
The complete mitochondrial genomes of four lagriine species (Coleoptera, Tenebrionidae) and phylogenetic relationships within Tenebrionidae
- Published
- Accepted
- Received
- Academic Editor
- Mudasir Ahmad Syed
- Subject Areas
- Entomology, Genomics, Taxonomy, Zoology
- Keywords
- Darkling beetles, Lagriinae, Mitogenome, Phylogenetic analysis
- Copyright
- © 2023 Wei and Shi
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2023. The complete mitochondrial genomes of four lagriine species (Coleoptera, Tenebrionidae) and phylogenetic relationships within Tenebrionidae. PeerJ 11:e15483 https://doi.org/10.7717/peerj.15483
Abstract
It is common to use whole mitochondrial genomes to analyze phylogenetic relationships among insects. In this study, seven mitogenomes of Tenebrionidae are newly sequenced and annotated. Among them, four species (Cerogira janthinipennis (Fairmaire, 1886), Luprops yunnanus (Fairmaire, 1887), Anaedus unidentasus Wang & Ren, 2007, and Spinolyprops cribricollis Schawaller, 2012) represent the subfamily Lagriinae. In this subfamily, the mitogenomes of the tribes Goniaderini (A. unidentasus) and Lupropini (L. yunnanus and S. cribricollis) were first reported; they were found to be 15,328–16,437 bp in length and encode 37 typical mitochondrial genes (13 PCGs, 2 rRNAs, 22 tRNAs, and a single noncoding control region). Most protein-coding genes in these mitogenomes have typical ATN start codons and TAR or an incomplete stop codon T–. In these four lagriine species, F, L2, I, and N are the most frequently used amino acids. In the 13 PCGs, the gene atp8 (Pi = 0.978) was the most diverse nucleotide, while cox1 was the most conserved gene with the lowest value (Pi = 0.211). The phylogenetic results suggest that Pimelinae, Lagriinae, Blaptinae, Stenochiinae, and Alleculinae are monophyletic, Diaperinae is paraphyletic, and Tenebrioninae appears polyphyletic. In Lagriinae, the tribe Lupropini appears paraphyletic because Spinolyprops is clustered with Anaedus in Goniaderini. These mitogenomic data provide important molecular data for the phylogeny of Tenebrionidae.
Introduction
The subfamily Lagriinae belongs to the family Tenebrionidae, which includes 11 subfamilies (Bouchard et al., 2011; Nabozhenko & Sadeghi, 2017; Kamiński et al., 2021), 11 tribes, and 273 valid genera that are widely distributed in the world (Bouchard et al., 2021). Most of the lagriine adults are herbivorous and mycophagous. Their larvae are forest-floor dwellers commonly found in leaf litter and other accumulations of dead plant matter (Matthews et al., 2010).
Although some taxonomists have made contributions to the higher-level classification of Lagriinae based on morphological characteristics (Doyen & Tschinkel, 1982; Doyen, Matthews & Lawrence, 1989; Bouchard et al., 2021) and molecular phylogeny (Kergoat et al., 2014; Aalbu, Kanda & Smith, 2017; Wu et al., 2022), the problems of classification remain unresolved. For example, the tribe Lupropini is paraphyletic. The genera Spinolyprops and Sphingocorse are placed in the tribe Lupropini; however, the morphological characteristics of this genus are more suitable for the tribe Goniaderini. Many genera of the tribe Lupropini are similarly situated and further studies are needed to resolve these discrepancies.
Mitogenomes have provided valuable data on phylogeny (Saccone et al., 1999; Cameron, 2014; Li et al., 2015; Qin et al., 2015; Nie et al., 2020; Tian, Yuan & Chen, 2020; Nie et al., 2021), evolution strategies (Krzywinski et al., 2011; Motyka et al., 2022; Nie et al., 2019), and species delimitation (Kim et al., 2021) over the past few decades. However, there are few studies about the higher-level phylogeny of Tenebrionoidea based on mitogenomes. Timmermans et al. (2010, 2016) used mitogenomes to explore the high-level phylogeny of Coleoptera, and their results suggested that Tenebrionoidea was monophyletic. Zhang et al. (2016) used ten mitogenomes of six subfamilies in Tenebrionidae to analyze the phylogenetic relationship of its subfamilies, and their results suggested that Lagriinae and Pimeliinae were sister groups, but that the relationships of Diaperinae and Tenebrioninae were inconclusive. Subsequently, Song et al. (2019) described the characteristics of the Gonocephalum outreyi mitogenome and reconstructed the higher-level relationship of Tenebrionoidea based on 26 tenebrionoid mitogenomes, showing that Tenebrionoidea is monophyletic, Tenebrionidae and Ciidae are sister groups, and Diaperinae and Tenebrioninae were non-monophyletic. Finally, Wu et al. (2022) reported on the characteristics of Alleculinae mitogenomes and reconstructed the high-level relationship of Tenebrionidae based on 36 mitogenomes, suggesting that Lagriinae, Pimeliinae, Stenochiinae, and Alleculinae were monophyletic, but that Diaperinae and Tenebrioninae were polyphyletic. Recently, genome data were also used to explore the phylogeny of beetles (McKenna et al., 2019; Cai et al., 2022).
In the present study, we sequenced and annotated the entire mitogenome sequences of four Lagriinae species, namely, Cerogira janthinipennis (Fairmaire, 1886), Luprops yunnanus (Fairmaire, 1887), Anaedus unidentasus Wang & Ren, 2007, and Spinolyprops cribricollis Schawaller, 2012. Of these, two tribes (Goniaderini and Lupropini) are the first to have their complete mitogenomes sequenced. We also conducted a preliminary analysis of genetic compositions and structural alterations to gain a better understanding of the phylogenetic relationships of Tenebrionoidea. Three newly-sequenced mitogenomes were provided (Crypsis chinensis Kaszab, 1946, Gonocephalum kochi Kaszab, 1952 and Morphostenophanes yunnanus Zhou, 2020) and data were obtained from the NCBI database to examine the phylogenetic relationship among species in the family Tenebrionidae. Mitogenomic data from these four lagriine species, as well as the evolutionary relationships within the subfamily Lagriinae, were reported in this study. The results of this work will provide a valuable basis for further evolutionary studies on beetles of Tenebrionoidea.
