
A novel alkane monooxygenase (alkB) clade revealed by massive genomic survey and its dissemination association with IS elements
- Published
- Accepted
- Received
- Academic Editor
- Joseph Gillespie
- Subject Areas
- Bioinformatics, Genetics, Genomics, Microbiology, Data Science
- Keywords
- Alkane monooxygenase, alkB, alk-gene clusters, Phylogenetic diversity, IS elements, Niches distribution
- Copyright
- © 2022 Wang et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2022. A novel alkane monooxygenase (alkB) clade revealed by massive genomic survey and its dissemination association with IS elements. PeerJ 10:e14147 https://doi.org/10.7717/peerj.14147
Abstract
Background
Alkanes are important components of fossil energy, such as crude oil. The alkane monooxygenase encoded by alkB gene performs the initial step of alkane degradation under aerobic conditions. The alkB gene is well studied due to its ubiquity as well as the availability of experimentally functional evidence. The alkBFGHJKL and alkST clusters are special kind of alkB-type alkane hydroxylase system, which encode all proteins necessary for converting alkanes into corresponding fatty acids.
Methods
To explore whether the alkBFGHJKL and alkST clusters were widely distributed, we performed a large-scale analysis of isolate and metagenome assembled genome data (>390,000 genomes) to identify these clusters, together with distributions of corresponding taxonomy and niches. The set of alk-genes (including but not limited to alkBGHJ) located near each other on a DNA sequence was defined as an alk-gene cluster in this study. The alkB genes with alkGHJ located nearby on a DNA sequence were picked up for the investigation of putative alk-clusters.
Results
A total of 120 alk-gene clusters were found in 117 genomes. All the 117 genomes are from strains located only in α- and γ-proteobacteria. The alkB genes located in alk-gene sets were clustered into a deeply branched mono-clade. Further analysis showed similarity organization types of alk-genes were observed within closely related species. Although a large number of IS elements were observed nearby, they did not lead to the wide spread of the alk-gene cluster. The uneven distribution of these elements indicated that there might be other factors affecting the transmission of alk-gene clusters.
Conclusions
We conducted systematic bioinformatics research on alk-genes located near each other on a DNA sequence. This benchmark dataset of alk-genes can provide base line for exploring its evolutional and ecological importance in future studies.
Introduction
Alkanes are widely distributed in nature, not only as the most abundant components of crude oil, but also low concentration alkanes produced by plants, insects, and microorganisms. It has been reported that marine algae could produce about 308 to 771 million tons of alkanes annually (Lea-Smith et al., 2015; Schirmer et al., 2010; Valentine & Reddy, 2015). Alkanes can serve as chemo-attractants as well as substrate. The capacity to use hydrocarbons as sole carbon and energy source is very common, and not restricted to any particular group of microorganisms, owing to the wide spread of alkane hydroxylase system (Nie et al., 2014). According to alkanes which could be catalyzed by responsible enzymes, they have been broadly classified into short-, mid-, and long-chain alkane degradation systems. The integral membrane non-heme iron monooxygenase, which catalyzes the initial hydroxylation of mid-chain alkanes, is a kind of widespread alkane degradation enzyme encoded by alkB (Rojo, 2009; Van Beilen et al., 2002). It has been well studied due to its ubiquity as well as the availability of experimentally functional evidence (Wang & Shao, 2013). To date, quite a few studies describing alkB sequences and their phylogenetic structure were elucidated (Nie et al., 2014; Smith et al., 2013; Wang et al., 2010; Wang & Shao, 2013). The alkB gene was generally considered as a molecular marker for alkane degradation as well (Christian et al., 2020).
The alkB-type alkane hydroxylase system consists of three core components: an integral membrane alkane monooxygenase and two soluble proteins (rubredoxin and rubredoxin reductase) which act as electron carriers between NADH and the hydroxylase (Nieder & Shapiro, 1975). The alkBFGHJKL and alkST clusters were a special kind of alkB-type alkane hydroxylase system. They were initially reported on OCT-plasmid of Pseudomonas putida PGo1 (Eggink et al., 1987a; Eggink et al., 1987b; Van Beilen et al., 1992; Van Beilen, Wubbolts & Witholt, 1994). The alk-gene clusters encode all proteins necessary for converting alkanes into corresponding fatty acids, endowing microorganisms with the ability to utilize alkanes as the sole carbon and energy source. At present, bioinformatics analysis of alkB genes on gene-cluster level has not been reported.
