
Exploration of severe early childhood caries microbiota through a novel developed nutrient enriched microbiological medium, high through-put 16S rRNA sequencing and culturomics
- Published
- Accepted
- Received
- Academic Editor
- Marisa Nicolás
- Subject Areas
- Genomics, Microbiology, Dentistry
- Keywords
- NEMM, SHI medium, Microbiota, S-ECC, Culturomics, Novel microbiological medium, Uncultured bacteria
- Copyright
- © 2024 Zhang et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2024. Exploration of severe early childhood caries microbiota through a novel developed nutrient enriched microbiological medium, high through-put 16S rRNA sequencing and culturomics. PeerJ 12:e18312 https://doi.org/10.7717/peerj.18312
Abstract
Introduction
Severe early childhood caries (S-ECC) is a widespread disease that harms children physically and mentally. Microorganisms are regarded as the dominant etiology of caries, however, S-ECC microbiome remains largely unknown, nearly 1/4 of them remained uncultivated. To explore S-ECC microbiota, a new bacterial medium, nutrient-enriched microbiological medium (NEMM) was designed in this study.
Methods
Eleven fresh S-ECC dental plaque samples were collected and cultivated in both NEMM and SHI medium (reference medium) for 0, 1, 3, 6, 9, 15, 21 and 28 days under aerobic and anaerobic conditions. Thereafter, the cultures were harvested, together with their corresponding clinical S-ECC dental plaque samples, for high through-put 16S rRNA sequencing and culturomics. The single colonies were cultured for further confirmation by sequencing the full length of the 16S rRNA gene after bacterial genomic DNA extraction and PCR amplification.
Results
Either NEMM or SHI medium showed a significant decrease in bacterial alpha diversity compared to clinical dental plaque samples by high throughput 16S rRNA sequencing analysis, indicating a larger room for the improvement of both media. NEMM displayed more living bacteria, abundant bacteria species, uncultured bacteria and capacities in carbohydrate transport and metabolism than SHI medium. The dynamic changes in bacterial community composition over time indicated that some bacteria tended to be enriched at specific time points. Culturomics and identification of bacterial species results were further confirmed by the high throughput 16S rRNA sequencing results.
Conclusion
We developed a new medium NEMM that could support S-ECC microbiota growth with a higher yield of living bacteria, higher abundance and capacity, and be suitable for cultivating oral uncultured bacteria via culturomics technology.
Introduction
Diverse microbiomes inhabit the human oral cavity, which are responsible for various infectious diseases, including tooth decay, periodontitis, and endodontic infection (Bhandary et al., 2024). Recent research also linked oral microorganisms with several systemic diseases, including type 2 diabetes (Sanz et al., 2018), Alzheimer’s disease (Wu et al., 2022), and cardiovascular disease (Sanz et al., 2020). Tooth decay is one of the most prevalent oral diseases globally (Peres et al., 2019). Early childhood caries (Grier et al., 2021) is defined as decay of the primary teeth in children younger than 71 months, which affects 23% of children in the United States of America. Force (2021) and more than 60% in China (Hu, Hong & Li, 2011). Early childhood caries (ECC) not only harms the physical and mental health of children but also causes a heavy burden to society as well (Grier et al., 2021; Hemadi et al., 2017). The presence of dental caries can lead to serious oral problems and general health, including dental pain, limiting oral functions such as chewing/eating, impaired nutritional status, and sleep disorder. Severe ECC (S-ECC) is an aggressive form of ECC. Based on the definition of S-ECC by the American Academy of Pediatric Dentistry, any sign of smooth-surface caries in a child younger than three years of age, and children aged 3–5 years who have one or more cavitated lesions, caries-caused missing or filled smooth surface in primary anterior teeth or decayed, missing or filled surfaces greater than or equal to four (age of 3), five (age of 4) or six (age of 5) are diagnosed as S-ECC patients (Anonymous, 2016).
The etiology of S-ECC is generally accepted as the pathogenic biofilms established as the consequence of complex, dynamic interactions among microorganisms, diet, and the host. The disease process was modulated through the polymicrobial interaction between pathogenic microorganisms and commensals within the highly structured biofilms (Bowen et al., 2018). The latest research assigns that the microorganisms associated with caries include Streptococcus mutans, Lactobacillus, Actinomyces, Bifidobacterium, and Scardovia species. More than 700 species of microorganisms have been identified in the oral cavity by next-generation sequencing technologies, however, a part of them remian uncultivated (Aas et al., 2005; Dewhirst et al., 2010; Paster et al., 2001). The 16S rRNA gene is the most commonly used bacterial taxonomy gene in culture-independent techniques to identify caries-associated pathogenic microorganisms (Pace, Olsen & Woese, 1986). However, owing to Koch’s postulates (the ground rules to determine the pathogenic agent of the disease (Fredricks & Relman, 1996)), the genetic heterogeneity of bacterial species and the strong influence of the environment on phenotype, species-level identification may not be sufficient (Wade, 2013). Hence, intensive studies of pure cultured fastidious bacteria may provide us with a new understanding of the role of microbiology in S-ECC.
Tian and his colleagues developed the SHI medium to sustain a higher diversity of the oral microbial community and more similar microbial profiles to the original saliva-derived oral microflora (Tian et al., 2010). Using SHI medium, He et al. (2015) revealed the TM7 phylum as “microbial dark matter”, which was recalcitrant to be cultivated, making it one of the most enigmatic phyla known. Furthermore, its potential pathogenic associations with immune suppression ability was found.
Considering that SHI medium only displayed a certain level of selectivity of microorganisms, thus, in this study, we developed a novel nutrient-enriched microbiological medium (NEMM), to cultivate more oral bacteria, especially for uncultivated microorganisms, and make the preparation for further investigating the characterization of the microbiome in S-ECC.
Materials & Methods
Subjects and sample collection
Dental plaque samples were collected from eleven patients with S-ECC. Three boys and six girls, aged from 2.53 to 5.33 years old, were diagnosed S-ECC with decayed, missing, and filled tooth surface (dmfs) scores were 14.11 ± 3.59 (Table S1). Participants were required to avoid eating, drinking and brushing their teeth at 2 h before dental plaque sampling. Ethical approval of the study was granted by Peking University School of Stomatology (PKUSSIRB-201839140), and written informed consent was obtained from all study participants’ guardians.
Eleven plaque samples were collected with a sterile curette from the labial/buccal smooth surfaces or the decayed cavity of the affected teeth. The dental plaque was transferred to a 1.5 mL sterile centrifuge tube full of pre-reduced medium (0.15 g sodium mercaptoacetate, 0.9 mL fresh 1% calcium chloride, and 100 mL distilled water, filtered for removing bacteria) and taken to the laboratory and processed for culture within 3 min. Samples were dispersed by vortexing for 30 s and diluted tenfold consecutively from 10−1 to 10−8.
Culture media
All samples were grown in duplicate at 37 °C under anaerobic conditions (80% N2, 10% H2, and 10% CO2) and normoxic conditions (air with 5% CO2). To mimic the nutritional environment of the dental plaque, a novel culture medium was designed. The basal liquid medium consisted of brain-heart infusion broth (OXOID, Basingstoke, UK) 24.5 g/L, agar (Solarbio, Beijing, China) 15 g/L, dextrose (Solarbio) 5 g/L, corn starch (Solarbio) 1.5 g/L, casein acid hydrolysate (Macklin, Shanghai, China) 17.5 g/L, hemin (Solarbio) 5 mg/L, VitK (Solarbio) 1 mg/L, VitB12 (Macklin) 100 µg/L, L-glutamine (Macklin) 0.1 g/L, adenine (Macklin) 10 mg/L, guanine hydrochloride (Macklin) 300 µg/L, p-aminobenzoic acid (Macklin) 130 µg/L, nicotinamide adenine dinucleotide (Macklin) 2.5 mg/L, thiamine pyrophosphate (Macklin) 1 mg/L, ferric nitrate (Macklin) 200 µg/L, cysteine hydrochloride (Solarbio) 0.259 g/L, L-cysteine (Solarbio) 11 mg/L, and sheep blood (Pingrui Biotechnology company) 5%. SHI medium was prepared as described before (Tian et al., 2010).