Materials and Methods
Sampling and DNA extraction
Cerogira janthinipennis and Crypsis chinensis specimens were collected in the Dayaoshan Mountains of the Guangxi Zhuang Autonomous Region, China. Luprops yunnanus and Spinolyprops cribricollis were collected using the sieve soil method in Jinghong City, Yunnan Province, China. A specimen of A. unidentasus was collected using the same method in Shiqian County, Guizhou Province, China. Gonocephalum kochi specimens were collected in Zunyi City, Guizhou Province, China, and Morphostenophanes yunnanus were collected in Yunlong County, Yunnan Province, China. The leg and thorax tissues were immediately preserved in 95% ethanol in the field and then were stored at −24 °C. The specimens were identified based on morphological characteristics. Total DNA was extracted from the muscle tissues using the Ezup Column Animal Genomic DNA Purification Kit (Shanghai, China) in accordance with the manufacturer’s instructions. This study was conducted under the approved guidelines of Animal Care and Use Committee of China West Normal University (approval number CWNU2022D021).
Sequencing, sequence assembly, annotation, and analysis
All of the mitogenomes in this study were sequenced with a whole genome shotgun strategy and Illumina sequencing technology using 150 bp paired-end reads. The adapter sequence and low-quality bases were deleted using Trimmomatic v. 0.36 (Bolger, Logse & Usadel, 2014). The target reads were assembled using IDBA UD v. 1.1.1 and Celera Assembler v. 8.3 (Crampton-Platt et al., 2015). The de novo assembly of the newly-generated sequences was conducted using Geneious 11.0.2 (Kearse et al., 2012). Protein-coding genes (PCGs) were identified as open reading frames. A total of 22 transfer RNA genes (tRNAs) were identified with the use of the MITOS Webserver, setting the default parameters with the Invertebrate Mito genetic code (Bernt et al., 2013). Their secondary structures were plotted manually from the MITOS predictions using Adobe Illustrator. Every sequence of tRNA genes was manually checked. The ribosomal RNA genes (rRNAs) and control region were identified by the boundaries of the tRNA genes. Mitogenome maps were produced using Organellar Genome DRAW COGDRAW (Greiner, Lehwark & Bock, 2019). The base composition and relative synonymous codon usage values of four Lagriinae mitogenomes were calculated using MEGA v 6.0 (Kumar, Stecher & Tamura, 2016). Strand asymmetry was calculated through the formula provided by Perna & Kocher (1995): AT-skew = (A – T)/(A + T), GC-skew = (G – C)/(G + C). The non-synonymous substitutions (Ka), synonymous substitutions (Ks), and Ka/Ks of PCG genes were calculated using DnaSP v. 5 (Rozas et al., 2003). Tandem repeat units in the control region of four Lagriinae species were identified using the Tandem Repeats Finder online tool (Benson, 1999).
Phylogenetic analyses
Phylogenetic trees for C. janthinipennis, L. yunnanus, A. unidentasus, S. cribricollis, and other tenebrionid beetles were reconstructed by aligning sequences of mitogenomes with those of 39 tenebrionid species (Table S1). Lymexylon navale and Hyloecetus dermestoides, belonging to Lymexyloidea, were chosen as outgroups, as they are phylogenetically closed to Tenebrionoidea in the Coleoptera (Timmermans et al., 2016; Cai et al., 2022). Phylogenetic trees were constructed using PhyloSuite v. 1.2.2 (Zhang et al., 2020) based on the maximum likelihood (ML) and Bayesian inference (BI) methods, respectively. The default parameters (ML: bootstrap: ultrafast; number of bootstrap: 5,000; maximum iterations: 1,000; minimum correlation coefficient: 0.90; replicates: 1,000; BI: generations: 2,000,000; sampling frequency: 100; number of runs: two; number of chains: four; burn-in fraction: 0.25) were used to construct phylogenetic trees. The default parameters (ML: bootstrap: ultrafast; number of bootstrap: 5,000; maximum iterations: 1,000; minimum correlation coefficient: 0.90; replicates: 1,000; BI: generations: 2,000,000; sampling frequency: 100; number of runs: two; number of chains: four; burn in fraction: 0.25) were used to construct phylogenetic trees. The resulting phylogenetic trees were edited and visualized using FigTree v. 1.43 (Horton et al., 2015).
Results
Genome organization and base composition
We sequenced and annotated the complete mitogenomes of four lagriine species Cerogira janthinipennis (ON303727, length: 15,328 bp), Luprops yunnanus (ON303728, length: 16,437 bp), Anaedus unidentasus (ON303730, length: 15,387 bp), and Spinolyprops cribricollis (ON303729, length: 15,454 bp), as well as three other tenebrionid species: Gonocephalum kochi (ON303729, length: 15,825 bp), Crypsis chinensis (ON303729, length: 16,724 bp), and Morphostenophanes yunnanus (MW822745, length: 15,616 bp). The complete mitogenomes of these seven species contained 13 protein-coding genes (PCGs), two ribosomal RNA genes (rRNAs), 22 transfer RNA genes (tRNAs), and a control region (CR). Four of the 13 PCGs (nad1, nad4L, nad4, nad5), two rRNAs, and eight of the 22 tRNAs (trnY, trnC, trnQ, trnV, trnL1, trnP, trnH, trnF) are encoded on the N-strand, while the other 23 genes (nine PCGs and 14 tRNAs) are encoded on the J-strand (Table S1; Figs. S1, S2). The mitogenome sequences of these four lagriine species were determined to be medium- to maximum-sized compared to those of the Tenebrionidae species as a whole, which ranged from 15,328 bp (C. janthinipennis) to 16,437 bp (L. yunnanus).
The AT nucleotide contents of the four lagriine mitogenomes were similar (79.81% in C. janthinipennis, 72.12% in L. yunnanus, 74.90% in A. unidentasus, and 75.43% in S. cribricollis). The entire mitogenome had high A + T contents (ranging from 72.12–79.81%; 69.86–78.54% for PCGs, 77.34–81.16% for tRNAs, 76.79–82.56% for rRNAs, and 77.62–87.33% for the CR) (Table 1). The AT-skews in the four mitogenome sequences were −0.048, 0.127, 0.091, and 0.044, respectively, of which the C. janthinipennis had a negative value. All of the GC skews were negative (Table 1), which indicated that the content of G is lower than C.
