
Phylogenetic relationship and comparative analysis of the main Bupleuri Radix species in China
- Published
- Accepted
- Received
- Academic Editor
- Alastair Culham
- Subject Areas
- Agricultural Science, Genomics, Plant Science, Taxonomy
- Keywords
- Bupleuri Radix, Genome skimming, Bupleurum, Plastome, Phylogeny
- Copyright
- © 2023 Wang et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2023. Phylogenetic relationship and comparative analysis of the main Bupleuri Radix species in China. PeerJ 11:e15157 https://doi.org/10.7717/peerj.15157
Abstract
Background
Bupleuri Radix (Chaihu) is a famous traditional Chinese medicine derived from Bupleurum, Apiaceae. The origin of cultivated Chaihu germplasm in China is unclear, which has led to unstable Chaihu quality. In this study, we reconstructed the phylogeny of the main Chaihu germplasm species in China and identified potential molecular markers to authenticate its origin.
Methods
Three Bupleurum species (eight individuals), B. bicaule, B. chinense, and B. scorzonerifolium, were selected for genome skimming. Published genomes from B. falcatum and B. marginatum var. stenophyllum were used for comparative analysis.
Results
Sequences of the complete plastid genomes were conserved with 113 identical genes ranging from 155,540 to 155,866 bp in length. Phylogenetic reconstruction based on complete plastid genomes resolved intrageneric relationships of the five Bupleurum species with high support. Conflicts between the plastid and nuclear phylogenies were observed, which were mainly ascribed to introgressive hybridization. Comparative analysis showed that noncoding regions of the plastomes had most of the variable sequences. Eight regions (atpF-atpH, petN-psbM, rps16-psbK, petA-psbJ, ndhC-trnV/UAC and ycf1) had high divergence values in Bupleurum species and could be promising DNA barcodes for Chaihu authentication. A total of seven polymorphic cpSSRs and 438 polymorphic nSSRs were detected across the five Chaihu germplasms. Three photosynthesis-related genes were under positive selection, of which accD reflected the adaptation fingerprint of B. chinense to different ecological habitats. Our study provides valuable genetic information for phylogenetic investigation, germplasm authentication, and molecular breeding of Chaihu species.
Introduction
Bupleuri Radix (Chinese name: Chaihu), the dried root of Bupleurum species, has been used to treat cold, fevers, influenza, menstrual disorders, and hepatitis for more than 2,000 years (Yang et al., 2017; Zhu et al., 2017). Its primary use is as the main constituent of Chinese medicine prescriptions for soothing the liver and relieving depression, such as the Xiaochaihu Decoction, Chaihu Liver-soothing Powder, and Xiaoyao Pill (Yang et al., 2017; Zhu et al., 2017). Bupleurum chinense DC. and B. scorzonerifolium Willd. have been authenticated as the official botanical origin of Chaihu in the Chinese Pharmacopoeia (National Pharmacopoeia Committee, 2020), and are referred to as “Bei Chaihu” and “Nan Chaihu”, respectively. More than 20 Bupleurum species are habitually utilized as Chaihu due to their morphological similarity (Pan et al., 2002). As one of the most popular medicines in China, the demand for Chaihu in prescriptions and exports is enormous, while the supply of wild Bupleurum herb is limited. Artificial planting of Chaihu has become the dominant agriculture industry in some regions, such as the Chencang district in Shaanxi Province (Ma et al., 2020). However, the use of multiple Bupleurum species in different districts has also contributed to the complexity and diversity of cultivated Chaihu germplasms.
Based on plantation surveys, the cultivated germplasms of Chaihu in China are mainly from five species, B. bicaule Helm, B. chinense, B. falcatum L., B. marginatum var. stenophyllum (Wolff) Shan et Y. Li, and B. scorzonerifolium (Xu et al., 2014; Zhang et al., 2021; Zhang et al., 2022). Among these species, B. chinense is the most common species cultivated in China (Ma et al., 2020; Zhang et al., 2021). As a widespread species, B. chinense exhibits broad intraspecific morphological variation and different levels of medicinal quality under different growing conditions. The unclear origins of wild and cultivated Chaihu resources result in somewhat unstable Chaihu qualities in the Chinese market. Previous studies have greatly improved our understanding of Bupleurum based on morphology (Pan et al., 2002; Sheh & Watson, 2005), chromosome counts (Ma et al., 2015), nuclear ribosomal internal transcribed spacer (nrITS) (Chao et al., 2014; Neves & Watson, 2004; Xie et al., 2009), and plastid DNA markers (Wang et al., 2008; Wang, Ma & He, 2011). However, because of limited genetic information, relationships among some closely related species could not be fully resolved, such as B. bicaule and B. scorzonerifolium.
With the development of high-throughput sequencing technologies, genetic information from both nuclear and organellar DNA has provided an opportunity for addressing problems that have remained unresolved using traditional molecular systematics approaches (Liu et al., 2021a; Liu et al., 2021b). Chloroplast, the plant organelle for photosynthesis and carbon fixation, provides valuable genetic information for phylogenies of plants due to its low nucleotide substitution rates and uniparental inheritance (Davis, Xi & Mathews, 2014; Delsuc, Brinkmann & Philippe, 2005; Thode, Lohmann & Sanmartín, 2020). Compared to plastid barcodes, whole-plastome sequences could generate higher phylogenetic resolution and improve species identification (Fu et al., 2022; Meng et al., 2021; Wikström, Bremer & Rydin, 2020). Considering conflicts between chloroplast and nuclear phylogenies, complementary phylogenetic trees inferred with different datasets are necessary (Meng et al., 2021). Some regions of the nrDNA repeat, like ITS are widely used as phylogenetic markers for lower-level plant identification (Baldwin et al., 1995; Liu et al., 2021a; Liu et al., 2021b; Yao et al., 2010). Genome skimming, which relies on next generation sequencing, is a kind of shallow genome sequencing that could capture the plastome, nuclear repeat, and mitogenome regions (Dodsworth, 2015). Benefitting from abundant datasets and cost-efficiency, genome skimming has been used successfully in phylogenetic reconstruction at various levels, especially for taxonomically complex groups (Dodsworth, 2015; Straub et al., 2012).
In this study, we used the genome skimming approach to obtain the complete chloroplast genomes and nrDNA of B. bicaule, B. chinense, and B. scorzonerifolium, together with published genomes of two other Bupleurum species for comparative analysis. We aimed to: (1) explore the phylogenetic relationships of the five Bupleurum species that are the germplasm sources for Chaihu in China; and (2) select potential markers for authentication of Chaihu germplasm by comparative analysis. This study will provide valuable genetic information and potential markers for authentication of cultivated Bupleurum species, which will be helpful for cultivation and quality control of Chaihu.
Materials & Methods
Sample material
Three wild Bupleurum species, B. bicaule (one, BB01), B. chinense (five, BC01-05), and B. scorzonerifolium (two, BS01-02) were collected from different districts in China (Table 1). Field experiments were approved by the Research Council of Xianyang Normal University (project number: 2019012). Fresh and healthy leaves were dried with silica gel, and then stored at −80 °C for DNA extraction. All species were authenticated by Director Xue Li (Xianyang Food and Drug Administration) and deposited at the Herbarium of Xianyang Normal University (http://www.xysfxy.cn/) accession Nos. XSYH202000101-XSYH202000801 (Table S1). Sampling information is shown in Table 1. To obtain the sequences of B. falcatum and B. marginatum var. stenophyllum for a broader comparison, we retrieved their raw sequenced reads from GenBank (B. falcatum, BF01: SRR12513791 and B. marginatum var. stenophyllum, BM01: SRR13195839), and used them for genome assembly and annotation as with the accessions sequenced in the study.
| Taxa | Code | Locality | Longitude, Latitude | Genome size (bp) | LSC length (bp) | SSC length (bp) | IR length (bp) | Unique genes | Unique CDS | tRNA | rRNA | Total GC content % | GC content of LSC (%) | GC content of SSC (%) | GC content of IR (%) | Herbarium accession |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B. chinense | BC01 | Heihe, Shaanxi | 108.1754, 34.0188 | 155659 | 85551 | 17508 | 26300 | 113 | 79 | 30 | 4 | 37.7 | 35.8 | 31.4 | 42.8 | XSYH2022000101 |
| B. chinense | BC02 | Danfeng,Shaanxi | 110.5020, 33.9004 | 155579 | 85505 | 17496 | 26289 | 113 | 79 | 30 | 4 | 37.7 | 35.8 | 31.4 | 42.8 | XSYH2022000201 |
| B. chinense | BC03 | Longde, Ningxia | 106.2090, 35.4868 | 155612 | 85461 | 17499 | 26326 | 113 | 79 | 30 | 4 | 37.7 | 35.8 | 31.4 | 42.8 | XSYH2022000301 |
| B. chinense | BC04 | Lanzhou, Gansu | 103.6524, 36.4385 | 155540 | 85426 | 17492 | 26311 | 113 | 79 | 30 | 4 | 37.7 | 35.8 | 31.4 | 42.8 | XSYH2022000401 |
| B. chinense | BC05 | Xizhou, Shanxi | 112.4409, 38.6399 | 155573 | 85449 | 17510 | 26307 | 113 | 79 | 30 | 4 | 37.7 | 35.8 | 31.4 | 42.8 | XSYH2022000501 |
| B. scorzonerifolium | BS01 | Ansai, Shaanxi | 109.2021, 36.7978, | 155828 | 85597 | 17597 | 26317 | 113 | 79 | 30 | 4 | 37.7 | 35.8 | 31.3 | 42.8 | XSYH2022000601 |
| B. scorzonerifolium | BS02 | Fugu,Shaanxi | 110.7683, 39.0646 | 155848 | 85612 | 17602 | 26317 | 113 | 79 | 30 | 4 | 37.7 | 35.8 | 31.3 | 42.8 | XSYH2022000701 |
| B. bicaule | BB01 | Hulunbuir, Neimeng | 118.1399, 47.6237 | 155866 | 85630 | 17602 | 26317 | 113 | 79 | 30 | 4 | 37.7 | 35.8 | 31.3 | 42.8 | XSYH2022000801 |
DNA extraction and sequencing
Total DNA was extracted using the CTAB method. Then, the extracted DNA (about 1 µg) was randomly sheared by Covaris (Brighton, UK), yielding fragments with an average size of 200–400 bp for library construction. Genome skimming sequencing was performed on the BGISEQ-500 platform with paired-end runs (BGI, Shenzhen, China).