Dental plaque culture
Samples were enriched aerobically and anaerobically using two liquid media SHI and NEMM designed by this study and subcultured in solid media after optimal dilution, whose single colonies numbers range from 100 to 500. The cultures were divided into four groups: (1) aNEMM (samples grown in NEMM anaerobically); (2) aSHI (samples grown in SHI medium anaerobically); (3) nNEMM (samples grown in NEMM under normoxic condition); and (4) nSHI (samples grown in SHI medium under normoxic condition). Consecutive tenfold dilutions of the plaque samples in two media were prepared by PBS and used to inoculate NEMM and SHI-agar plates in triplicate. Plates were incubated and photographed at 0, 1, 3, 6, 9, 15, 21 and 28 days. Meanwhile, 500 µL suspension cultures at these different time points were collected for 16S rRNA gene sequencing analysis and bacterial isolation. For clinical isolates, suspension cultures were gradient diluted from 10−1 to 10−8, and then the diluted bacterial solutions were plated on the NEMM- and SHI-agar plates. The clinical isolates were picked up from the plates according to morphology and then identified by sequencing the full length of the 16S rRNA gene as described below.
High through-put 16S rRNA gene sequencing
Nine clinical dental plaque samples and their cultures in NEMM and SHI media under anaerobic and normoxic conditions at eight indicated time points (9 clinical samples + 144 SHI cultures +144 NEMM cultures) were respectively subjected to high through-put 16S rRNA gene sequencing at Biomarker Technologies (Biomarker Technologies, Beijing, China) where the total DNA was isolated, amplified and sequenced according to their standard procedures. In brief, microbial DNA was extracted from the bacterial suspensions according to the manufacturer’s protocols. DNA concentration was assessed by a Nanodrop (Thermo Scientific) and quality was determined by agarose gel electrophoresis. PCR was used to amplify the variable regions 4 and 5 (V4-V5) of bacterial 16S rRNA gene by using 515F (ACTCCTACGGGAGGCAGCA) and 907R (GGACTACHVGGGTWTCTAAT, H = A/C/T, V = A/C/G, W = A/T) PCR primers. The PCR procedure was set as previously published research (Li et al., 2017). Purified amplicons were pooled in equimolar solutions and paired-end sequenced (2 × 300) on an Illumina MiSeq platform. After passing data quality check, the original high-throughput sequencing data were converted into FASTQ format files for bioinformatics analysis. The high-quality tag data were obtained after filtering and compared with sequences in the SILVA database (http://www.arb-silva.de).
Bioinformatics analysis
The bioinformatics analysis was conducted using QIIME2 2020.6 (Fig. S1). The alpha diversity indices of Chao1, ACE, Shannon, Simpson and phylogenetic diversity (PD whole tree) were calculated using Mothur software (version v.1.30) with coverage over 99%. Beta diversity analysis was performed by non-metric multi-dimensional scaling (NMDS) analysis based on the weighted unifrac and unweighted unifrac algorithms. Linear discriminant analysis (LDA) effect size (LEfSe) was conducted to define the biomarkers of these groups. The threshold on the logarithmic LDA score for the distinguishing features was set to 4.0. Microbial functions were predicted using PICRUSt2 (v1.0.0) software following the online protocol and aligned to the Clusters of Orthologous Groups of proteins (COG) database. The profiles of Kyoto Encyclopedia of Genes and Genomes (KEGG) levels were generated based on the KEGG database. Analysis of Variance (ANOVA) is mainly used to test whether the mean of multiple samples is equal, so as to judge the significant difference of the average of two or more samples. Level of statistical significance (P value) was set as <0.05 for all comparisons. The feature sequences of Top 80 abundance ratio at OTU/ASV taxonomic level were selected, multiple sequence comparison was performed with QIIME command line and a taxa tree was constructed. Then the R package was used to combine the taxa tree and species taxonomic abundance data.
Species identification using full-length 16S rRNA gene amplicon sequence
A total of 1,344 single colonies from NEMM-agar plates and 1,312 single colonies from SHI-agar plates were picked up and inoculated into NEMM and SHI liquid media, cultured in aerobically or anaerobically, respectively. DNA from these clinical isolates was extracted using hexadecyl trimethyl ammonium bromide (CTAB) methods (Zheng, Gao & Deng, 2012). The full-length of 16S rRNA gene was amplified via PCR reaction. 27F (AGAGTTTGATCCTGGCTCAG) and 1492R (GGTTACCTTGTTACGACTT) served as forward and reverse primers, respectively. PCR reaction conditions were set as follows: predenatured at 94 °C for 5 min, 35 cycles, including denaturation at 94 °C for 30s, annealing at 54 °C for at 30s, elongation 72 °C for 90s, and extended at 72 °C for 10 min. The full-length of 16S rRNA gene amplicons were sequenced using the Sanger method. The sequences of 16S rRNA gene were blasted in either NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi) or EzBioCloud (https://www.ezbiocloud.net/) to establish taxonomic relationships based on currently recognized prokaryotic reference species. Sequences with homology below 98.65% were defined as potential new strains (Kim et al., 2014).
Statistical analysis
The t-test was performed on the species abundance data between the groups using Metastats software. Principal component analysis was used to analyze the composition of the microbiome. BugBase was used to evaluate microbial phenotype followed by the Mann–Whitney U test. The species leading to the composition difference between the two groups of samples were determined by P value or Q value (correcting P value). The level of statistical significance (P value or Q value) was set as <0.05.
Results
Comparison of bacterial community between S-ECC clinical dental plaque samples and their cultured bacteria
To investigate the difference in bacterial community of S-ECC among the original dental plaque, and cultured bacteria, we recruited nine S-ECC patients, half of the samples were used for culture and the other half for 16S rRNA gene high-throughput sequencing directly without culture. Alpha diversity indices of ACE, Chao1, Shannon and Simpson showed clinical dental plaque samples had greater alpha diversity than either all their cultured counterparts (Fig. 1A) or the cultured bacteria under normoxia and anaerobic conditions (Fig. 1B), although the Venn diagram showed that the OTU numbers of cultured bacteria at species level under anaerobic and normoxia conditions were higher than that in clinical samples (Fig. 2A). Core flora of clinical S-ECC samples had 14 common features, including two unclassified Streptococcus, one unclassified Granulicatella, two unclassified Veillonella, one unclassified Fusobacterium, one unclassified Aggregatibacter, one unclassified Kingella, one unclassified Lachnoanaerobaculum, one unclassified Lactobacillales, one unclassified Abiotrophia, one unclassified Rothia, one Rothia aeria, and one Capnocytophaga endodontalis (Fig. S2).
