∧G), parC (73S∧R/I, 227Q ∧H, 379L∧I, 578C∧Y) and parE (551T∧A) occurred less frequently (Zhang et al., 2013). However, the parE mutation in A. pleuropneumoniae is possibly not involved in enrofloxacin resistance (Wang et al., 2010). In our study, most strains possessed gyrA mutations, and six strains possessed a gyrA mutation (D87H) not been previously reported. However, we do not know whether this mutation is directly related to fluoroquinolone resistance. We also identified four parC mutations. Unlike other studies, we found the parC 578 mutation in both resistant and sensitive strains, suggesting this mutation is not involved in resistance (Zhang et al., 2013). Overall, the QRDR analysis in our study suggested that the mutations at codon 83 or 87 of gyrA were responsible for fluoroquinolone resistance and that gyrB and parE were not.
Interestingly, our PFGE results indicated that almost 70% of our H. parasuis were genetically diverse, similar to a recent report (Guo et al., 2012). These results are in contrast to a previous study presenting evidence for the clonal spread of β-lactam resistance (San Millan et al., 2007). Our data suggests that resistance genes are spread via transferable elements such as plasmids and transposons in addition to clonal spread. Therefore, research on mechanisms for the spread of antimicrobial resistance in H. parasuis needs further investigation.
Conclusions
In this study, we comprehensively and systematically investigated for the first time the distribution of the most common resistance genes in H. parasuis in China. These genes included tetB, tetC, sul1, sul2, ermA, ermB, blaTEM−1, blaROB−1, catl, flor, rmtB, rmtD, aadA1 and aac (3′)-IIc. The gyrA mutations S83F/Y and D87Y/N/H/G correlated with fluoroquinolone resistance in H. parasuis. These strains were also genetically diverse as judged by PFGE. These data suggest that antimicrobial resistance in H. parasuis is primarily the result of transferable determinants and multiple target gene mutations. The exact roles for these detected resistance determinants in H. parasuis await further study.
Supplemental Information
Additional Information and Declarations
Competing Interests
Lili Guo is an employee of Qingdao Yebio Biological Engineering Co., Ltd.
Author Contributions
Yongda Zhao conceived and designed the experiments, prepared figures and/or tables, authored or reviewed drafts of the paper, approved the final draft.
Lili Guo performed the experiments, analyzed the data, prepared figures and/or tables, authored or reviewed drafts of the paper, approved the final draft.
Jie Li performed the experiments, analyzed the data, authored or reviewed drafts of the paper, approved the final draft.
Xianhui Huang contributed reagents/materials/analysis tools, prepared figures and/or tables, authored or reviewed drafts of the paper, approved the final draft.
Binghu Fang conceived and designed the experiments, contributed reagents/materials/analysis tools, authored or reviewed drafts of the paper, approved the final draft.
Animal Ethics
The following information was supplied relating to ethical approvals (i.e., approving body and any reference numbers):
The animal research committees of the South China Agriculture University granted approval for this research.
Data Availability
The following information was supplied regarding data availability:
The raw data are provided in a Supplemental Information 1.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31372479), Science and Technology Planning Project of Guangdong Province, China (No. 2015B090901059), and the National Key Research Program of China (grant 2017YFD0501404). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.

{kind=link}