Genome assembly and annotation
High-throughput sequencing produced approximately 2 Gb raw data per sample. Raw reads were filtered and then assembled de novo into contigs using SOAPdenovo2 (Luo et al., 2012). To isolate plastid sequences, the generated contigs were pooled and mapped to the reference plastome (B. chinense: MN337347) using Bowtie2 (Langmead & Salzberg, 2012), then assembled to scaffolds by SPAdes v.3.11.1 (Bankevich et al., 2012). Geneious Prime (Kearse et al., 2012) was used to align the scaffolds with the aforementioned reference, resulting in the final plastid genomes. Annotation of the assembled sequences was also performed in Geneious Prime with B. chinense (MN337347) as a reference. All plastomes were deposited in GenBank with the accession numbers shown in Table S1.
Comparative analysis and sequence divergence analysis
For comparative analysis, MAFFT v.7 (Katoh et al., 2002) was used to align the plastomes of Bupleurum individuals. The nucleotide substitution numbers and sequence identities of Bupleurum plastomes were calculated with Geneious Prime. The IR expansion/contraction of five Bupleurum species was tested by comparing the IR borders and neighboring genes of ten chloroplast genomes using an IRscope online program (Amiryousefi, Hyvönen & Poczai, 2018). To detect and characterize the divergence hotspots, mVISTA (Frazer et al., 2004) was used to compare the complete plastid genomes of five Bupleurum species with B. chinense (BC01) as a reference. The nucleotide diversity (Pi) among the plastid genome sequences was calculated using sliding window analysis in DnaSP v.5 (Librado & Rozas, 2009). The window size was 600 bp, and the step size was 200 bp. The single nucleotide polymorphisms (SNPs) and insertions-deletions (InDels) were identified using MEGA v.6 (Kumar et al., 2018) and visualized using Circos 0.64 (Krzywinski et al., 2009).
Simple sequence repeats in the chloroplast genome sequences (cpSSRs) were identified via Perl script in MISA (Thiel et al., 2003) with respective thresholds of 10, 5, 4, 3, 3, 3 for mono-, di-, tri-, tetra-, penta-, and hexanucleotides. The candidate polymorphic cpSSRs and nuclear SSRs (nSSRs) were identified using CandiSSR (Xia et al., 2016). The nuclear contigs were used to generate nSSRs by removing the plastid and mitochondrial contigs from the original contigs using Bowtie2 with B. chinense as a reference (GenBank accession numbers MN337347 and OK166971 for chloroplast and mitochondrial genomes, respectively). The parameters implemented in CandiSSR were as follows: flanking sequence length of 100, blast e-value cutoff of 1e−10, BLAST identity cutoff of 95, and BLAST coverage cutoff of 95.
Gene selective pressure analysis
To detect whether plastid genes were under selection pressure, the ratio of the non-synonymous (Ka) to the synonymous substitution rate (Ks) was calculated for all protein-coding genes by DnaSP. Genes were considered to undergo positive selection when the value of Ka/Ks was higher than 1, while Ka/Ks < 1 indicated that the genes were under neutral selection (Yang & Nielsen, 2002).
Phylogenetic analysis
For phylogenetic inference, maximum likelihood (ML) and Bayesian inference (BI) analyses were performed for three different datasets: (1) complete plastome sequences, (2) all shared protein-coding sequences (CDS), and (3) nrDNA sequence data (ITS). For phylogenetic analysis based on plastid data, we selected 32 plastid genomes, including eight accessions of three species we sequenced, two accessions (MT797174 and MT075712) we reassembled and reannotated, 20 Bupleurum accessions retrieved from NCBI, together with two accessions as outgroups (Chamaesium paradoxum MK780227, Pleurospermum rivulorum MW147504).
To obtain nrDNA data, the original contigs of the accessions studied in the plastid phylogeny were mapped to the ITS sequence of B. chinense (MH710800) and extracted by Bowtie2. The species used in the plastid and nrDNA datasets were kept largely consistent. For species without nrDNA data, the ITS sequences were downloaded according to Wang, Ma & He (2011). Finally, 49 nrITS sequences of Bupleurum accessions, together with Pleurospermum hookeri var. thomsonii HQ824799 and P. rivulorum HQ824798, were selected for the phylogenetic reconstruction.
The software jModelTest v.2.1.4 (Posada, 2008) was used to determine the best-fit nucleotide substitution model, resulting in GTR+I+G for the plastid datasets and GTR+G for the nrITS data under the Akaike Information Criterion (AIC). ML analyses were conducted using RAxML v.8.2.11 (Stamatakis, 2014) with 1,000 bootstrap iterations. BI analyses were performed in MrBayes v.3.2.6 (Ronquist & Huelsenbeck, 2003). Four Markov chains starting with a random tree were run simultaneously for 5,000,000 generations, followed by a sampling point at every 1,000 generations. The resulting trees were visualized using FigTree v.1.4.4 (Rambaut, 2018). Bootstrap support (BS) value and posterior probability (PP) were used to evaluate the feasibility of each branch for ML and BI, respectively.
Results
Chloroplast genome structure and characteristics analyses
Plastid sequences were successfully assembled for ten accessions, of which B. falcatum (MT797174, BF01) and B. marginatum var. stenophyllum (MT075712, BM01) were reassembled. For Bupleurum species sequenced in this study, the complete plastomes ranged from 155,540 bp in BC04 to 155,866 bp in BB01. Sequences of the B. chinense plastomes ranged from 155,540 bp to 155,659 bp, while those from B. scorzonerifolium ranged from 155,828 bp to 155,848 bp. All chloroplast genomes exhibited a typical quadripartite structure, consisting of a large single-copy (LSC) region (85,426–85,630 bp) and a small single-copy (SSC) region (17,492–17,602 bp) separated by two inverted regions (IRs, each with 26,289–26,326 bp) (Table 1, Fig. 1). The overall GC content of each plastome was 37.7%, whereas the GC contents in the LSC, SSC, and IR were 35.8%, 31.3–31.4%, and 42.8%, respectively.
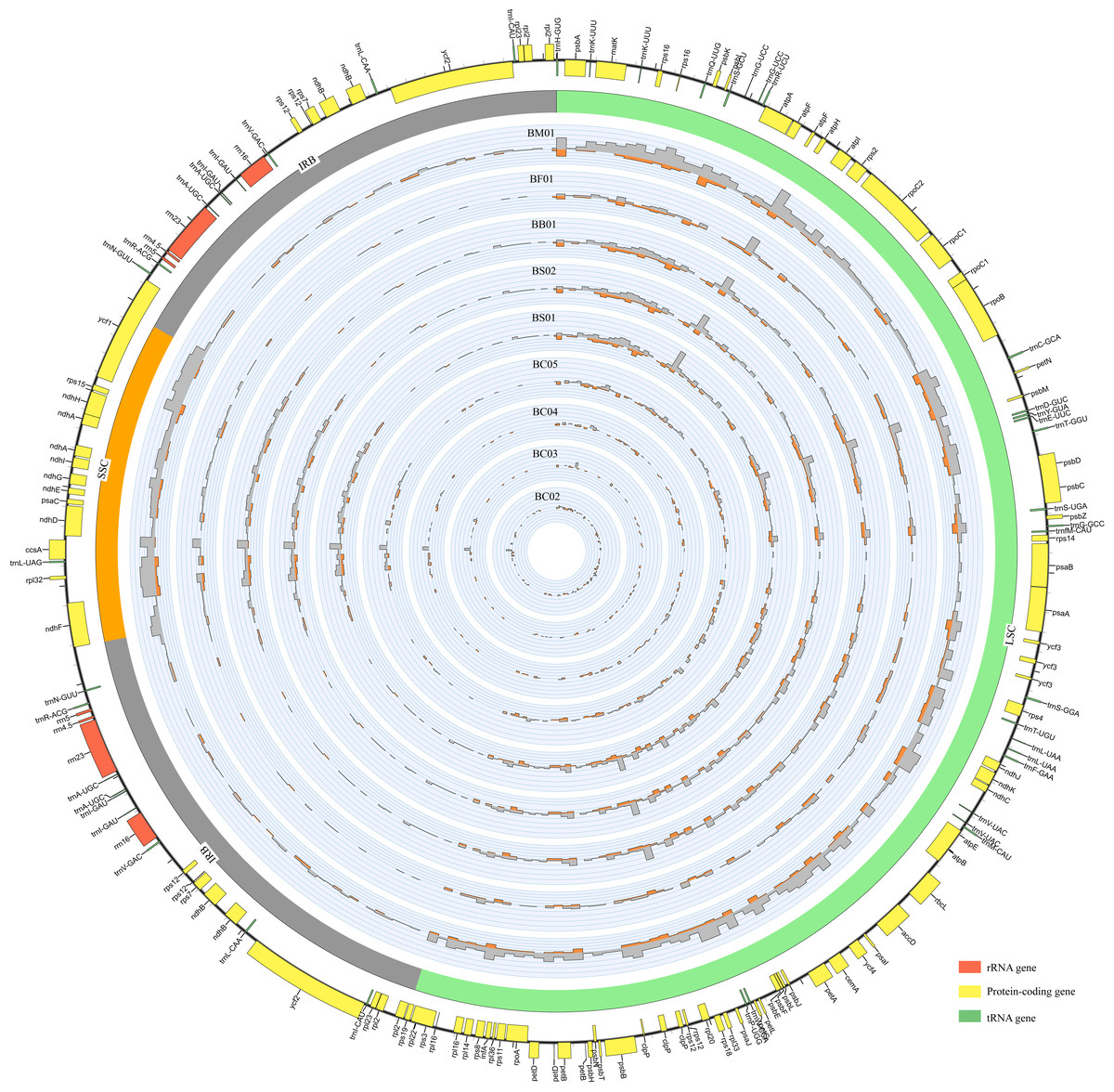
Figure 1: An overview of gene distribution and genome variation among Bupleurum plastomes using Circos.