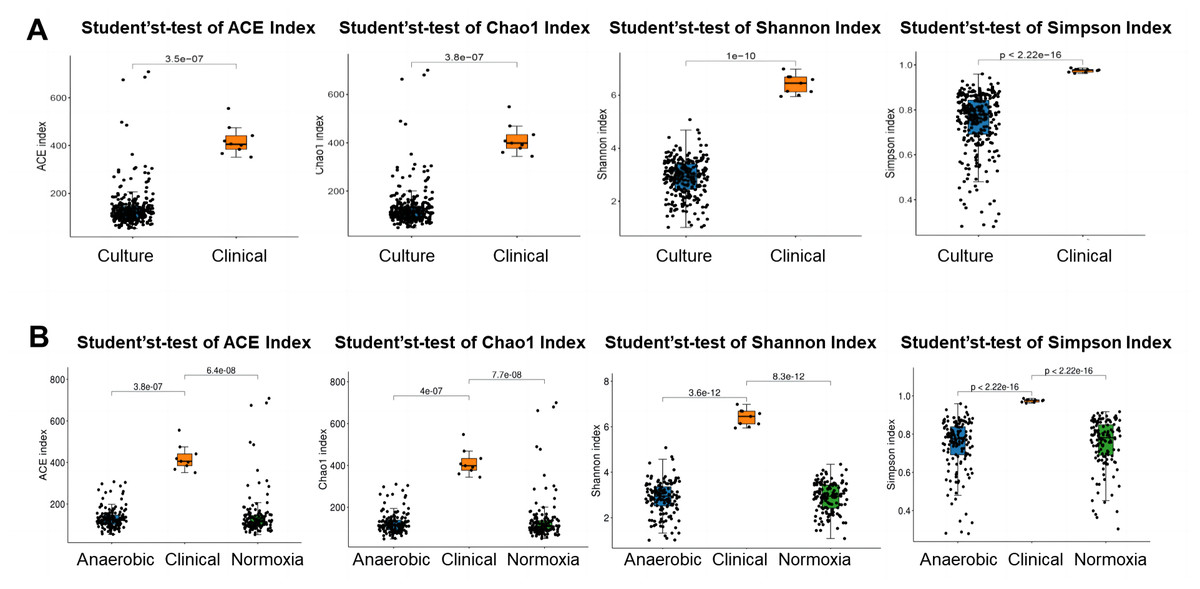
Figure 1: Alpha diversity of bacterial communities among aerobic culture, anaerobic culture and clinical.
(A) Alpha diversity was measured with ACE index, Chao1 index, Shannon index and Simpson index between clinical samples and liquid media cultured bacteria. (B) Alpha diversity was measured with ACE index, Chao1 index, Shannon index and Simpson index between clinical sample group and liquid media cultured bacteria group among clinical sample group, two liquid media cultured bacteria under anaerobic condition group and under normoxia condition group.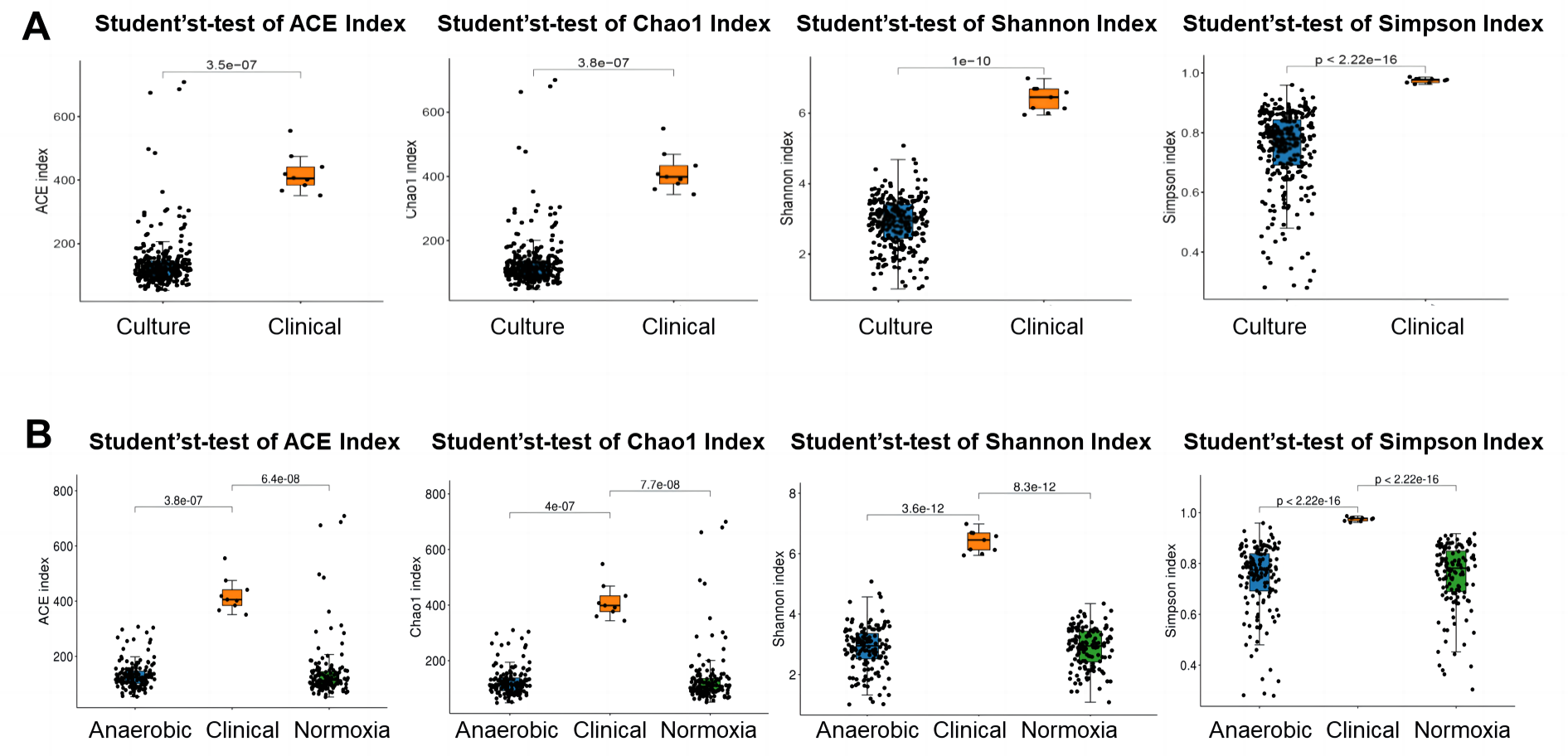{kind=link}
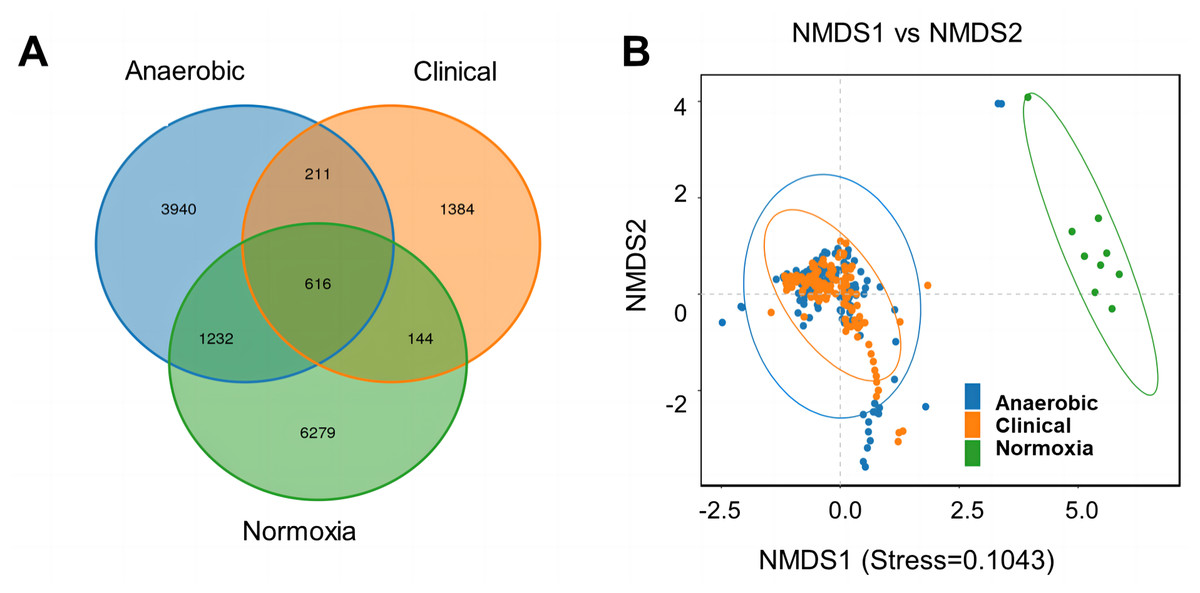
Figure 2: Venn diagram and NMDS based on the abundance of OTUs.