| Gene | Strand | Position from |
To | Start codons | Stop condons | Intergenic nucleotides |
|---|---|---|---|---|---|---|
| trnI | J | 1/1/1/1 | 62/63/66/63 | 0/0/0/0 | ||
| trnQ | N | 60/61/68/64 | 128/129/136/132 | -3/-3/1/0 | ||
| trnM | J | 129/130/136/132 | 191/198/199/194 | 0/0/-1/-1 | ||
| nd2 | J | 192/199/200/195 | 1,196/1,203/1,195/1,187 | ATT/ATA/ATT/ATT | TAA/TAA/TAG/TAA | 0/0/0/0 |
| trnW | J | 1,195/1,202/1,228/1,191 | 1,257/1,267/1,294/1,254 | -2/-2/32/3 | ||
| trnC | N | 1,258/1,267/1,300/1,254 | 1,318/1,327/1,360/1,313 | 0/-1/5/-1 | ||
| trnY | N | 1,319/1,328/1,365/1,315 | 1,381/1,391/1,430/1,378 | 0/0/4/1 | ||
| cox1 | J | 1,383/1,393/1,432/1,380 | 2,916/2,923/2,962/2,910 | -/-/-/- | TAA/TAA/TAA/TAA | 1/1/1/1 |
| trnL2 | J | 2,917/2,924/2,963/2,911 | 2,979/2,986/3,025/2,973 | 0/0/0/0 | ||
| cox2 | J | 2,980/2,987/3,026/2,974 | 3,660/3,665/3,704/3,652 | ATT/ATA/ATA/ATA | TAG/TAA/TAA/TAA | 0/0/0/0 |
| trnK | J | 3,662/3,666/3,705/3,653 | 3,731/3,735/3,774/3,722 | 1/0/0/0 | ||
| trnD | J | 3,731/3,736/3,777/3,727 | 3,794/3,799/3,839/3,789 | -1/0/2/4 | ||
| atp8 | J | 3,795/4,326/3,842/3,847 | 3,953/4,481/4,033/3,993 | ATT/ATT/ATA/ATA | TAA/TAG/TAA/TAA | 0/526/2/55 |
| atp6 | J | 3,947/4,475/4,027/3,987 | 4,616/5,146/4,695/4,655 | ATG/ATG/ATG/ATG | TAA/TAA/TAA/TAA | -7/-7/-7/-7 |
| cox3 | J | 4,617/5,146/4,695/4,655 | 5,399/5,929/5,480/5,438 | ATG/ATG/ATG/ATG | TAA/TAA/TAA/TAA | 0/-1/-1/-1 |
| trnG | J | 5,401/5,930/5,480/5,439 | 5,462/5,992/5,541/5,500 | 1/0/-1/0 | ||
| nad3 | J | 5,463/5,993/5,542/5,501 | 5,816/6,346/5,895/5,854 | ATA/TTG/ATT/ATA | TAG/TAG/TAG/TAG | 0/0/0/0 |
| trnA | J | 5,815/6,345/5,894/5,853 | 5,866/6,409/5,956/5,915 | -2/-2/-2/-2 | ||
| trnR | J | 5,877/6,409/5,956/5,915 | 5,939/6,472/6,019/5,977 | 10/-1/-1/-1 | ||
| trnN | J | 5,939/6,472/6,019/5,977 | 6,004/6,537/6,081/6,040 | -1/-1/-1/-1 | ||
| trnS1 | J | 6,005/6,538/6,082/6,041 | 6,063/6,595/6,138/6,099 | -1/-1/-1/-1 | ||
| trnE | J | 6,064/6,596/6,139/6,100 | 6,125/6,657/6,201/6,161 | 0/0/0/0 | ||
| trnF | N | 6,124/6,656/6,200/6,160 | 6,186/6,717/6,262/6,221 | 0/0/0/0 | ||
| nd5 | N | 6,187/6,718/6,262/6,222 | 7,891/8,416/7,968/7,926 | ATA/ATT/ATT/ATT | TAA/TAA/TAA/TAA | 0/0/-1/0 |
| trnH | N | 7,892/8,417/7,972/7,927 | 7,957/8,484/8,033/7,988 | 0/0/3/0 | ||
| nad4 | N | 7,958/8,485/8,033/7,989 | 9,284/9,811/9,358/9,312 | ATG/ATG/ATG/ATG | TAA/TAA/TAA/TAA | 0/0/-1/0 |
| nad4L | N | 9,278/9,805/9,352/9,306 | 9,565/1,0092/9,627/9,584 | ATG/ATG/ATT/ATA | TAA/TAA/TAA/TAA | -7/-7/-7/-7 |
| trnT | J | 9,568/10,098/9,630/9,587 | 9,631/10,160/9,692/9,649 | 2/5/2/2 | ||
| trnP | N | 9,632/10,161/9,693/9,650 | 9,693/10,223/9,756/9,711 | 0/0/0/0 | ||
| nad6 | J | 9,697/10,226/9,759/9,714 | 10,179/10,717/10,247/10,202 | ATG/ATC/ATC/ATA | TAA/TAA/TAA/TAA | 3/2/2/2 |
| cytb | J | 10,179/10,717/10,247/10,202 | 11,315/11,850/11,377/11,332 | ATG/ATG/ATG/ATG | TAA/TAA/TAA/TAA | -1/-1/-1/-1 |
| trnS2 | J | 11,314/11,849/11,377/11,331 | 11,380/11,915/11,440/11,394 | -2/-2/-1/-2 | ||
| nad1 | N | 11,398/12,105/11,469/11,415 | 12,348/13,055/12,419/12,359 | TTG/TTG/TTG/TTG | TAG/TAG/TAG/TAA | 17/89/28//10 |
| trnL1 | N | 12,349/13,056/12,420/12,360 | 12,410/13,117/12,482/12,422 | 0/0/0/0 | ||
| rrnL | N | 12,411/13,118/12,483/12,423 | 13,675/14,395/13,750/13,685 | 0/0/0/0 | ||
| trnV | N | 13,676/14,396/13,751/13,686 | 13,740/14,464/13,813/13,752 | 0/0/0/0 | ||
| rrnS | N | 13,741/14,465/13,814/13,753 | 14,436/15,168/14,556/14,486 | 0/0/0/0 | ||
| A+T rich region | 14,437/15,169/14,557/14,487 | 15,328/16,437/15,387/15,454 | 0/0/0/0 |
Protein-coding regions and codon usage
In these four lagriine complete mitogenomes, the lengths of PCGs ranged from 11,030 to 11,087 bp, accounting for 67.36–72.22% of the full mitogenomes. In 13 PCGs, the nad5 (1,699–1,707 bp) and atp8 (147–192 bp) were the largest and smallest genes, respectively. All the PCGs had a typical ATN start codon, except nad1, which began with TTG. For stop codons, most PCGs terminated with TAR (TAA/TAG) and few had an incomplete stop codon T- (the TAA stop codon is completed by the addition of 3′ A residues to the mRNA) (Table S1).
In PCGs, the numbers of amino acids and relative synonymous codon usage (RSCU) were calculated. The 13 PCGs of A. unidentasus, C. janthinipennis, L. yunnanus, and S. cribricollis contained 3,684, 3,682, 3,681, and 3,667 codons, respectively, excluding stop codons. In these four species, the most frequently used codons were UUU (276, 379, 224, 280), UUA (299, 427, 274, 348), AUU (334, 401, 266, 350), and AUA (223, 243, 200, 231) (Fig. 1). Accordingly, F, L2, I, and N are the most frequently used amino acids in these four species.