Genes located outside and inside of the outer layer circle represent clockwise and counterclockwise transcription, respectively. The inner circles exhibit the SNPs and InDels every 600 bp across five Bupleurum species with B. chinense (BC01) as a reference. The histograms of SNPs are outside the circles, while the histograms of InDels are inside the circles.{kind=link}
Each Bupleurum chloroplast genome encoded a total of 132 genes, of which 113 were unique, containing 79 protein-coding genes (CDS), 30 transfer RNA genes (tRNA), and four ribosomal RNA genes (rRNA). The different functional genes were divided into three categories, namely self-replication, photosynthesis, and other (Table S2). A total of 19 genes were duplicated in the IRs, including six CDSs (rps 12, rps 7, ndh B, ycf 2, rpl 23, and rpl 2), seven tRNAs (trn N-GUU, trn R-ACG, trn A-UGC, trn l-GAU, trn V-GAC, trn L-CAA, and trn l-CAU), four rRNAs (rrn 4.5, rrn 5, rrn 23, and rrn 16), and two pseudogenes (ycf 1 and rps 19) with incomplete copies. The rps 12 gene was a particular trans-splicing gene with the 5′-end exon located in the LSC region, while two copies of the 3′-end exon were situated in the IR. There were 18 genes containing introns, including six tRNAs and 12 CDSs. Among these, ycf 3, clp P, and rps 12 had two introns. The gene trn K-UUU had the largest intron with 2,531 bp, where the mat K gene was located.
Comparisons of the Bupleurum plastomes
Differences and evolutionary divergences were tested based on ten Bupleurum plastomes, including five B. chinense, two B. scorzonerifolium, one B. bicaule, one B. falcatum, and one B. marginatum var. stenophyllum. The ten Bupleurum plastomes exhibited a high level of sequence similarity and structure (Table 2). The percentage of identity was 96.72–99.97%, with 49-5,160 nucleotide differences. Among B. chinense plastomes, the identity narrowly ranged from 99.4% to 99.81%. At the interspecies level, B. chinense (BC01) and B. marginatum var. stenophyllum (BM01) exhibited the most significant sequence difference with 96.72% identity (Table 2).
| BC01 | BC02 | BC03 | BC04 | BC05 | BS01 | BS02 | BB01 | BF01 | BM01 | |
|---|---|---|---|---|---|---|---|---|---|---|
| BC01 | 451 | 293 | 522 | 930 | 2131 | 2155 | 2165 | 1499 | 5160 | |
| BC02 | 99.71 | 415 | 438 | 869 | 2082 | 2114 | 2126 | 1441 | 5121 | |
| BC03 | 99.81 | 99.73 | 456 | 869 | 2062 | 2098 | 2106 | 1476 | 5141 | |
| BC04 | 99.67 | 99.72 | 99.71 | 924 | 2126 | 2159 | 2167 | 1436 | 5142 | |
| BC05 | 99.4 | 99.44 | 99.44 | 99.41 | 2034 | 2064 | 2078 | 1517 | 5139 | |
| BS01 | 98.64 | 98.67 | 98.68 | 98.64 | 98.7 | 224 | 243 | 1733 | 5062 | |
| BS02 | 98.62 | 98.65 | 98.66 | 98.62 | 98.68 | 99.86 | 49 | 1731 | 5050 | |
| BB01 | 98.62 | 98.64 | 98.65 | 98.62 | 98.67 | 99.84 | 99.97 | 1745 | 5065 | |
| BF01 | 99.04 | 99.08 | 99.06 | 99.08 | 99.03 | 98.89 | 98.89 | 98.89 | 4947 | |
| BM01 | 96.72 | 96.74 | 96.73 | 96.73 | 96.73 | 96.78 | 96.79 | 96.78 | 96.85 |
To further observe the contraction and expansion of IR, the exact IR border positions and their adjacent genes among the Bupleurum plastomes were compared (Fig. 2). Genes rps 19, rpl 2, and trn H were in the junctions of LSC/IR, while ycf 1, ndh F, and trn N flanked the junctions of SSC/IR. Genes in the junction regions were the same length, except the ycf 1 gene, which ranged from 5,483 bp to 5,490 bp. In all ten Bupleurum plastomes, rps 19 crossed the LSC/IRb region with 279 bp in length, of which 70 bp occurred in the IRb region. The pseudogene fragment of rps 19 was located aside the IRa/LSC junction with 70 bp. The rpl 2 gene was separated from the LSC/IRb junction with 126–127 bp in B. chinense; 127 bp in B. bicaule, B. falcatum, and B. scorzonerifolium; and 130 bp in B. marginatum var. stenophyllum. The trn H gene was 4 bp away from the IRa/LSC region in all ten Bupleurum plastomes. In the SSC/IR junctions, ycf 1 crossed the SSC/IRa junction, and the pseudogene fragment narrowly ranged from 1,876–1,877 bp in IRb. The length of the ycf 1 gene was 5,484 bp in B. chinense and B. falcatum, while it was 5,483 bp in B. bicaule and B. scorzonerifolium, and 5,490 bp in B. marginatum var. stenophyllum. The ndh F gene was 26–34 bp away from the IRb/SSC junction, and the trn N gene was 2,204–2,228 bp away from the SSC/IRa junction.

Figure 2: Comparison of the IR border regions among the ten plastid genomes of five Bupleurum species.