(A) Venn diagram displays the number of common and unique characteristics among the groups. Different colors represent different groups. The overlaps represent the common taxa between groups, and the non-overlapping portions represent unique taxa in each group. (B) NMDS was measured with weighted UniFrac. Each sample is represented by a dot.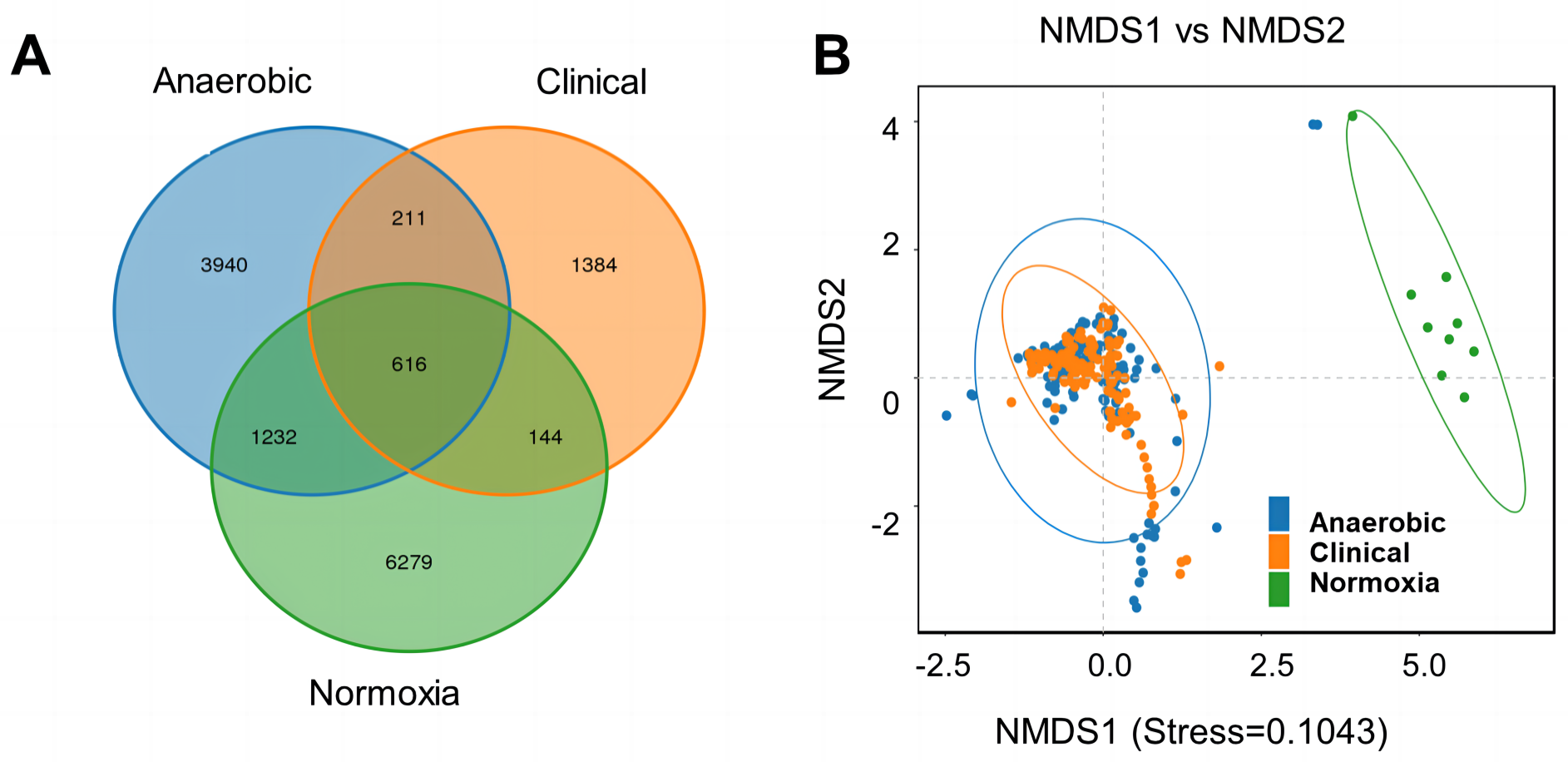{kind=link}
To visualize the distribution of samples in coordinate system, NMDS analysis showed that anaerobic cultured bacteria and normoxia cultured bacteria samples had similar culture features. However, both had a markedly different bacterial structure in comparison with clinical sample group (Fig. 2B).
To display the composition of dominant bacteria, the stacked column plot showed the percentage of each high-abundance bacteria. In clinical samples, except Veillonella, Leptotrichia and Streptococcus, other bacteria occupied the most of bacterial community. No matter under anaerobic and normoxia conditions, the percentages of other bacteria, Veillonella and Leptotrichia were clearly decreased. Meanwhile, there were observed increases in Desemzia, Actietobacter, Lactobacillus, Limosilactobacillus, Ligilactobacillus, Vagococcus and Streptococcus (Fig. 3A). The results indicated that there were more bacterial categories in clinical samples than cultures by media. NEMM and SHI media only could cultivate part of clinical bacteria in S-ECC plaques. There is still a lot of room for improvement in the culture medium.

Figure 3: Bacterial community structure analysis.
(A) The distributions of the predominant bacteria at genus level. (B) The relative abundance of differential predominant bacteria among aerobic culture, anaerobic culture and clinical groups analyzed at genus level by ANOVA.{kind=link}
To detect the difference among the groups, ANOVA was used to display the relative difference of bacteria at the genus level. The results showed that both anaerobic and normoxia cultures were beneficial for the growth of Streptococcus, Bacillus (including other unclassified Bacillus) and Bifidobacterium. The relative higher abundance of most other bacteria, including Leptotrichia, Veillonella, Corynebacterium, Actinomyces, Fusobacterium, Neisseria, Campocytophaga, Prevotella, Selenomonas, Campylobacter and Aggregatibacter in clinical samples presented marked lower percentage in both anaerobic and normoxia cultures. It suggested that most of the above-mentioned bacteria categories could not be successfully enriched by NEMM and SHI media (Fig. 3B).
To assess the predicted function of the bacterial community in clinical samples and the cultured bacteria samples, microbial phenotype prediction was evaluated by BugBase followed by the Mann–Whitney U test. The results showed that there were decreases in Gram-positive bacteria, facultatively anaerobic bacteria and stress tolerance ability, but increases in Gram-negative bacteria, anaerobic respiration, biofilms forming and potential pathogenicity in clinical samples compared with cultured samples under aerobic and anaerobic conditions (Fig. 4). The results indicated that uncultured Gram-negative, anaerobic, potentially pathogenic and biofilm-forming bacteria may contribute to S-ECC development.

Figure 4: BugBase analysis among different groups.