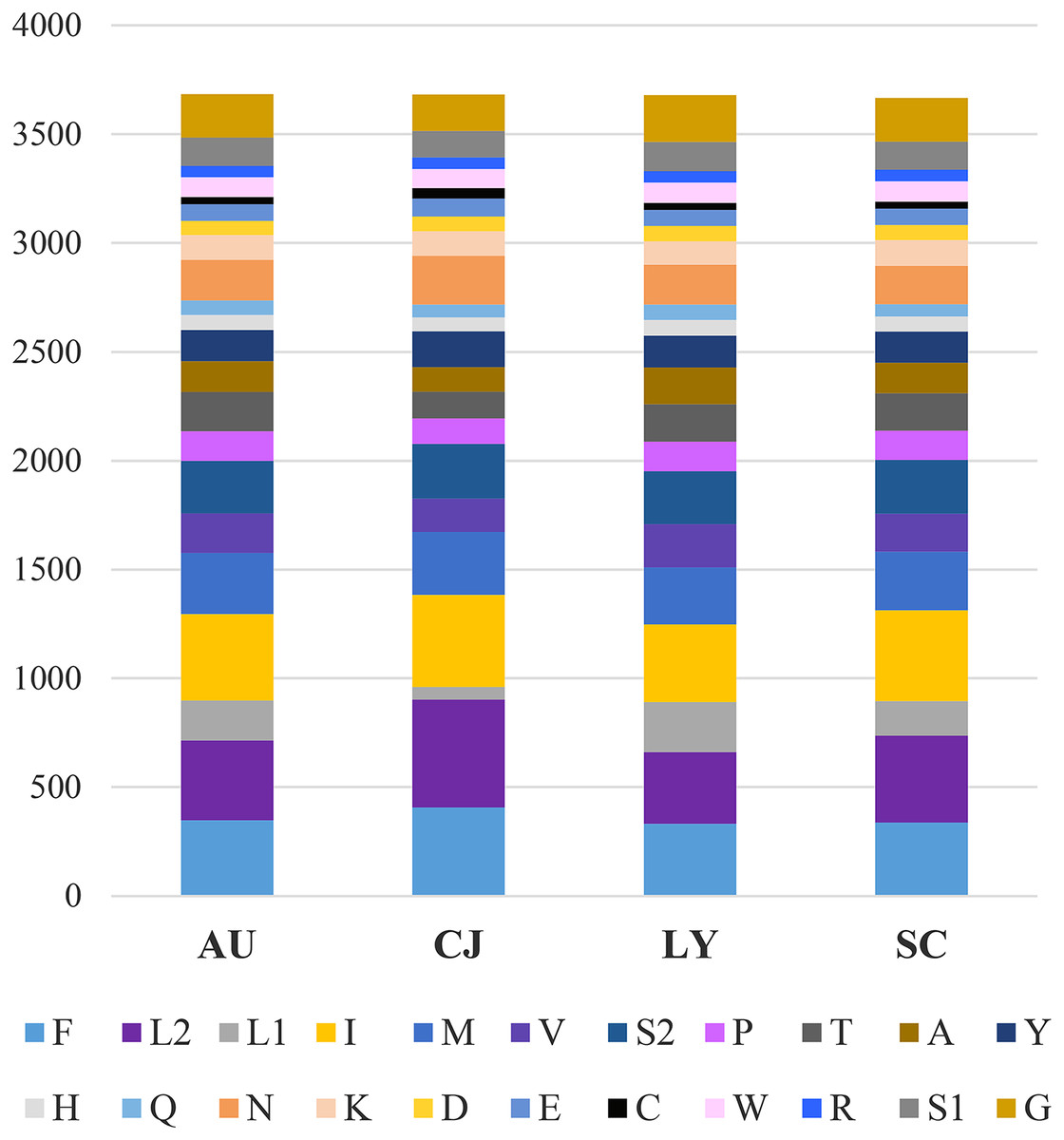
Figure 1: Numbers of different amino acids in the mitogenomes of four lagriine species; the stop codon is not included.
Abbreviations: AU, Anaedus unidentasus; CJ, Cerogira janthinipennis; LY, Luprops yunnanus; SC, Spinolyprops cribricollis.{kind=link}
The ratio of nonsynonymous/synonymous (Ka/Ks) of 13 PCGs was also calculated; the result is representative of mutations and the evolutionary rate (Hurst, 2002). The Ka/Ks of nad1 (3.157) are distinctly higher than 1 (Fig. 2), indicating that it is under strong positive selection (Mori & Matsunami, 2018), whereas cox1 (0.088), cox2 (0.114), and cytb (0.129) had values lower than the other genes (Fig. 2). These ratios suggest that the cox1, cox2, and cytb genes can be used as barcodes for deducing the phylogenetic relationships of Tenebrionidae.
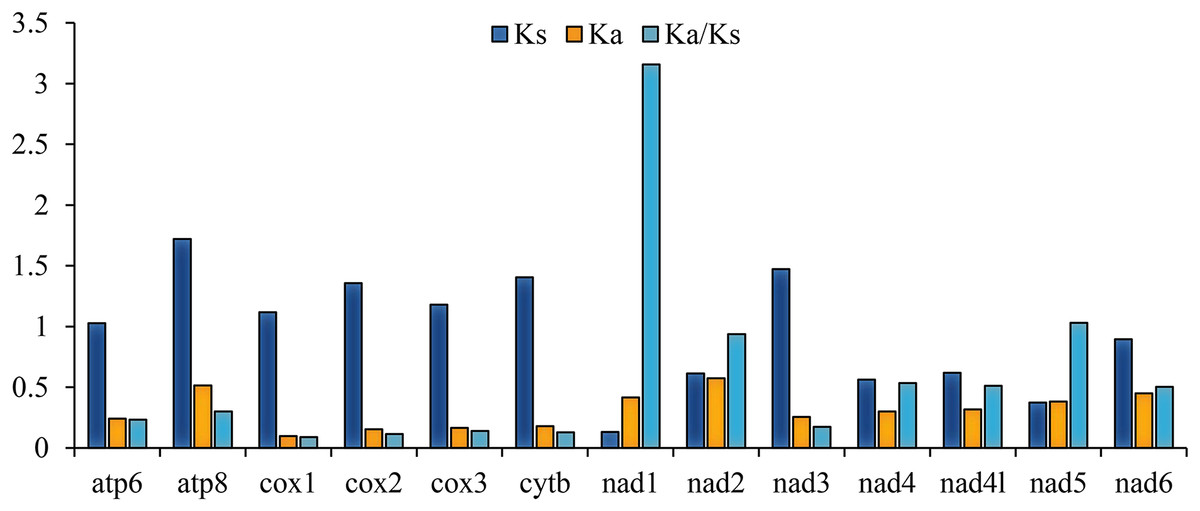
Figure 2: The ratio of Ka/Ks of 13 PCGs among the four lagriine mitogenomes.