{kind=link}
Sequence variation analysis
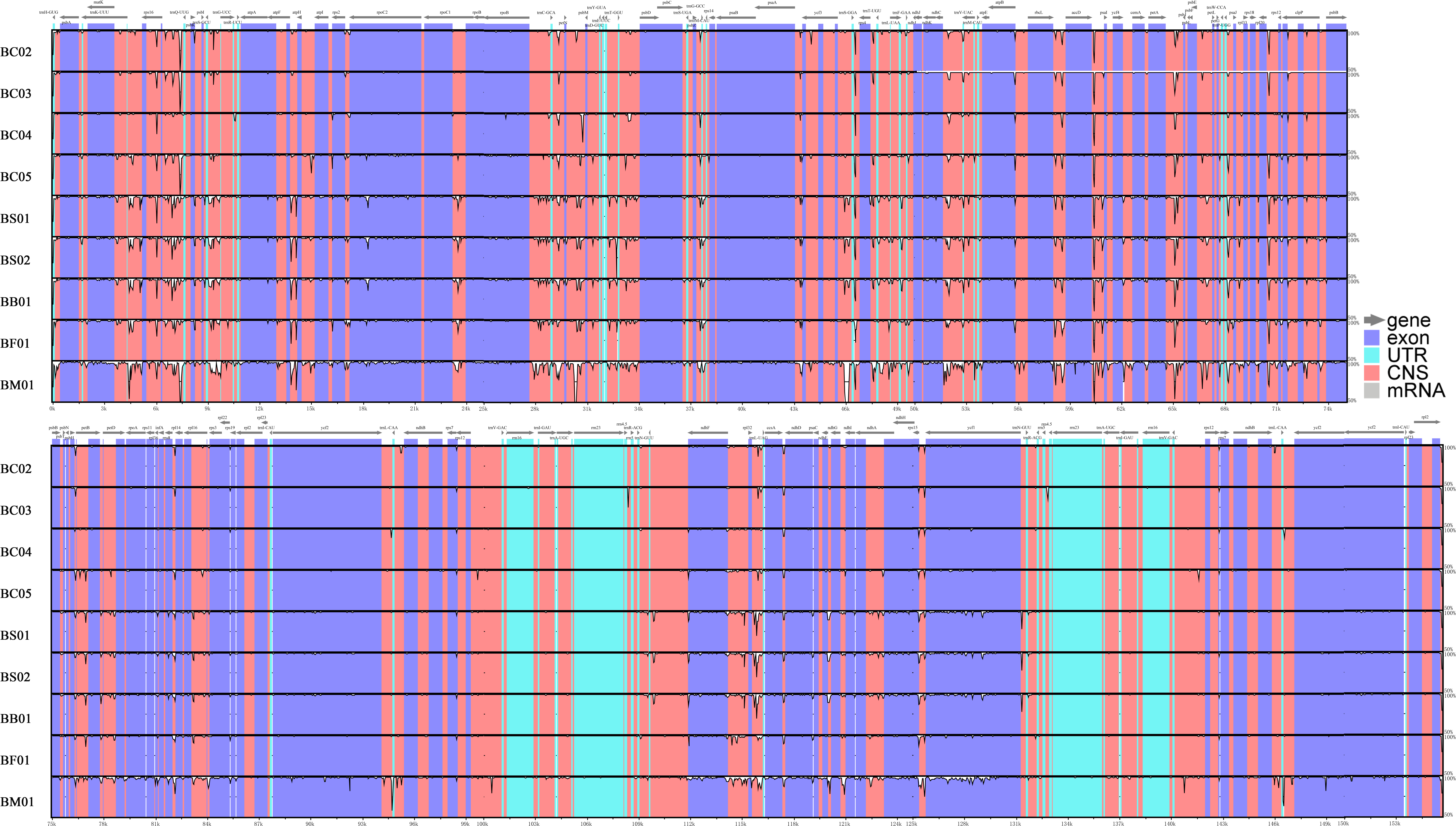
Abundant polymorphic sites were observed among the five Bupleurum species. In addition, B. chinense sequences also exhibited intraspecies divergence. With B. chinense (BC01) as a reference, the mVISTA result indicated that noncoding sequences exhibited a higher level of divergence than CDS, and most of the sequences were situated in the LSC and SSC regions (Fig. 3). We characterized genomic polymorphisms of the Bupleurum plastomes, including SNPs and InDel-variable loci (Fig. 1, Table S3). At the interspecies level, the number of SNPs varied from 420 (BF01 vs. BC01) to 2,091 (BM01 vs. BC01), and the InDel loci varied from 163 (BF01 vs. BC01) to 340 (BM01 vs. BC01). At the intraspecies level, the numbers of SNPs and InDels among B. chinense plastomes ranged from 70–303 and 39–114, respectively. The LSC regions harbored the largest number of genomic polymorphism sites, followed by SSC regions (Fig. 4). The IRs were conserved among Bupleurum species. The nucleotide diversity (Pi) of the chloroplast genomes was calculated to assess the sequence divergence level. Among all ten Bupleurum species, atp F-atp H, pet N-psb M, rps 16-psb K, pet A-psb J, ndh C-trn V/UAC, and ycf 1 had relatively higher divergence values (Pi >0.015). Chloroplast genome diversity at the intraspecies level of B. chinense was also detected, of which rps 16 intron, ccs A-ndh D, rbc L-acc D, rps 16-psb K, and ndh C-trn V/UAC had high Pi values.
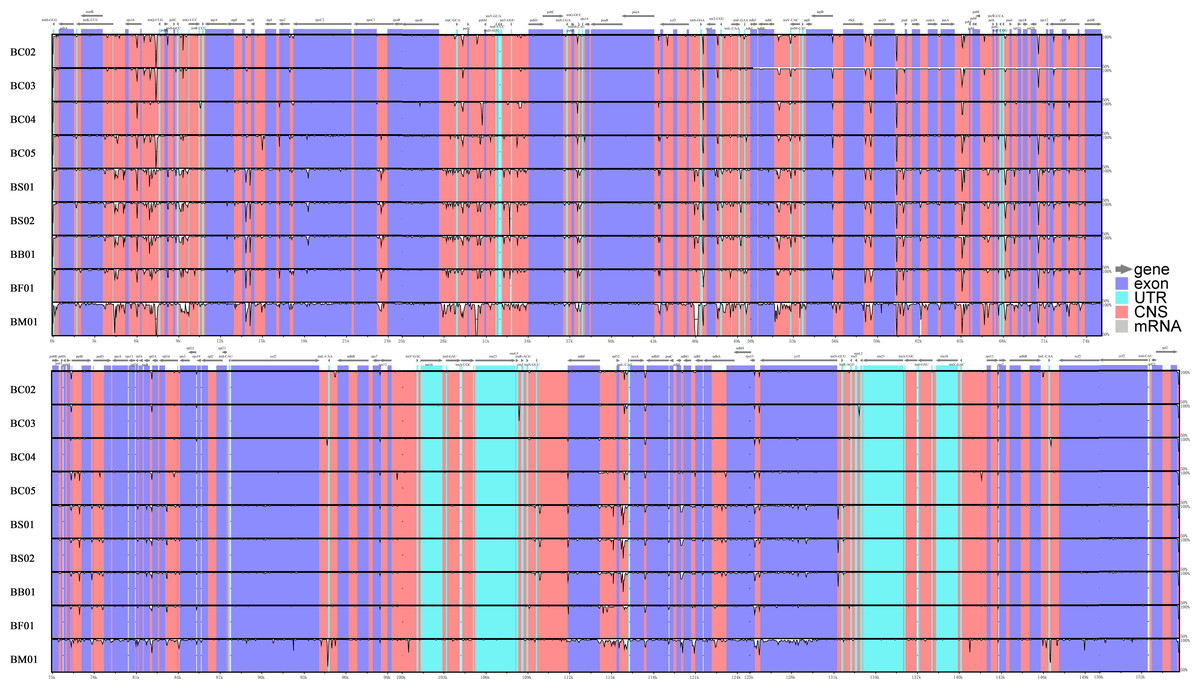
Figure 3: MVISTA-based sequence identity among the plastid genomes of five Bupleurum species with B. chinense (BC01) as a reference.
{kind=link}
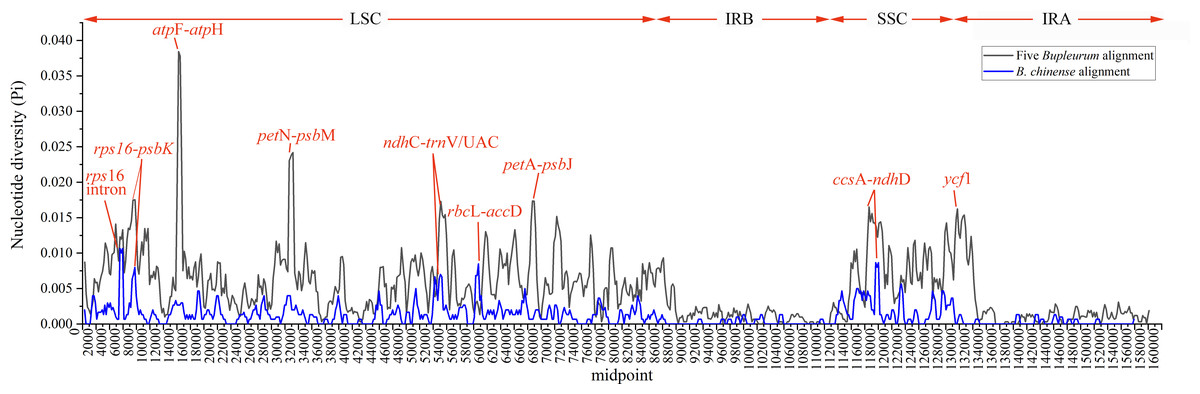
Figure 4: Nucleotide diversity (Pi) among the plastomes with sliding window analysis (window length: 600 bp).