The three lines from the bottom up are the lower quartile, the mean and the upper quartile in BugBase analysis.{kind=link}
NEMM cultivates more abundant bacteria species compared to SHI medium
All cultured microbiota were examined in terms of both species proportion and abundance, with a specific focus on the enrichment of species. Of the organisms presented in the inoculum, all 32 phyla, 87 classes, 260 orders, 501 families, 1,107 genera and 1,502 bacterial species were captured. We identified 3,719, 5,605, 3,705, and 3,460 OTUs in the aNEMM, nNEMM, nSHI and aSHI groups, respectively. Among them, 608 OTUs were uniform, occupying 4.9% of all the OTUs detected, indicating that a steady facultative anaerobe microorganism composition was found in all four fluid media. The other OTUs were not shared in all groups. Among these variable microbiomes, 2,061 OTUs were unique to the aNEMM group, while 1,848 OTUs could be detected only in aSHI. Moreover, 4,010 OTUs could be found only in nNEMM groups, while 2,096 OTUs could be found only in nSHI groups.
To compare NEMM with SHI, which are suitable for cultivating specific bacterial types, we further analyzed 16S rRNA gene high-throughput sequencing data. Alpha diversity indices of ACE and Chao1 showed no significant difference between NEMM and SHI under the normoxia condition. However, under anaerobic culture conditions, NEMM had greater alpha diversity indices of ACE and Chao1 than SHI (Fig. 5A). NMDS analysis showed that regardless of whether anaerobic or normoxia, cultured bacteria samples had similar features (Fig. 5B). Venn diagram showed that the OTU numbers of NEMM cultured bacteria under anaerobic and normoxia conditions were higher than that in SHI cultured bacteria under the same conditions (Fig. 5C).

Figure 5: General bacterial community structure analysis between NEMM and SHI media during aerobic (normoxia) and anaerobic culture.
(A) Alpha diversity was measured with ACE index and Chao1 index. P < 0.05 was considered statistically significant. (B) NMDS was measured with Unweighted UniFrac. Each sample is represented by a dot. (C) Venn diagram displays the number of common and unique characteristics among the groups.{kind=link}
Differential predominant bacteria between NEMM and SHI were analyzed in the relative abundance of bacteria at the genus level by ANOVA. Of the 19 most predominant bacterial genera found in NEMM and SHI media, Ligilactobacillus, Limosilactobacillus, Lactobacillus, and Desemzia showed the highest abundance levels. Based on the relative abundance of cultured bacteria, NEMM was suitable for cultivating bacteria including Lactobacillus, Desemzia, Veillonella, Psychrobacterium and Leptotrichia, while SHI was good for culturing Ligilactobacillus, Limosilactobacillus, Bacillus, Neisseria, Leuconostoc, Cryptobacterium, Aggregatibacter and Kingella (Fig. 6).

Figure 6: The relative abundance of differential predominant bacteria between NEMM and SHI analyzed at genus level by ANOVA.
{kind=link}
Bugbase was used to predict the function of bacterial community in NEMM and SHI under anaerobic and normoxia culture conditions. The results showed that the microbiota developed in NEMM were predicted stronger anaerobic ability, whether under anaerobic conditions or normoxic conditions. Under normoxic culture conditions, there were no significant differences in Gram-positive bacteria among the four groups (aNEMM, aSHI, nNEMM and nSHI). However, under anaerobic conditions, NEMM supported the growth of more Gram-negative bacteria and less Gram-positive bacteria. Dental plaque samples cultured in NEMM were more likely to be stress tolerant and less likely to be potential pathogens and form biofilms. Moreover, it had weaker facultatively anaerobic ability when under anaerobic conditions (Fig. 7).

Figure 7: BugBase analysis among different groups.
Note: X-axis, group name; Y-axis, Relative abundance in percentage. The three lines from bottom to top are lines indicating lower quartile, average and upper quartile.{kind=link}
To assess the effects of the two media on the predicted gene categories (COGs), we compared the predicted COGs between aNEMM and aSHI, nNEMM and nSHI (Fig. 8A). NEMM showed more ability in carbohydrate transport and metabolism than SHI medium under both anaerobic conditions and normoxia conditions. Additionally, those in NEMM displayed an increased relative abundance of protein functions linked to defense metabolism, inorganic ion transport and metabolism under anaerobic conditions. Furthermore, pathways of membrane transport and cellular community-prokaryotes were associated with aNEMM group (Fig. 8B).

Figure 8: Function prediction by PICRUSt2.
The differential analysis of COG function (A) and KEGG pathway (B) show the abundance ratio of different functions between groups, whose middle figures show the difference ratio of functional abundance within the 95% confidence interval.{kind=link}
NEMM and SHI media were suitable for culturing specific bacteria respectively
The predominant bacteria were largely consistent among the four groups (aNEMM, aSHI, nNEMM, nSHI), but different relative abundances could be observed (Fig. S3). More genera, species and uncultured bacteria were captured in aNEMM groups at day 1 (Table S2) under anaerobical condition compared with those in aSHI Day1 group. For normoxia conditions, there was a significant difference of genera and uncultured bacteria between NEMM and SHI media (Table 1). Table S3 showed that genera, species and uncultured bacteria detected in nNEMM were higher compared to nSHI at each time point.
| aNEMM vs aSHI | nNEMM vs nSHI | |||||
|---|---|---|---|---|---|---|
| mean ± SD (aNEMM) | mean ± SD (aSHI) | P value | mean ± SD (nNEMM) | mean ± SD (nSHI) | P value | |
| Genus | 268.43 ± 15.76 | 253.57 ± 47.13 | 0.5 | 369.86 ± 61.63 | 255.57 ± 65.01 | 0.0469* |
| Species | 345.86 ± 14.97 | 327.71 ± 55.23 | 0.5781 | 453.43 ± 83.62 | 317.86 ± 80.56 | 0.0781 |
| Uncultured bacteria | 18.29 ± 2.60 | 19.00 ± 6.97 | 0.875 | 30.14 ± 8.06 | 17.71 ± 7.26 | 0.0469* |
Notes:
The nonparametric Wilcoxon test was performed to evaluate differences.
- SD
-
standard deviation
Principal component analysis of the development of the oral microbiome shows distinct shifts in the composition of the microbiome. We could see the changes in bacterial community structure over time in Figs. 9A and 9B. The proportions of Streptococcus were reduced along with time in the four groups. In this study, the NEMM as a growth and colonization substrate resulted in a dynamic microbial succession, characterized by an initial proportional reduction in Veillonella, corresponding to an increase of Lactobacillus and unclassified_Bacilli proportions under anaerobic conditions. Bifidobacterium reached the highest percentage on the 15th day and then decreased in aNEMM group. In the aSHI group, the proportions of Lactobacillus and unclassified_Bacilli increased gradually before the 15th day. While Ligilactobacillus increased rapidly from day one to day three in both aSHI and nSHI groups. In nNEMM group, Ligilactobacillus, unclassified_Bacilli, Lactobacillus, Lacticaseibacillus and Lactiplantibacillus showed the time-dependent accumulation, while Acinetobacter exhibited the opposite tendency.

Figure 9: Bacterial genus change between NEMM and SHI medium over time under anaerobic condition (A) and normoxia condition (B).
{kind=link}
The taxa that most likely explains the differences between NEMM and SHI media from different time points are defined by LEfSe. Figures 10A and 10B showed cladograms representing the potential biomarkers of different groups. At the genus level, Veillonella was significantly enrichment in the aNEMM cultured Day1 group (aNEMM-Day1), while Streptococcus, Gardiobacterium and Granulicatella exhibited relatively higher abundance in the aSHI-Day1 group. Bifidobacterium, Akkermansia and Desemzia were significantly more abundant in the aNEMM-Day15, aNEMM-Day21 and aNEMM-Day28 groups, respectively. While Ligilactobacillus, unclassified_Bacilli and Lacticaseibacillus were most abundant in aSHI-Day6, aSHI-Day21 and aSHI-Day28 groups, respectively. For samples grown aerobically, significant bacterial differences were also detected, with Veillonella, Desemzia and Lacticaseibacillus were remarkably enriched in the nNEMM-Day1, nNEMM-Day9 and nNEMM-Day28 groups at the genus level, while Streptococcus exhibited higher abundance in the nSHI-Day1 group (Figs. 11A and 11B, LDA > 4, P < 0.05).