{kind=link}
In PCGs, the nucleotide diversity (Pi) with different data ranged from 0.211 (cox1) to 0.978 (atp8) (Fig. 3). Among the 13 PCGs, the gene atp8 (Pi = 0.978) is the most diverse nucleotide, followed by nad2 (Pi = 0.407) and nad6 (Pi = 0.375), however, cox1 is the most conserved gene with the lowest value (Pi = 0.211).
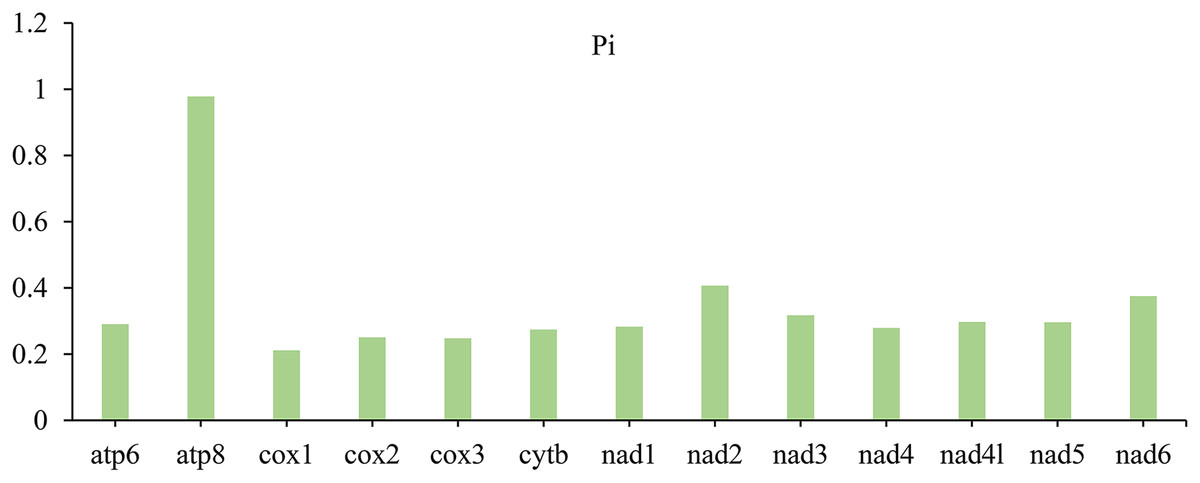
Figure 3: Nucleotide diversity (Pi) of 13 PCGs among four lagriine mitogenomes.
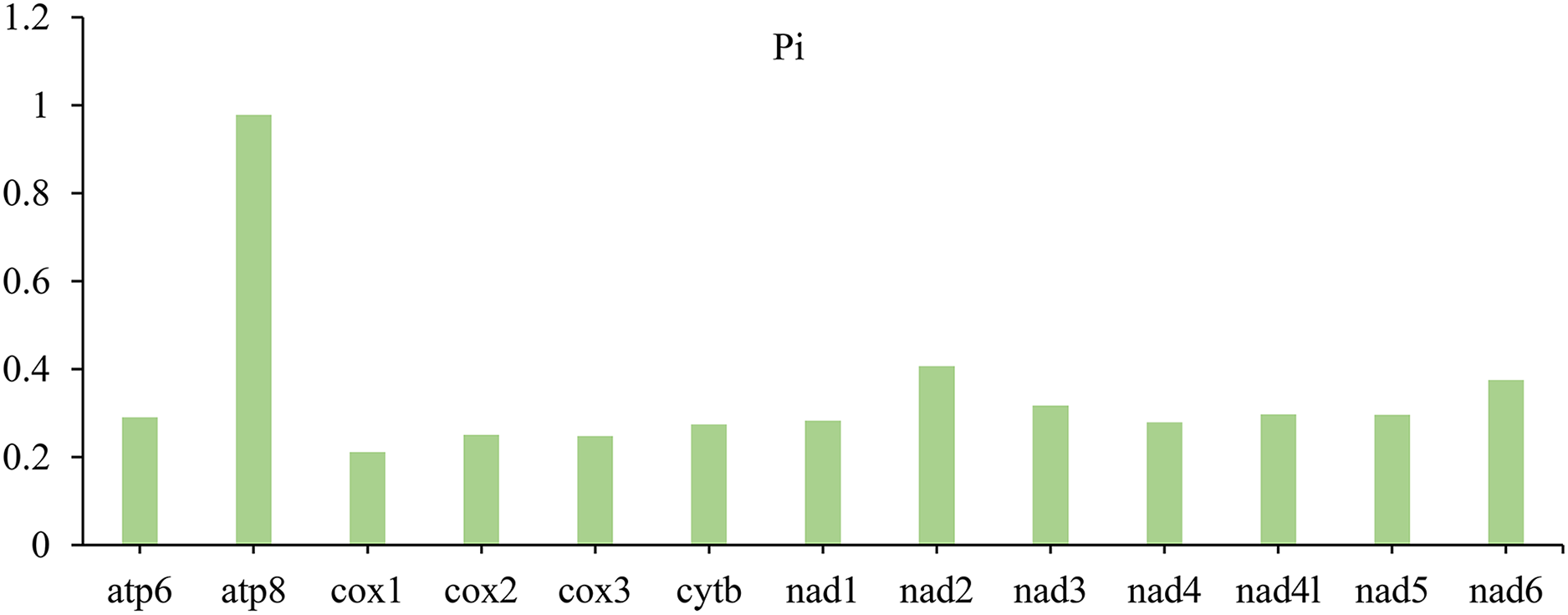{kind=link}
Transfer and ribosomal RNA genes
The total tRNAs lengths of A. unidentasus, C. janthinipennis, L. yunnanus, and S. cribricollis were 1,403, 1,391, 1,421, and 1,395 bp, respectively. Eight of the 22 tRNAs (trnY, trnC, trnQ, trnV, trnL1, trnP, trnH, trnP) were encoded on the N-strand, while the other 14 tRNAs were on the J-strand (Table 1). The tRNAs of four species were between 52 and 70 bp in length. The trnK is the longest of four species, while trnS1, trnA, trnS1, and trnS1 are the shortest of A. unidentasus, C. janthinipennis, L. yunnanus, and S. cribricollis, respectively. Among the 22 tRNAs, 21 tRNAs showed the typical clover-leaf secondary structure (Figs. S2–S5), however, trnS1 had a simple loop (Fig. 4). In L. yunnanus, a 526 bp insertion was present at trnD and atp8 junction.