The y-axis represents nucleotide diversity of the alignment of five Bupleurum species (in black) and the B. chinense alignment (in blue), while the x-axis represents the position of the window midpoint.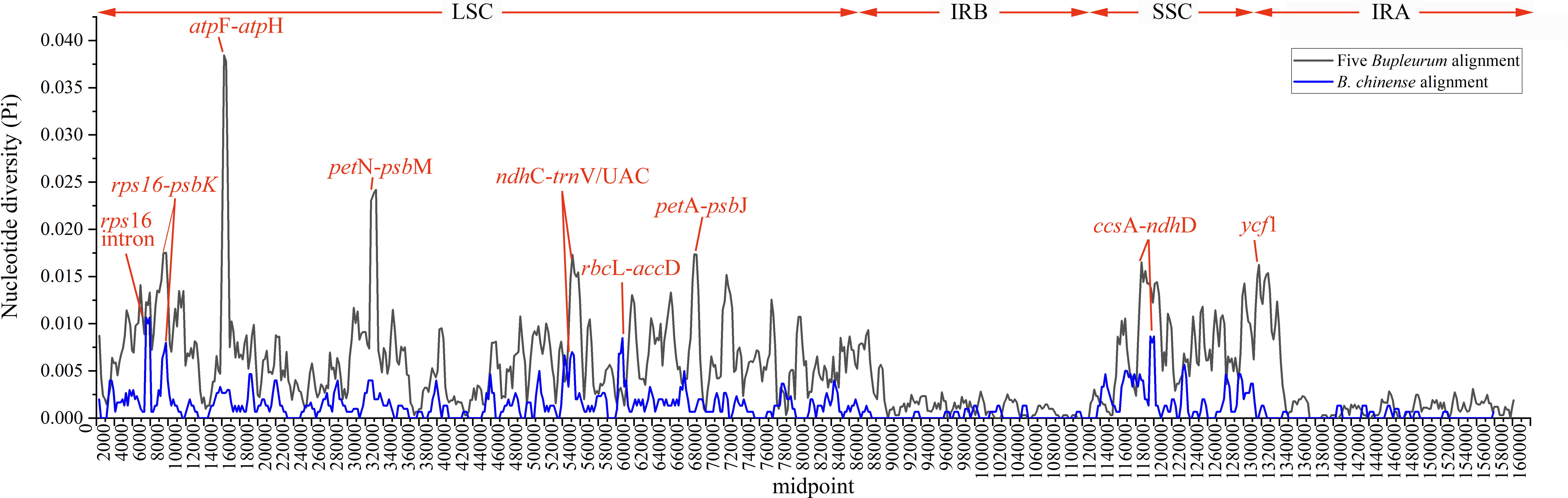{kind=link}
Simple sequence repeats (SSRs) analysis showed that the total number of cpSSR loci in plastomes ranged from 57 (BC05) to 81 (BM01) (Fig. 5A, Table S4). As shown in Fig. 5A, the SSRs were mainly located in the intergenic spacer (IGS), followed by CDS and intron regions. Monomers were the primary type, accounting for 60–68.9% of the SSRs in ten Bupleurum plastomes. As the patterns of SSR distribution were similar among the ten Bupleurum plastomes, BC01 was chosen as an example to exhibit the SSR characters. A total of 60 SSRs were detected in BC01, of which 58.3% were monomers, 18.3% were dimers, 13.4% were trimers, 6.7% were tetramers, 3.3% were pentamers, and no hexamers were present. Hexamers were only found in BF01 and BM01. Moreover, 65% of the SSRs were located in the IGS region, followed by the CDS region (20%) and the intron (15%). The genes rps 16-psb K, rps 2-rpo C2, trn T/GGU-psb D, and ycf 1 harbored more than two SSRs in each Bupleurum plastome. By comparing the cpSSRs across the five Bupleurum species using CandiSSR, a total of seven polymorphic cpSSRs were identified (Fig. 5B, Table S5). Specifically, four were dimers, two were trimers, and one was a pentamer. All polymorphic cpSSRs were detected in noncoding regions, of which trn T/GGU-psb D harbored two polymorphic cpSSRs. Additionally, 438 candidate polymorphic nSSRs were identified from the nuclear contigs of the five Bupleurum species (Fig. 5C, Table S6). More specifically, dimers were the primary type of polymorphic nSSR (55%), followed by trimers (22.6%) and tetramers (18%). Polymorphic pentamers and hexamers accounted for 3% and 1.4%, respectively.
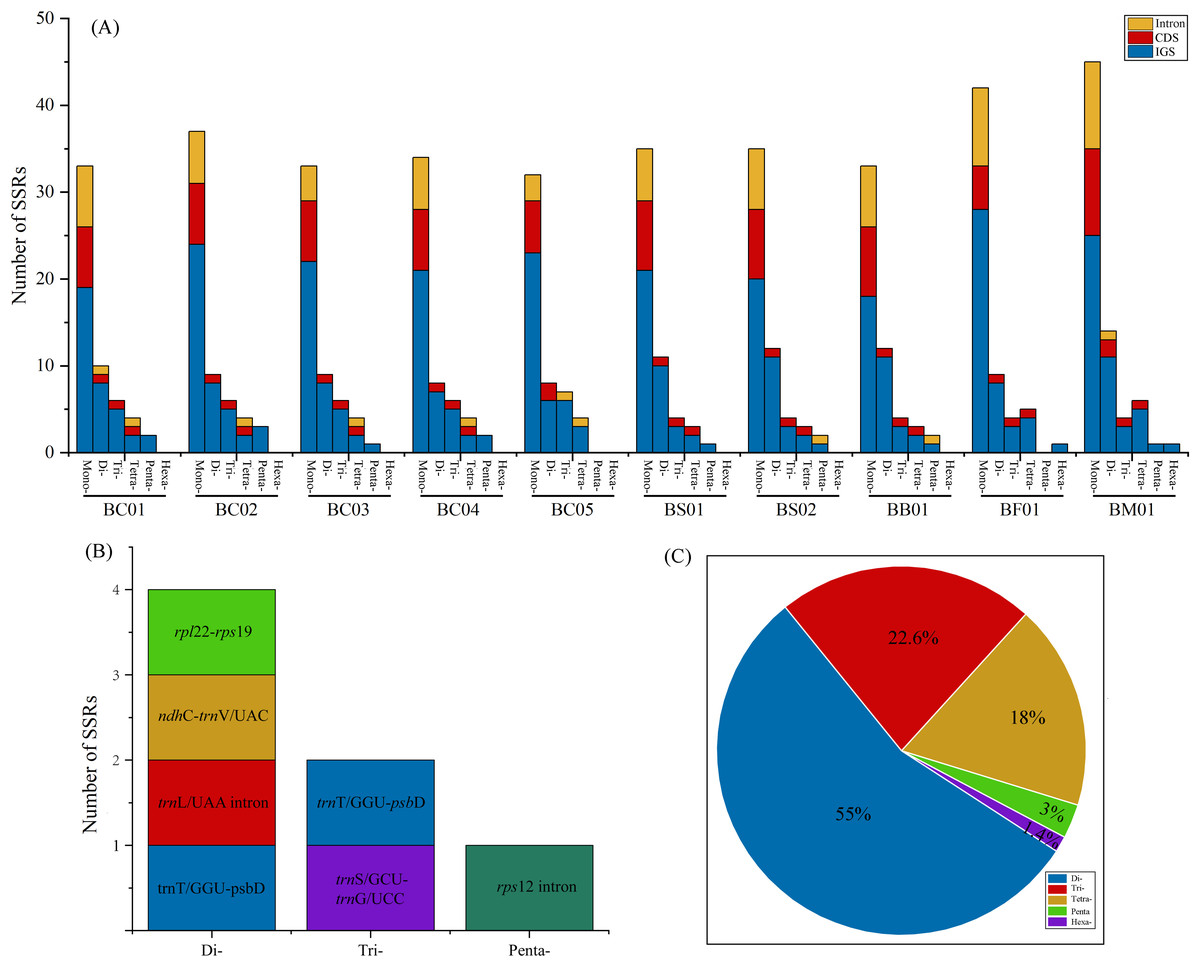
Figure 5: Analysis of SSRs in the five Bupleurum species.
(A) Number and location of six types of cpSSRs in ten plastomes of the five Bupleurum species. (B) Seven polymorphic cpSSRs across five Bupleurum species identified using CandiSSR. (C) Polymorphic nSSRs across five Bupleurum species identified using CandiSSR.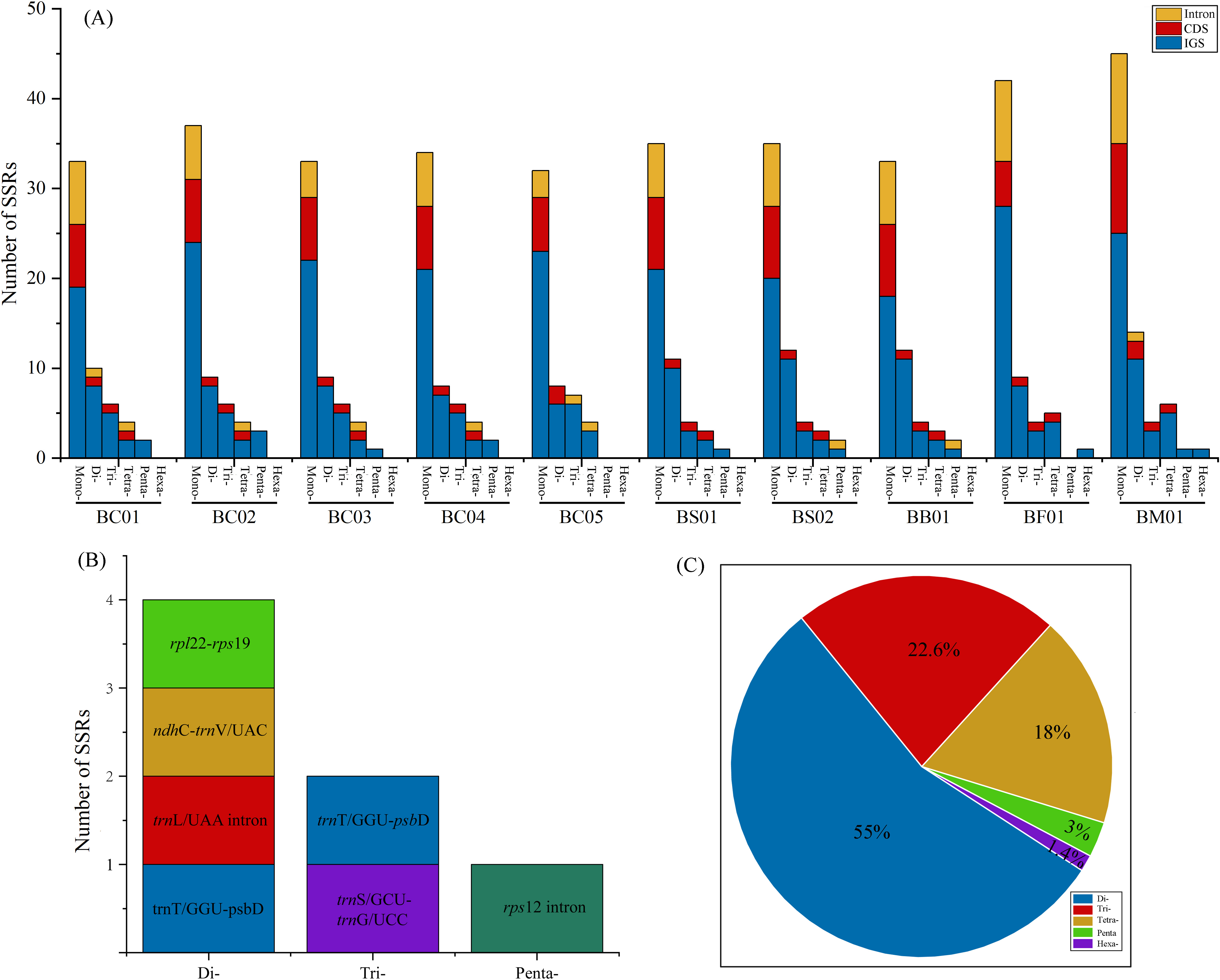{kind=link}
Selective pressure analysis
To pinpoint whether genes underwent adaptive evolution in Bupleurum plastomes, the Ka/Ks values of the protein-coding genes were calculated. At the interspecies level, the Ka/Ks values of psa J and ndh B were greater than 1, indicating that the corresponding genes experienced positive selection (Fig. 6). The Ka/Ks values of the photosynthetic genes and self-replication genes were 0.1853 ± 0.3804 and 0.1926 ± 0.2377, respectively. Both were lower than those of other genes (0.4127 ± 0.2489). Among the B. chinense plastomes, acc D related to the subunit of acetyl-CoA-carboxylase had the highest of Ka/Ks (1.076), followed by rpo C2 (Ka/Ks = 0.827) which is related to self-replication (Fig. 6).