Figure 10: Cladogram and LDA score for taxonomic representation of significant differences among groups under anaerobic condition.
The colored nodes from the inner to the outer circles represent taxa from the phylum to genus level. The significantly different taxa are signified by different colors representing the four groups through cladogram (A) and LDA score (B).{kind=link}

Figure 11: Cladogram and LDA score for taxonomic representation of significant differences among groups under normoxia condition.
The colored nodes from the inner to the outer circles represent taxa from the phylum to genus level. The significantly different taxa are signified by different colors representing the four groups through cladogram (A) and LDA score (B).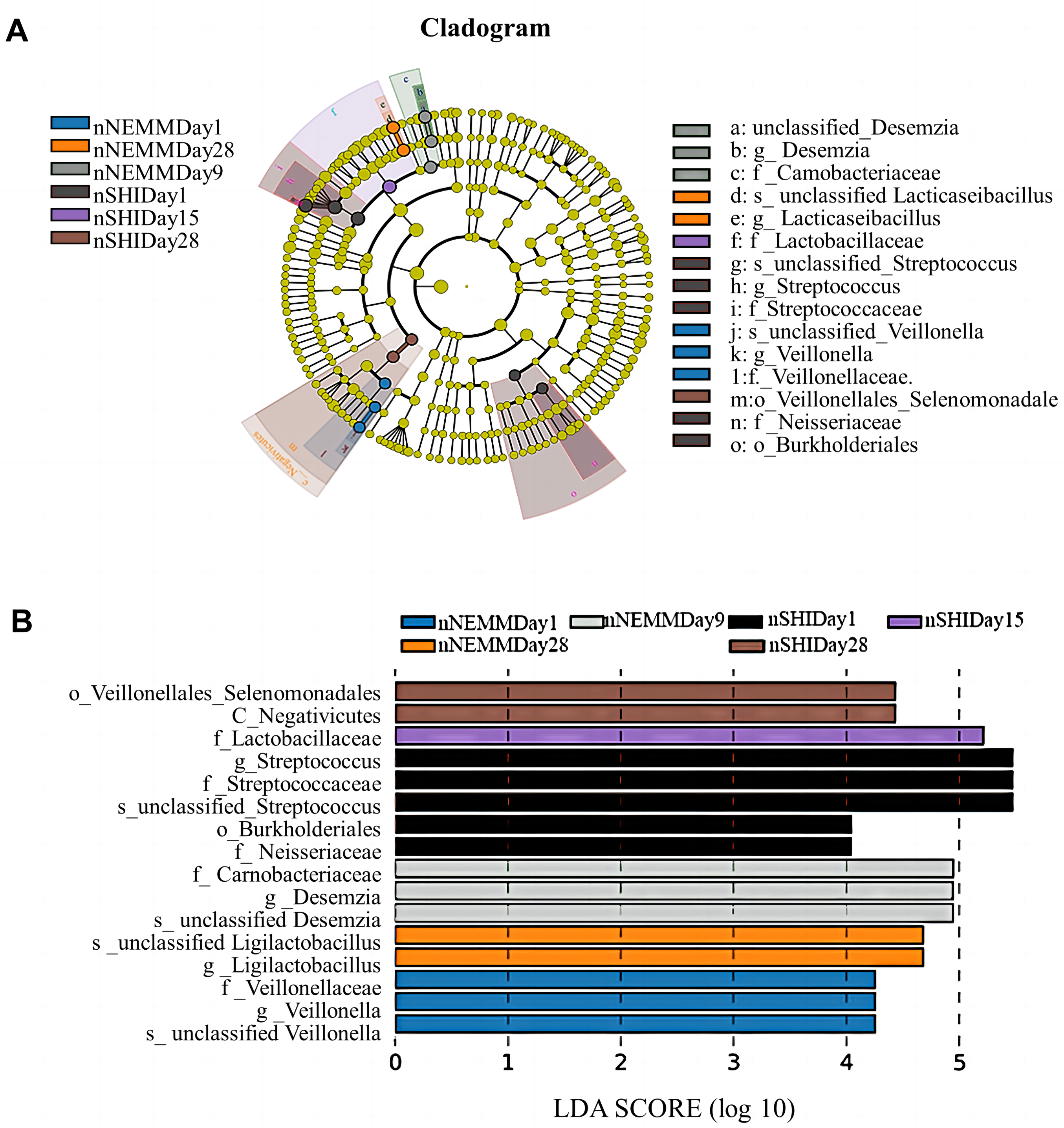{kind=link}
To study the difference in microbial community abundance between two groups of samples, Metastats software was used to conduct t-test on genus richness data between groups (Fig. 12). At the genus level, Actinobacillus, Pseudoxanthomonas were significantly enriched in aSHI medium while Bilophila was significantly enriched in aNEMM medium (Fig. 12A). Unclassified Clade III, NS3a marine group and unclassified Saprospiraceae were more abundant in nNEMM suspension (Fig. 12B). The same tendency can also be seen in Fig. S4. From the taxa tree, aNEMM was suitable for culturing Anaerolinea, Aerococcus, Lactobacillus and Negativicutes. Planococcus, Desemzia and Psychrobacter were enriched in NEMM no matter under normoxia or anaerobic condition. Geobacillus, Lactococcus, Clostridiaceae, Hyphomicrobium, Pseudolabrys, Paracoccus, Rhodobacter, Rickettsiaceae, Sphingobium, Legionella, Moraxella, Luteimonas and Lysobacter were suitable for growth in nNEMM. Bacillus, Pediococcus, Brevundimonas, Caulobacter, Phenylobacterium, Sphingomonas and Pseudoxanthomonas were suitable for growing in aSHI. Blastomonas were enriched in nSHI. Abiotrophia grew well in SHI both under normoxia and anaerobic conditions (Fig. S4).