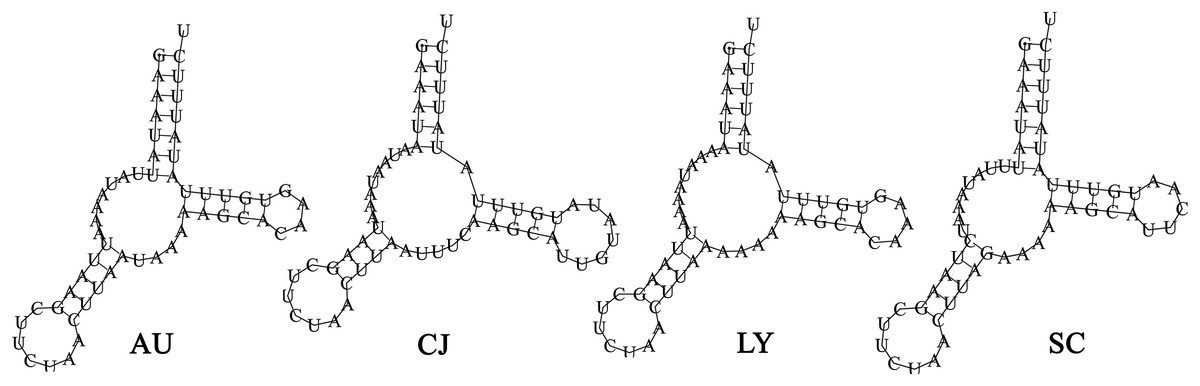
Figure 4: The predicted secondary structures of the trnS1 in the mitogenomes of the four lagriine species.
Abbreviations: AU, Anaedus unidentasus; CJ, Cerogira janthinipennis; LY, Luprops yunnanus; SC, Spinolyprops cribricollis.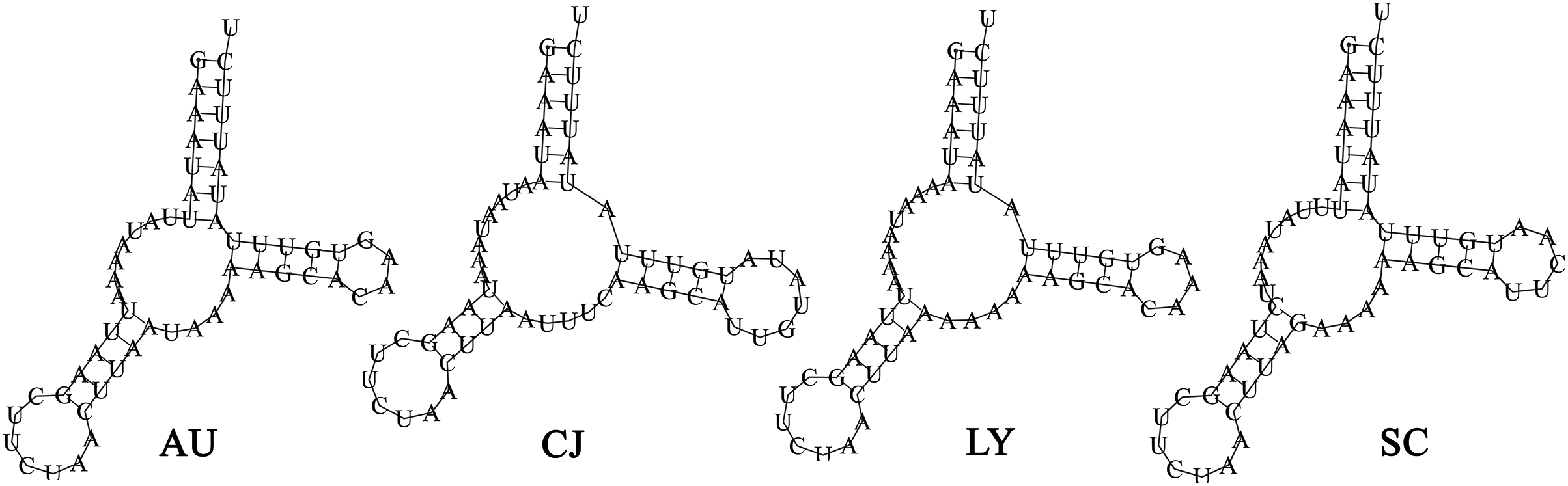{kind=link}
The total length of rRNAs ranged from 1,961 bp (C. janthinipennis) to 2,011 bp (A. unidentasus). The AT content of four species ranged from 76.79% (L. yunnanus) to 82.56% (C. janthinipennis) (Table 2). The two rRNAs (rrnL and rrnS) of four species are encoded on the N-strand. The rRNAs of four species were located between the control region and trnL1, and separated by trnV.
| Species | Full mitogenome | PCGs | rRNAs | tRNAs | CR | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Length (bp) | A% | C% | A + T% | AT-skew | GC-skew | A + T% | A + T% | A + T% | A + T% | |
| Cerogira janthinipennis | 15,328 | 38.00 | 10.49 | 79.81 | −0.048 | −0.039 | 78.54 | 82.56 | 81.16 | 87.33 |
| Luprops yunnanus | 16,437 | 40.65 | 18.12 | 72.12 | 0.127 | −0.300 | 69.86 | 76.79 | 77.34 | 77.62 |
| Anaedus unidentasus | 15,387 | 40.85 | 16.77 | 74.90 | 0.091 | −0.336 | 73.33 | 79.41 | 77.62 | 80.99 |
| Spinolyprops cribricollis | 15,454 | 39.37 | 16.29 | 75.43 | 0.044 | −0.326 | 73.75 | 78.97 | 78.42 | 82.33 |
Control region
The control region (CR) was the longest non-coding region. It was located between trnI and rrnS. The length of CR ranged from 831 bp (A. unidentasus) to 1,269 bp (L. yunnanus). The AT content of the four species ranged from 77.62% (L. yunnanus) to 87.33% (C. janthinipennis), which is distinctly higher than that of the full mitogenome, PCGS, rRNAs, and tRNAs in the same species. These four sequences had a positive AT skew. The tandem repeat element numbers of A. unidentasus, C. janthinipennis, L. yunnanus, and S. cribricollis are 1, 5, 3, and 3, respectively.