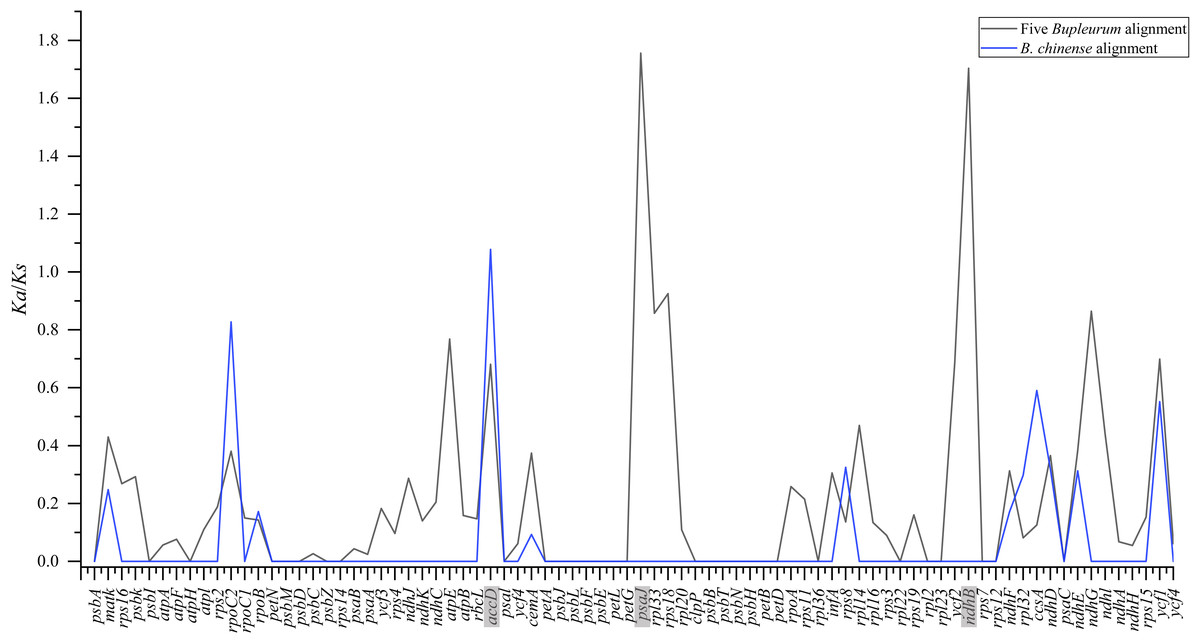
Figure 6: Ka/Ks values of 79 shared coding genes in the alignment of five Bupleurum species (in black) and the B. chinense alignment (in blue).
The genes with Ka/Ks values larger than one are in grey.{kind=link}
Phylogenetic analysis
Phylogenetic trees based on complete plastid genomes and the CDS showed the same topology, therefore, only the tree based on the complete plastid genomes with a slightly higher bootstrap support value was shown. All Bupleurum accessions were clustered into two main groups with absolute support (Fig. 7). B. marginatum var. stenophyllum (MT075712) and the other three congeneric species that are mainly distributed in Southwestern China formed a monophyletic group, appearing as an early branching clade within Bupleurum (Clade I). The remaining accessions could be divided into two subgroups, Clade II and Clade III (Fig. 7). B. chinense accessions and the B. falcatum accession were both in Clade II. B. chinense grouped with B. commelynoideum and B. yinchowense forming a clade with absolute support. Within Clade III, morphologically similar species B. angustissimum, B. bicaule, and B. scorzonerifolium were clustered together, sister to B. rockii and B. euphorbioides.
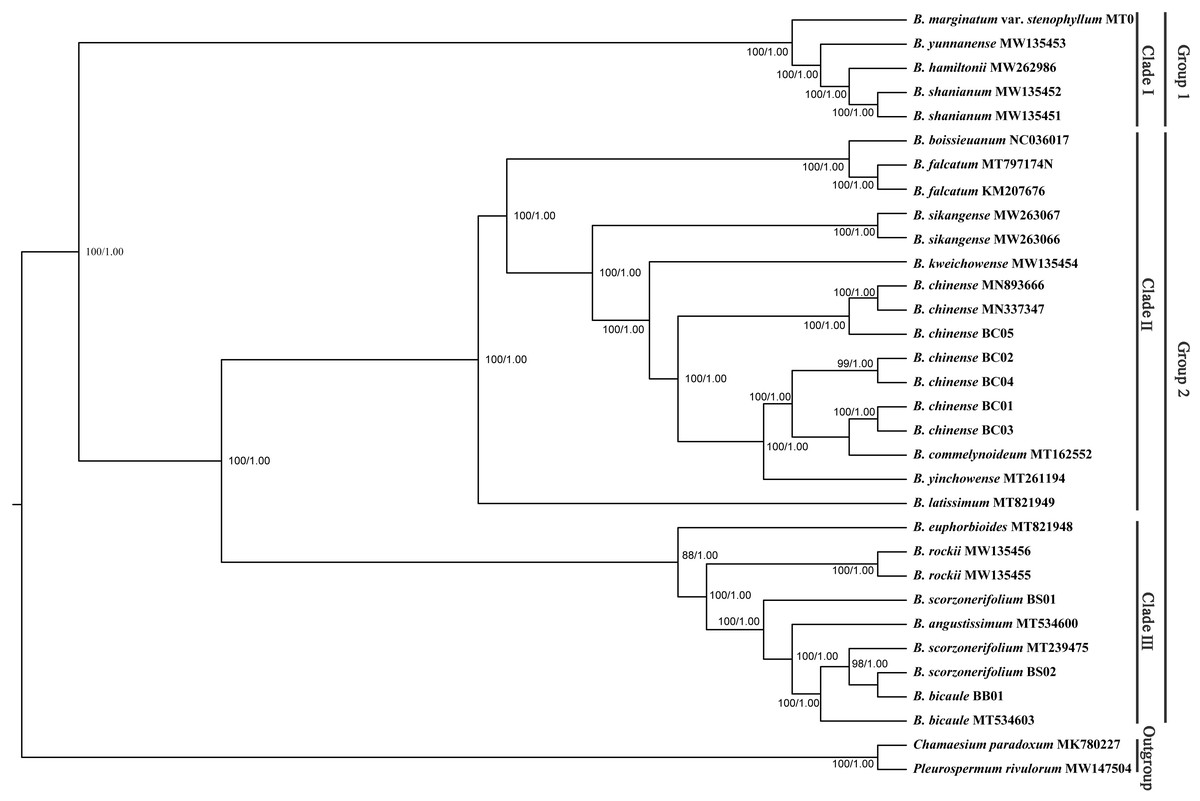
Figure 7: Phylogenetic tree reconstructed using maximum likelihood (ML) and Bayesian inference (BI) based on 32 complete plastid genomes.
Numbers shown at the corresponding nodes represent ML bootstrap support (BS) values and Bayesian posterior probabilities (PP), respectively.{kind=link}
The Bupleurum accessions were also clustered into two groups based on ITS data (Fig. 8). Though some of the branch nodes were poorly supported, each Bupleurum species studied formed a monophyletic lineage with high support, except for B. chinense, B. angustissimum, and B. scorzonerifolium. However, incongruences were observed between the phylogenies based on nuclear ITS and chloroplast datasets. Based on ITS data, B. chinense and its forms were nested with B. yinchowense, while distinct from B. falcatum. The B. scorzonerifolium accessions from northwestern China were grouped with B. angustissimum as shown in the tree based on the chloroplast dataset. B. bicaule represented an independent lineage in the nuclear dataset.