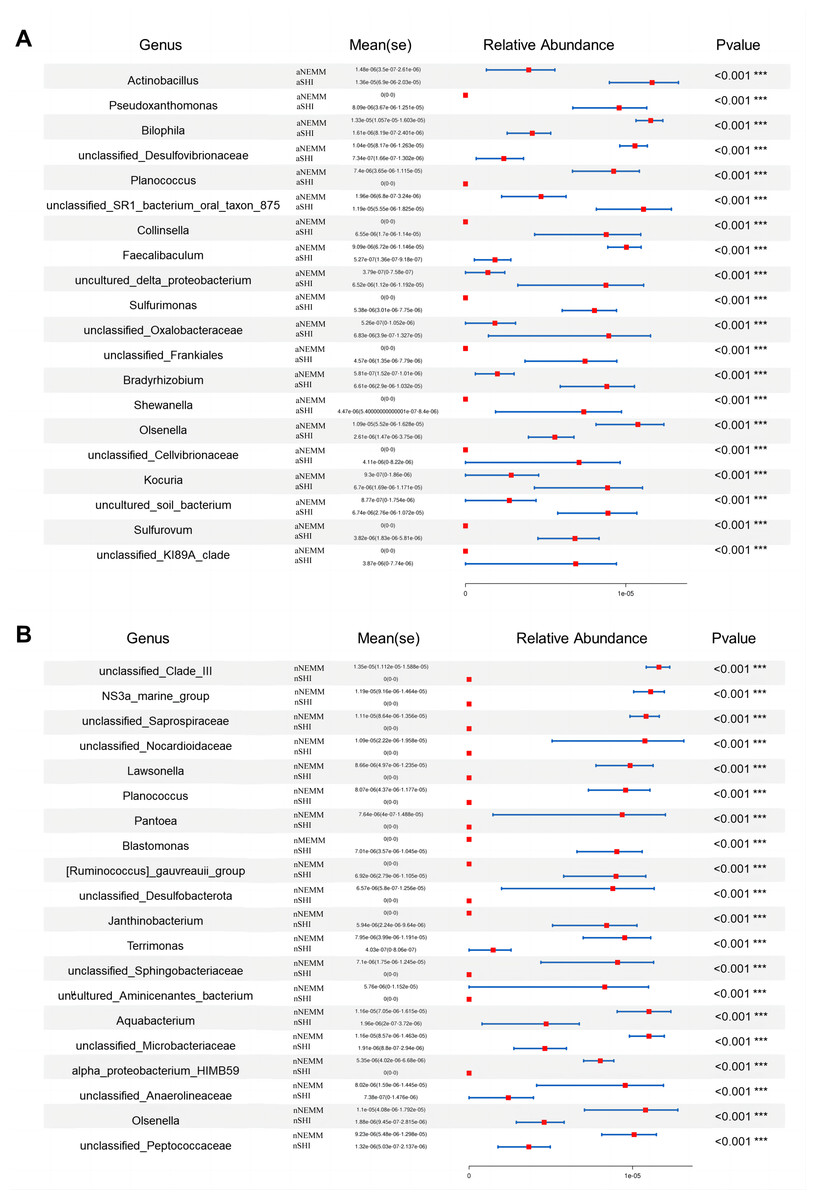
Figure 12: Metastats statistical analysis at genus level under anaerobic condition (A) and normoxia condition (B).
{kind=link}
Microbiota developed in NEMM and SHI media, under anaerobic and normoxia conditions exhibited distinct community structures. According to the relative abundance ratios of genera shown on taxa tree (Fig. S4), we constructed Table 2 for clearly showing the different microbial profiles cultured in NEMM medium and SHI medium. At the genus level, Anaerolinea, Planococcaceae, Acerococcus, Desemzia, Sphongopyxia, Psychrobacter and some not assigned species were significantly enriched in aNEMM. Planococcaceae, Geobacillus, Facklamia, Desemzia, Lactococcus, Blastomonas, Sphongopyxia, Legionella, Psychrobacter, Luteimonas, Lysobacter and Pseudoxanthomonas were more abundant in nNEMM conditions. The aSHI condition was suitable for Pseudograclibacillus, Sphingomonas and Pseudoxanthomonas, while Lactococcus, Blastomonas, Sphingomonas, Legionella, Luteimonas, Lysobacter and Pseudoxanthomonas were more abundant in nSHI condition.
| Bacteria type | aNEMM | aSHI | nNEMM | nSHI |
|---|---|---|---|---|
| Anaerolinea | ✓ | |||
| Planococcaceae | ✓ | ✓ | ||
| Pseudograclibacillu | ✓ | |||
| Geobacillus | ✓ | |||
| Acerococcus | ✓ | |||
| Facklamia | ✓ | |||
| Desemzia | ✓ | ✓ | ||
| lactococcus | ✓ | ✓ | ||
| Blastomonas | ✓ | ✓(dominant) | ||
| Sphingomonas | ✓ | ✓ | ||
| Sphongopyxia | ✓ | ✓ | ||
| Legionella | ✓ (dominant) | ✓ | ||
| Psychrobacter | ✓ | ✓ | ||
| Luteimonas | ✓ (dominant) | ✓ | ||
| Lysobacter | ✓ (dominant) | ✓ | ||
| Pseudoxanthomonas | ✓(dominant) | ✓ | ✓ | |
| Not assigned | ✓ | ✓ (dominant) | ✓ |
Notes:
Note that ✓ means significantly enriched.
NEMM helps to culture previously uncultivated oral bacteria
NEMM supported the growth of many oral species that have not been cultured from S-ECC clinical samples so far. All 128 previously uncultivated species were detected by 16S rRNA gene sequencing using SILVA as the reference database.
To study the difference in microbial community abundance between groups of samples, Metastats analysis was used to conduct t-test on species richness data between groups. Uncultured bacteria including methanogenic_archaeon, Truepera_sp., Acidobacterium_sp., Desulfovibrionaceae_bacterium and Nitrosomonadaceae_bacterium could only be detected in aNEMM group. Comparing relative abundance of these uncultured species between aNEMM and aSHI media, uncultured bacteria including forest_soil_bacterium, Clostridiales_bacterium, Desulfuromonadales_bacterium, Firmicutes_bacterium and Alphaproteobacteria_bacterium were observed higher in aNEMM group (Table S4). Discrepancies were also detected between the nNEMM and nSHI groups, with higher abundances of uncultured bacteria including Desulfuromonadales_bacterium, Bacteroidetes_bacterium, Clostridium_sp., Mollicutes_bacterium, Bacteroidales_bacterium, Holophaga_sp. and Acidobacteria_bacterium in the nNEMM group. Moreover, uncultured bacteria including methanogenic_archaeon, Sphingobacteriales_bacterium, Aminicenantes_bacterium, marine_bacterium, forest_soil_bacterium, Acidobacterium_sp., Xanthomonadaceae_bacterium, Sorangiineae_bacterium, marine_microorganism and candidate_division_SBR1093_bacterium were only observed in the nNEMM group (P < 0.05) (Table S5).
The serial diluted culturomics samples derived from above broth medium conditions were inoculated into the two media-agar plates and incubated under normoxia and anaerobic, respectively, for 1-5 days (Fig. S5). The bacterial diversity gradually decreased over time. With the extension of culture time, the difference of dominant bacteria in the two media gradually decreased, and from the morphological point of view, SHI and NEMM agar plates tended to cultivate similar bacterial diversity. After this process, the slow-growing colonies were subcultured and identified to isolate previously uncultivated phylotypes. Through culturomics, uncultivated bacteria belonging to the Prevotella, Actinomyces, Pseudomonas, Granulicatella, Lactobacillus, Campylobacter, Streptococcus and Bacillus genera were identified in the biofilm growth media. The oral taxa identified under the four conditions were shown in Fig. S6, displaying that both NEMM and SHI media could culture the uncultivated bacterium in aerobic and anaerobic conditions.
Also, it turned out that Achromobacter spanius, Lactobacillus delbrueckii subsp and Actinomyces naeslundii in aNEMM, Pseudomonas sp. and Veillonella parvula in aSHI, Brevibacillus agri in nNEMM, Leuconostoc lactis, Chryseobacterium indologenes, Cardiobacterium hominis and Kingella bonacorsii in nSHI may tend to be enriched in each growth condition.
Discussion
Due to different nutritional requirements, factors produced by other microbes, strict inter-species interactions, slow growth, competition/inhibition and dormancy in diversified bacterial species (Overmann, Abt & Sikorski, 2017; Tidjani Alou et al., 2020), developing a medium for cultivating microbiota, especially oral microbial flora, has always been a challenge. In previous studies, SHI medium has been widely used for oral bacteria research, but it was originally designed to support different sub-populations within the original oral microbial community derived from pooled saliva (Edlund et al., 2013; Tian et al., 2010). Specific nutrient or chemical requirements may be necessary for the growth of some fastidious bacteria. Some studies regarding functional alteration associations with S-ECC indicated that carbohydrate metabolism, amino acid metabolism, and metabolism of cofactors and vitamins are the major pathways of KEGG in caries and healthy groups (Tang et al., 2022; Wang et al., 2019). Thus, we designed a novel bacterial medium attempting to culture the fastidious bacteria, which were nutritionally demanding and were difficult to grow on ordinary medium, especially for dental plaque in a diseased state of S-ECC.