Phylogenetic analyses
The phylogenetic relationships among 39 Tenebrionidae species (Table S1) were reconstructed based on the nucleotide sequences of mitogenomes using the maximum likelihood and Bayesian inference methods. The outgroup species, Mylabris sp. (Meloidae) and Oedemera virescens (Oedemeridae), were the first to be separated from the tenebrionid clade. Phylogenetic trees based on the ML and BI methods showed approximately identical topologies (Figs. 5, S10). All the Tenebrionidae species were clustered together, which suggested that Tenebrionidae is monophyletic. The target species (A. unidentasus, C. janthinipennis, L. yunnanus, and S. cribricollis) and other lagriine species were clustered into single branch with high support (Fig. 5, BI = 0.87) suggesting that Lagriinae is monophyletic. These results are consistent with previous studies (Doyen & Tschinkel, 1982; Doyen, Matthews & Lawrence, 1989; Kergoat et al., 2014; Gunter et al., 2014; Timmermans et al., 2016; Aalbu, Kanda & Smith, 2017; Wu et al., 2022). The three other target species, Crypsis chinensis, Gonocephalum kochi, and Morphostenophanes yunnanus, were clustered together with Diaperinae, Blaptinae, and Stenochiinae species, respectively. In this study, the monophyly of the subfamilies Pimelinae (BI = 1), Blaptinae (BI = 0.89), Stenochiinae (BI = 1), Alleculinae (BI = 1) was also supported (Fig. 5); the subfamily Diaperinae was paraphyletic, and Tenebrioninae appears polyphyletic. The subfamily Pimelinae was the sister group to the remaining subfamilies, which was consistent with the results of Kergoat et al. (2014) but inconsistent with the results of other studies (Doyen & Tschinkel, 1982; Gunter et al., 2014; Timmermans et al., 2016; Wu et al., 2022). The results also suggested that the classification of Tenebrioninae and Diaperinae needs to be further studied.
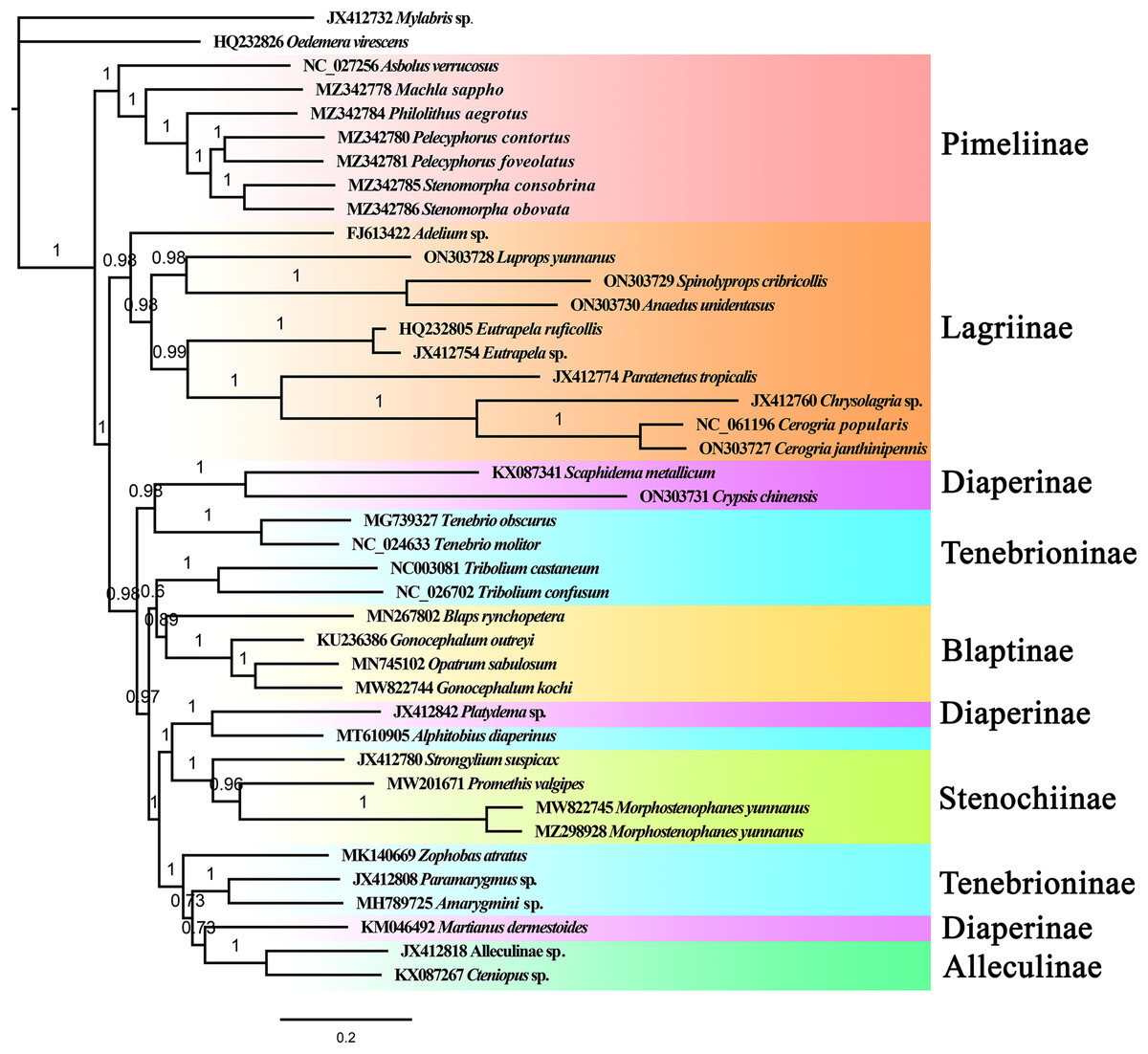
Figure 5: The BI tree of Tenebrionidae inferred based on 13 PCGs.