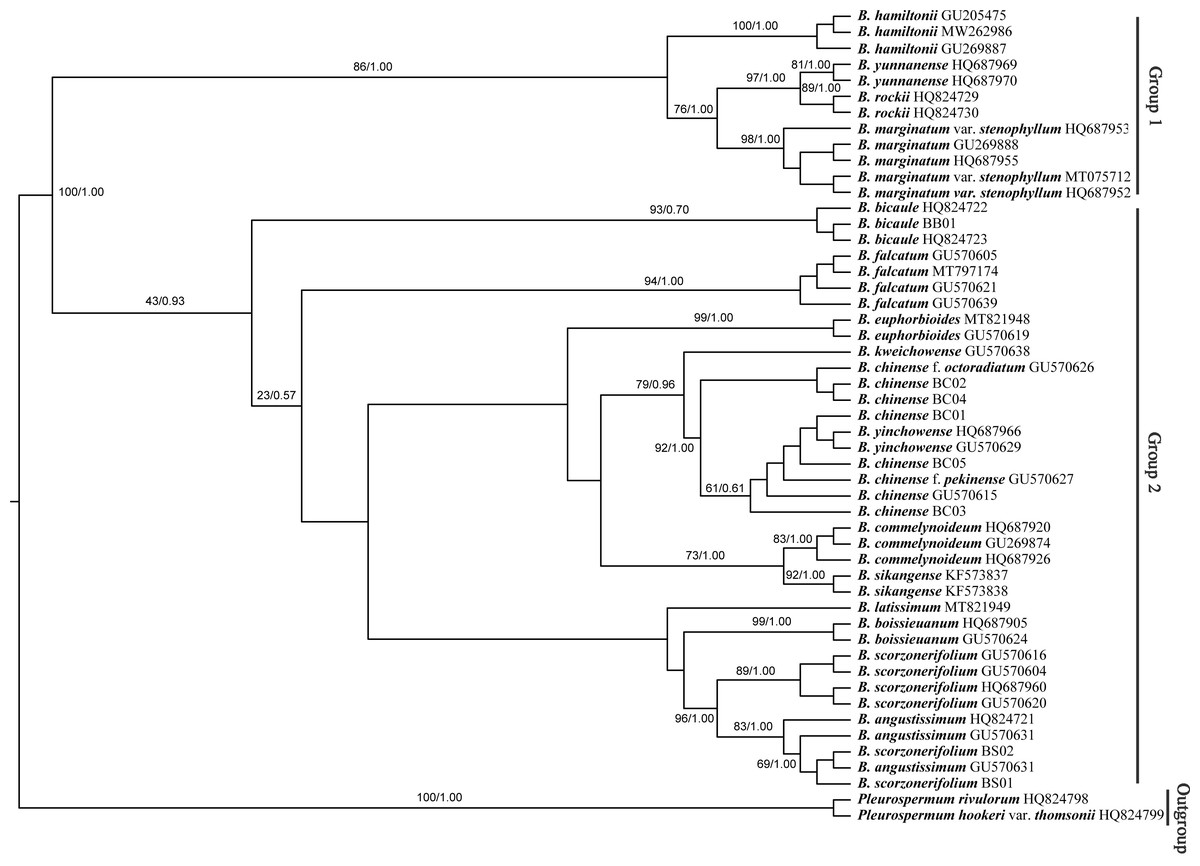
Figure 8: Phylogenetic tree reconstructed using maximum likelihood (ML) and Bayesian inference (BI) based on ITS.
Numbers shown at the corresponding nodes represent BS/PP.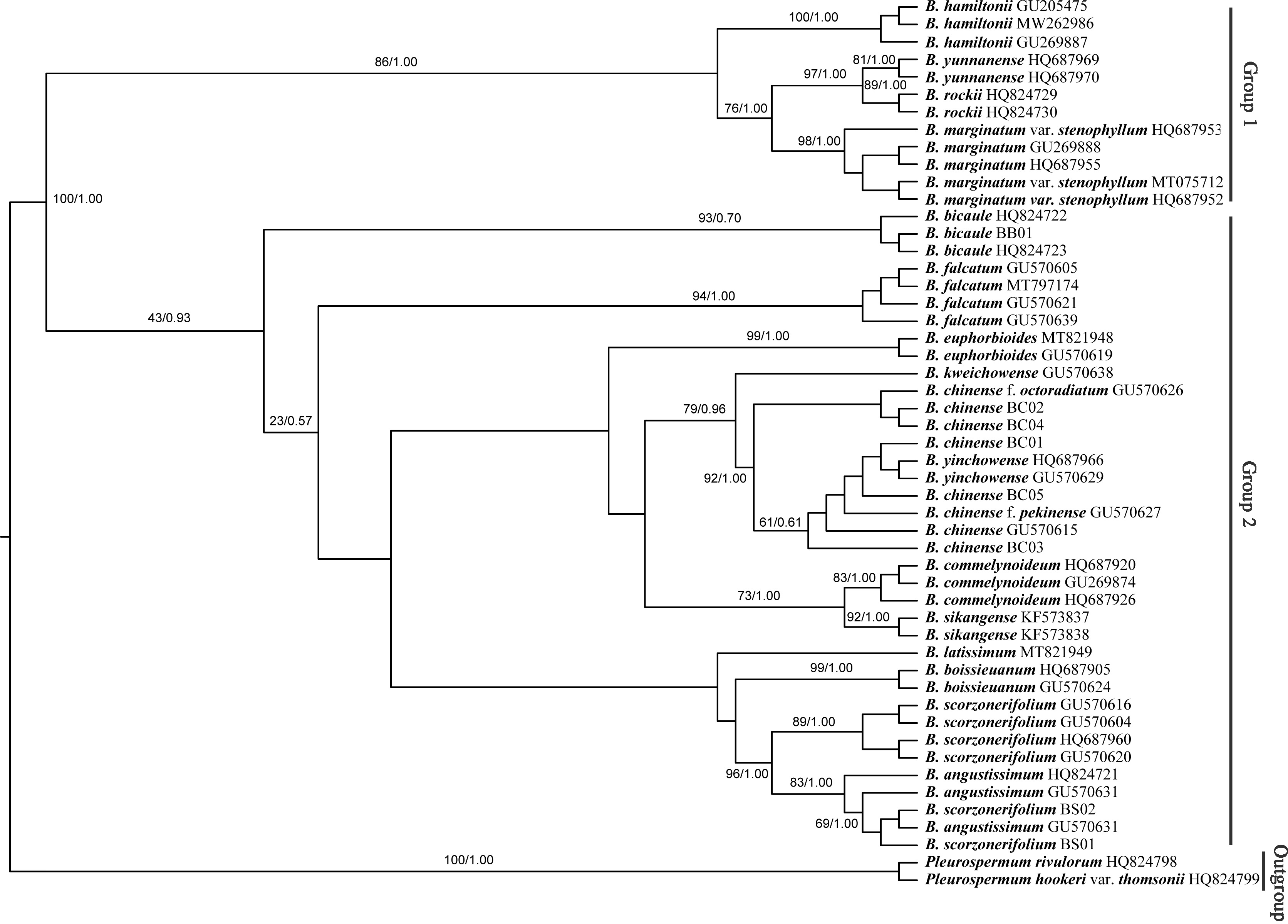{kind=link}
Discussion
Infrageneric relationships within Bupleurum and taxonomic implications
Bupleurum L. is one of the largest genera in Apiaceae, represented by 150–180 species, of which nearly 50 species occur in China (Sheh & Watson, 2005). Due to the similar morphological characteristics and broad intraspecific morphological variation under different ecological habitats, species delimitation based on traditional classification systems is extremely difficult in this genus. The complicated infrageneric relationships also result in problems with cultivation. Based on more extended sampling, Wang, Ma & He (2011) proposed to divide the Chinese Bupleurum into two groups. Specifically, one group contained species from the southwest, while another consisted of the species mainly distributed in northern China. However, the statistical support of the main clade was relatively low based on the combined dataset of mat K and trn H-psb A. In this study, the phylogenetic topology based on complete chloroplast genomes was congruent to the framework of Wang, Ma & He (2011), but with much higher internal resolution. Among the five Chaihu germplasm resources, B. marginatum var. stenophyllum clustered in the early branching group of the phylogenetic trees, separated from the other four Bupleurum species. Due to the high yield and high content of saikosaponins, B. marginatum var. stenophyllum was introduced from Tibet to Gansu and Shanxi Provinces in China with widespread planting (Liu et al., 2021a; Liu et al., 2021b). Modern pharmacological studies have shown that saikosaponins are not only the main bioactive components of Chaihu, but also the main toxic components (Huang & Sun, 2010; Lv et al., 2009; Zhou et al., 2021). Wang et al. (2020) demonstrated that B. marginatum var. stenophyllum contained higher content of saikosaponins A and D, and had a higher level of acute toxicity. Xia et al. (2021) compared the overall chemical components of B. marginatum var. stenophyllum and B. chinense, and showed clear species-clustering. Thus, the use of B. marginatum var. stenophyllum as Chaihu should be done with caution.