We designed NEMM medium based on S-ECC bacterial metabolism and the role of nutrients in bacterial culture and proliferation. Bacteria must obtain nutrients from their surrounding environment to survive. Carbohydrates are the primary energy and carbon sources for most bacteria during growth in laboratory media. In bacteria, proteins in the outer membrane surrounding the cell actively transport carbohydrates and trace nutrients like iron into the cell’s interior (Zinke et al., 2024). In this study, NEMM meets the needs of previous studies’ findings that multiple pathways involving carbohydrate metabolisms are obviously up-regulated in the S-ECC group compared to caries free group (Tang et al., 2022; Wang et al., 2019). It should be emphasized that the upregulation of carbohydrate transport and metabolism in NEMM might reflect more bacteria with higher cariogenic potential.
In the metabolism of S-ECC, several pathways involving carbohydrate metabolism were deferentially regulated (Tang et al., 2022; Wang et al., 2019). Dextrose and corn starch served as carbon sources. β-Nicotinamide adenine dinucleotide (NAD), thiamin pyrophosphate (TPP) and L-glutamine play key roles in glycolysis. As a cofactor in over 300 redox reactions, NAD (NAD(+)) and its reduced form NADH (reduced nicotinamide-adenine dinucleotide) play a critical role in cellular metabolism due to their function (Zhou et al., 2011). TPP, an active form of thiamine (Vitamin B1), is an essential molecule for all living organisms (Goyer, 2017). TPP acts as a co-factor for key enzymes of glycolysis such as pyruvate dehydrogenase, transketolase, and pyruvate decarboxylase (Llavero-Pasquina et al., 2022; Strobbe & Van Der Straeten, 2018). Many bacteria rely on the TPP to lose the ability to produce thiamine de novo (Llavero-Pasquina et al., 2022).
Cofactors and vitamins are vital for bacteria growth and function as well. Glutamine comprises more than 50% of the body’s free amino acid pool and is a precursor for the synthesis of nucleic acids and glutathione (Heys & Ashkanani, 1999). Glutamine might initiate the signaling pathways related to the metabolism of nitrogenous compounds in bacteria (Dai et al., 2013). A study demonstrated that L-glutamine supplements in basal salt medium with carbon and nitrogen sources along with requisite inoculum size yielded the best quantities (Patel et al., 2021). Cobalamin (vitamin B12) has regulatory involvement in gene expression, microbial folate, ubiquinone, and methionine processes (Romine et al., 2017; Rosnow et al., 2018), suggesting a requirement for this co-factor. Cobalamin was demonstrated to be the essential nutrilite (Lochhead & Thexton, 1951) because of the lack of the ability to biosynthesize cobalamin for most bacteria (Lu et al., 2020). Recent research reported that Porphyromonas gingivalis growth relies on vitamin B12 (Saiki et al., 2022).
Adenine and guanine have various functions in bacteria. Kiyoshi and colleagues added adenine to culture Clostridium saccharoperbutylacetonicum strain N1-4 at 37 °C. The addition of adenine exhibited the maintenance of cell viability originating from the maintenance of ATP levels (Kiyoshi et al., 2017). Torino et al. (2005) compared the growth and extracellular polysaccharides(EPS) production by Lactobacillus helveticus ATCC 15807 in the different media, and found that the addition of p-amino benzoic acid did not affect either growth or the EPS production, while guanine displayed a dual effect, a decrease in cell growth and an increase in EPS formation.
To date, although nearly 1/4 of bacteria are still uncultured in the oral cavity, researchers have known the existence of these uncultured bacteria through next-generation sequencing, third-generation sequencing or metagenomics technologies. However, some species were likely present at a low abundance in the inoculum and were therefore not detected via sequencing. Cultivating these “dark matter” in S-ECC plaque will help to explore their contributions to health and their role in disease etiology, and pave the way for prevention of dental caries. In this study, we combined 16S rRNA gene sequencing and culturomics to prove that NEMM can support the development of more bacteria from S-ECC dental plaque and help to cultivate uncultured or targeted bacteria.
For example, Veillonella, which belongs to Gram-negative obligate anaerobic coccus, is recognized to play an important role in the S-ECC-causing process and identified as a candidate biomarker in S-ECC with low abundance or without S. mutans (Gross et al., 2012; Zhang et al., 2022). The potential biomarkers of different groups were presented by LEfSe. Among these significantly different genera, Veillonella was one of the most specific biomarkers in aNEMM group. Moreover, we exhibited the changing process of bacterial community structure through 16S rRNA gene sequencing and culturomics. It indicated a better capture ability for Veillonella before the first day in aNEMM group. Growth development and bacterial activity are important aspects that need to be addressed. It will be helpful for isolating targeted strains at proper time points. The relative abundance of the bacterial genus shifted in biofilms over 28 days of growth in NEMM and SHI medium. It is likely that inhibition of metabolism, due to low pH and possibly carbohydrate limitation, starts to impact growth of some community members and leads to a shift in the community structure. Vagococcus, which is uncommon in the oral bacteria community, was found enriched in NEMM and SHI medium. Actually, there has been some articles that reported their existence in the oral cavity such as Vagococcus fluvialis (Al-Ahmad et al., 2008). We can rule out the possibility of medium contamination from Fig. S7. Therefore, we speculate some of them may come from the food eaten such as milk or food additives or non-food substances such as soil and toys contaminated by bacteria entering the children’s mouths through accidental events (Wullschleger et al., 2018).
Conclusions
In conclusion, NEMM medium was superior in yielding more biomass when used to cultivate dental plaque-derived biofilm. The 16S rRNA gene sequencing results strongly supported that NEMM was able to maintain an in vitro oral microbial community with high diversity. It also supported the growth of many oral species that have not been cultured so far, which may be helpful in capturing more pathogenic bacteria and will contribute to real strains for research via culturomics.
Supplemental Information
{kind=link}
Venn diagram displays the number of common and unique characteristics among the clinical samples
Different colors represent different groups. The core microbial community contains 14 bacteria at species level. C:clinical sample.
{kind=link}
The distributions of the predominant bacteria at the genus level
The predominant taxa (the top ten abundance levels) are shown.
{kind=link}
Taxa tree among different groups at genus level
Visualization is carried out to display genera in the form of evolutionary tree and pie chart, which reflects:(1) Evolutionary relationship between genera; (2) Information such as the relative abundance ratios of genera between different groups.
{kind=link}
The morphology change of the bacteria community with time photographed by camera at NEMM and SHI solid medium(take sample S-ECC2 for example)
{kind=link}
The genera of bacteria isolated from the four culture conditions. (A) aNEMM; (B) aSHI; (C) nNEMM; (D) nSHI. Top 20 genera were list on the right of chart
{kind=link}
Horizontal agarose gel electrophoresis of chosen bacteria colonies
Blank controls are the PCR product generated from two liquid media as PCR templates, respectively. M:marker; NC: blank control.
{kind=link}
The clinical characteristics of S-ECC patients in this study
Count estimates of genus, species and previously uncultured bacteria detected in NEMM and SHI at anaerobic condition (aNEMM and aSHI) at different time points
Count estimates of genus, species and previously uncultured bacteria detected in NEMM and SHI at normoxia condition (nNEMM and nSHI) at different time points
Metastats analysis of previously uncultured bacteria detected in aNEMM and aSHI biofilms
Bacteria significantly higher in aNEMM compared to aSHI (P < 0.05).
Metastats analysis of previously uncultured bacteria detected in nNEMM and nSHI biofilms
Bacteria significantly higher in nNEMM compared to nSHI (P < 0.05).