The value on each branch shows its posterior probability.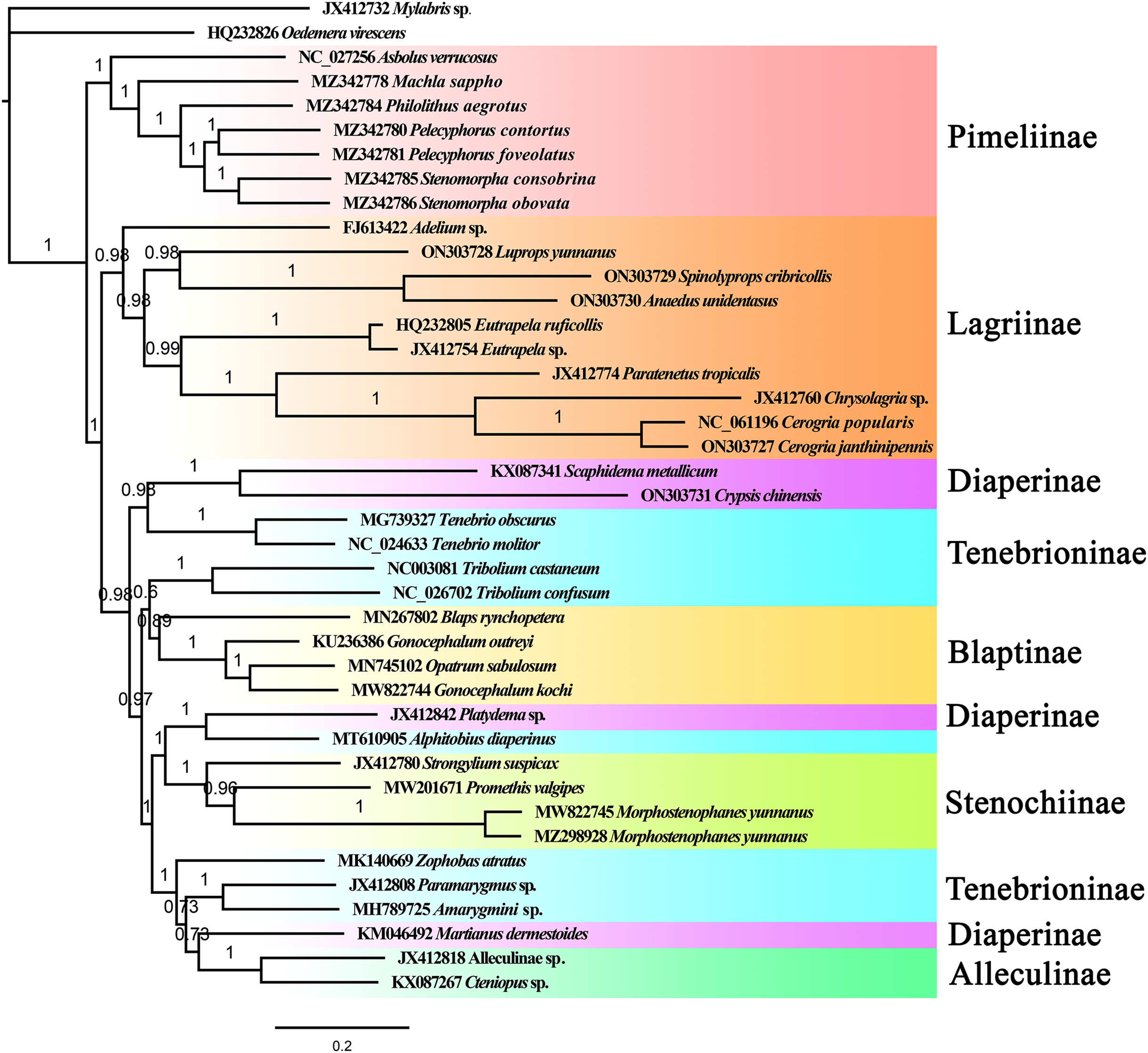{kind=link}
Discussion
The genes composition and arrangement of four Lagriinae species were found to be the same as those in most other Tenebrionidae species (Liu & Wang, 2014; Rider, 2016; Song et al., 2019; Yang et al., 2019; Hong et al., 2020; Bai et al., 2018, 2019, 2021; Smith et al., 2021; Liu et al., 2022; Wu et al., 2022). In these four lagriine species, all complete mitogenomes had high A + T contents, which is consistent with the typical base bias of the Tenebrionidae mitogenomes. Compared to the subfamily Alleculinae (Wu et al., 2022), all PCGs of the subfamily Lagriinae had typical start codons and the ratio of Ka/Ks was lower than 1. In this study, the CR was located between trnI and rrnS, which is consistent with previous studies (Liu & Wang, 2014; Rider, 2016; Song et al., 2019; Yang et al., 2019; Hong et al., 2020; Bai et al., 2021; Wu et al., 2022).
In the subfamily Lagriinae, the genera Spinolyprops (Lupropini) and Anaedus (Goniaderini) formed a single clade; the results of Aalbu, Kanda & Smith (2017) suggested that the tribe Lupropini was paraphyletic, which was consistent with our findings. Based on the BI and ML tree topologies, the position of the genus Spinolyprops in the tribe Lupropini was not suitable. In terms of morphology, the genus Spinolyprops is more consistent with Goniaderini (Matthews & Lawrence, 2019). This is supported by the following characteristics: (1) concealed clypeolabral membrane; (2) pronotum with sides explanate; and (3) defensive glands absent (adult species from China without defensive glands). Based on mitogenome data and morphological characteristics, we recommend that the genus Spinolyprops should be transferred from Lupropini to Goniaderini. However, more samples are needed to confirm the present results. Compared to the tribes Lupropini, Goniaderini, and Lagriini, the tribe Adeliini is an original group.
In this study, the subfamily Tenebrioninae and Diaperinae were polyphyletic and paraphyletic, respectively. In the subfamily Tenebrioninae, the genus Tenebrio was closed to Diaperinae (Scaphidema + Crypsis). In the subfamily Diaperinae, the genus Platydema and the genus Alphitobius (Tenebrioninae) were sister groups. These results were consistent with a previous study by Wu et al. (2022). This phylogenetic question needs to be further addressed with more samples and more genes, as well as mitogenomic and nucleotide genes.
In the future, more mitogenome samples are needed to resolve the phylogeny of the Tenebrionidae to better understand the natural evolutionary processes.
Conclusion
In this study, the mitogenomes of the tribes Goniaderini and Lupropini were first reported and the compositional features were analyzed. The mitogenomes were 15,328–16,437 bp in length and encoded 37 typical mitochondrial genes. Comparisons of the four newly generated lagriine mitogenome to all available mitogenomes of Tenebrionidae revealed no significant differences among them in terms of the AT content of different genome regions, amino acid composition, and relative synonymous codon usage. In the PCGs, the atp8 (Pi = 0.978) was the most diverse nucleotide, while cox1 was the most conserved gene with the lowest value (Pi = 0.211). The phylogenetic results suggest that Pimelinae, Lagriinae, Blaptinae, Stenochiinae, and Alleculinae are monophyletic, Diaperinae is paraphyletic, and Tenebrioninae appears polyphyletic. In Lagriinae, the tribe Lupropini appears paraphyletic because Spinolyprops is clustered with Anaedus in Goniaderini. These mitogenome sequences provide valuable molecular data for the phylogenetic studies of Tenebrionidae in the future.