In the plastid and ITS trees, B. chinense accessions were clearly distinct from those of B. scorzonerifolium. Morphologically, the two species are distinguished by a combination of characteristics, including leaf veins, fibrous remnant sheaths, and color of the root bark. Notably, phylogenetic trees based on complete plastid genomes and nuclear ITS datasets both supported close affinities of B. yinchowens and B. chinense, and B. angustissimum and B. scorzonerifolium. The delimitation confusion has long been an issue in these species (Wang, Ma & He, 2011). Based on similar morphology and overlapping ecological distributions (Sheh & Watson, 2005), we proposed B. yinchowens as conspecific to B. chinense, and B. angustissimum as conspecific to B. scorzonerifolium, but more evidence was needed. B. falcatum showed a closer phylogenetic relationship with B. chinense in the plastid trees, while the species was resolved as a distinct clade not closely related to B. chinense and B. scorzonerifolium but to B. bicaule. B. falcatum is a cultivated species originating from Japan, where it is the official botanical origin of Bupleuri Radix in the The Society of Japanese Pharmacopoeia (2011). Consequently, we speculated that introgressive hybridization during years of cultivation might contribute to the conflicts between plastid and nuclear ITS phylogenies. Theoretically, evolutionary processes including incomplete lineage sorting, introgressive hybridization, and paralogy conflation might contribute to phylogenetic incongruence among different datasets (Liu et al., 2020; Wendel & Doyle, 1998). Many studies suggested that introgressive hybridization was a prominent factor for phylogenetic incongruence at lower taxonomic levels. Based on the plastid datasets, B. bicaule and B. scorzonerifolium were clustered with B. angustissimum. Morphologically, these three species are highly similar, with fibrous remnant sheaths around the stem. However, B. bicaule accessions did not cluster with B. angustissimum and B. scorzonerifolium in the nrDNA tree. Considering the corresponding node had low support, we cannot exclude the possibility that insufficient information from ITS sequences resulted in the incongruence. Moreover, B. bicaule was considered to be involved in hybridization events to coexist with other Bupleurum species. The delimitation of B. bicaule needs further study.
Conserved plastid genome structure
For the five Bupleurum species examined here, the chloroplast genomes displayed a typical quadripartite structure, as in most angiosperms (Palmer, 1985). The chloroplast genome structures were found to be largely conserved with lengths varying from 155,540 bp to 155,866 bp. Previous studies have reported that gene loss (Wakasugi et al., 1994; Wolfe, Morden & Palmer, 1992), expansion and contraction of the IRs (Lin et al., 2003; Parks, Cronn & Liston, 2009), and intergenic region variation (Tang et al., 2004; Wu et al., 2011) are three important factors driving size variation in the plastid genome. Across the five Bupleurum species in this study, the genes distributed around the junction boundaries slightly varied in length. Among these, the SSC/IR borders showed a higher level of differences than LSC/IR, in contrast to the study of Huang et al. (2021) in a broader comparison of Bupleurum species. As expected for the intraspecific divergence inferred from plastid genomes, the junction boundaries in B. chinense accessions were more convergent. We proposed that size variations in the chloroplast genome within Bupleurum were due to variation of intergenic regions.
Variable regions for potential molecular markers
In this study, abundant sequences with SNPs and SSRs were detected across the five Bupleurum species. As expected, large-scale sequencing provided abundant variable sequences, which could be used for species authentication and phylogenetic inference. Our results showed that the majority of variable sequences were located in noncoding regions at either the interspecific level or the intraspecific level. Previous studies have demonstrated that noncoding sequences of the chloroplast genome perform well as phylogenetic markers, and in phylogenetic construction and population genetic studies (Shaw et al., 2007; Borsch & Quandt, 2009). In addition, the majority of noncoding sequences with high nucleotide diversity were situated in single-copy regions. Perry & Wolfe (2002) showed that the nucleotide mutation rate of single-copy regions was 2.3 times higher than that of the inverted repeats.
In this study, atp F-atp H, pet N-psb M, rps 16-psb K, pet A-psb J ndh C-trn V/UAC, and ycf 1 regions had high values of nucleotide diversity across the five Bupleurum species. Atp F-atp H has been proposed as a potential DNA barcode for identifying plant taxa at the species level (Lahaye et al., 2008; Thakur et al., 2019). The high nucleotide diversity of the remaining five genes has been reported in Bupleurum before (Huang et al., 2021; Li et al., 2020; Xie et al., 2021). In particular, ycf 1 has been proposed as the most promising plastid DNA barcode of land plants (Dong et al., 2015). The Pi values of the corresponding genes were slightly lower than that in Huang et al. (2021), mainly because of the limited Bupleurum species involved in our study.
Repeat units, distributed in genomes with high frequency, are essential in the rearrangement and divergence of the genome (Weng et al., 2014). Microsatellites (SSRs), containing repetitive sequences of 1–6 bp in length, are used as molecular markers for population genetics, phylogeography, and systematics (George et al., 2015). Owing to the rapid development of next-generation sequencing, abundant genomic microsatellites with a high level of polymorphisms are available for plant species (Xia et al., 2016). Based on complete plastomes, numerous cpSSRs were detected for the five Chaihu germplasm resources, varying from 51 (B. chinense BC05) to 81 (B. marginatum var. stenophyllum). The mononucleotide category was the most abundant, followed by di-, tri-, tetra- and pentanucleotides. Further, seven cpSSRs were found to be polymorphic across the five Bupleurum species, and were located in noncoding regions. One polymorphic cpSSR was located in ndh C-trn V/UAC, which also exhibited high nucleotide diversity. We propose that ndh C-trn V/UAC might be candidate molecular markers for cultivated Bupleurum species. At the same time, a total of 438 candidate polymorphic nSSRs were detected, most of which were dimers. Among polymorphic cpSSRs and nSSRs, the di- and trinucleotide repeats were dominant categories and no monomers were detected, consistent with the characteristics of the identified polymorphic SSRs in other taxonomic studies (Bhandari et al., 2020; Dabral et al., 2021). Notably, all regions with high variability possessed SSRs. The identified SSRs are helpful in intraspecific phylogeographic and population-level genetic studies of Chaihu germplasms as in Angelica heterocarpa (Revardel & Lepais, 2022), Grevillea robusta (Dabral et al., 2021), and Erysimum teretifolium (del Valle, Herman & Whittall, 2020).
Adaptive evolution among Bupleurum species
Plants might leave fingerprints in plastid genomes in response to environmental changes, within which some genes exhibit positive selection. The value of Ka/Ks is used to measure the selective pressure of the coding genes in angiosperms (Yang & Nielsen, 2002). The chloroplast functions directly in photosynthesis and carbon fixation, and three genes with essential roles in photosynthesis showed positive selection in this study. Among five Bupleurum species, the Ka/Ks values of psa J and ndh B were greater than one. Among the plastomes of B. chinense, we identified acc D as a positively selected gene. The substitution rates of the functional genes at intraspecies and interspecies levels were different, which might be caused by the diverse evolutionary history of Bupleurum species. Interestingly, the nucleotide diversity of psa J, ndh B, and acc D in coding regions was not significant (Figs. 3 and 4), presumably due to the close affinity among the Bupleurum species. Similar results were found in Huang et al. (2021) and Li et al. (2020). The psa J gene is involved in the excitation of photosystem I, which has been essential during the life history of plants (Schöttler et al., 2007). The ndh B gene encodes for the subunits of NAD(P)H dehydrogenase complex involved in photosystem I cyclic and chlororespiratory electron transport in higher plants (Martín & Sabater, 2010). Horváth et al. (2000) showed that ndh B-inactivated tobacco plants cause a moderate decline in photosynthesis via stomatal closure under humidity stress conditions. The acc D gene, encoding the β-carboxyl transferase subunit of acetyl-CoA-carboxylase, has been demonstrated as an essential gene in plastid genome evolution (Kode et al., 2005). Madoka et al. (2002) confirmed that acc D affected leaf longevity and seed yield.
B. chinense is a widespread species in China, within which three forms were recognized by Sheh & Watson (2005) with differences in leaves and bracteoles. Environmental variation drives leaf morphology variation within species (Byars, Papst & Hoffmann, 2007). We also observed leaf variations in the B. chinense populations under different ecological conditions during field sampling. Han et al. (2006) identified three types of B. chinense based on leaf shapes, namely broad, long, and medium. The mutation accumulation of functional genes affects photosynthesis efficiency, which may cause ecological diversity of plants (Zheng et al., 2017). Consequently, positively selected genes provide important insights into adaptive molecular evolution of Bupleurum species.
Conclusions
In this study, the complete chloroplast genomes and nuclear ITS of five Bupleuri Radix germplasm resources, namely B. bicaule, B. chinense, B. falcatum, B. marginatum var. stenophyllum, and B. scorzonerifolium, were obtained using genome skimming. All plastomes we sequenced were conserved and exhibited a typical quadripartite structure, encoding 113 identical genes including 79 protein-coding genes, 30 tRNA genes, and four rRNA genes. Phylogenetic analysis inferred using the chloroplast genomes and nrITS resolved the relationships of the five Bupleurum species and their closely related species. B. marginatum var. stenophyllum was separate from the other four Bupleurum species studied. B. chinense and B. scorzonerifolium, which are the official botanical origins of Chaihu in the Chinese Pharmacopoeia, were in two separate clades. However, the phylogenetic delimitation of B. bicaule and B. falcatum needs further study, because introgressive hybridization might be involved. By comparison analysis, molecular markers, including plastid hotspots and polymorphic SSRs, were generated for authentication of Chaihu germplasms. Photosynthesis related genes psa J, ndh B, and acc D were found to be under positive selection as a response to adapting to diverse environments.