The alkB-type alkane hydroxylase system from the plasmid of Pseudomonas putida GPo1 was unusual not only because of its compact gene organization, but also as its n-alkane substrate range, from C3 to C12 (alkanes in gasoline range), while many other related hydroxylases typically oxidize alkanes longer than C10 (Tsai et al., 2017). Schneiker et al. (2006) reported alkSBGHJ operon and alkK nearby in Alcanivorax borkumensis SK2, a plasmid free strain containing only a single circular chromosome, suggesting alk-gene clusters were not restricted in plasmid and neither in P. putida.
So far, many comprehensive studies have been carried out to investigate alk-genes, including functional characterization (Smits, Witholt & Van Beilen, 2003), regulation mechanism (Canosa et al., 2000), fusion and transfer (Chakrabarty, 1973), heterologous expression (Liu et al., 2011; Liu et al., 2010; Minak-Bernero et al., 2004; Sameshima et al., 2008; Smits et al., 2001; Wang et al., 2017; Whyte et al., 2002), promoter (Staijen Ivo, Marcionelli & Witholt, 1999) and biotransformation application (Lu et al., 2021; Van Nuland et al., 2017). Sequence analysis of alk-gene clusters had observed several insertion sequences (IS) located in its flanking regions. Together with its lower G-C content compared with the host strain, previous studies suggested these genes were part of a large mobile element, which may be responsible for horizontal transfer across different species (Van Beilen et al., 2001). Although it was supposed to spread horizontally, there was still a lack of relevant research on whether it is widely distributed.
To fill this knowledge gap, we performed a large-scale analysis of isolate and metagenome assembled genome data to identify alk-gene clusters, together with distributions of corresponding taxonomy and niches. A comprehensive bioinformatics analyses, including phylogenetic analysis, taxonomy distribution, niches distribution, organization structure, chromosome location and IS elements analyses, were performed to provide detailed information of alkB genes on gene-cluster level.
Materials and Methods
Identification of alk-gene cluster
An initial database of AlkBFGHJKL, alkN and alkST (alk-genes hereafter) was constructed using protein sequences described in the study by Van Beilen et al. (2001). All available prokaryotes genomes (both bacteria and archaea) were downloaded from NCBI (http://www.ncbi.nlm.nih.gov) on March 2, 2022, representing for 200 phyla (146 out of which were candidatus). More specifically, the genome sequences of over 390,000 isolates or MAGs from 57,335 species belonging to 3,687 genera were included in this analysis. At the same time, the taxonomy and niches information corresponding to these genomes were also downloaded. The sequences from genomes with assembly levels of “Complete” or “Chromosome” were marked as chromosomal or plasmid according to their corresponding description. The rest genome sequences were marked as “Contig/Scaffold”. Subsequently, gene prediction was performed using Prokka v1.13 (Seemann, 2014). The presence of each gene in the genome was screened using blastp with an e-value of 1e−10 (Camacho et al., 2009).
Identification of putative gene clusters was performed using a two-step method. For the first, a set of four genes, that were alkBGHJ were used as markers for identification of putative alk-fragment (DNA fragment containing alk-genes) location using criteria as follows: (1) alkB must exist, (2) alkGHJ were present within 10 kb flanking region of alkB gene, (3) sequence of DNA segments flanking the alkB gene, ranging from 10 kb upstream to 28 kb downstream of the starting codon, were extracted from genome sequence for next step. Second, alk-fragments were re-predicted using prokka (Seemann, 2014), and then screened by each gene of AlkBFGHJKL, alkN and alkST using blastp in blast package v2.9.0 (Camacho et al., 2009). All alk-fragments were verified manually and the fake ones were deleted. The alk-fragments at the end of contigs, which contain truncated gene clusters, were excluded in this analysis. The IS elements in alk-fragments were identified using ISEScan v1.7.2.2 (Xie & Tang, 2017). Organization of alk-genes and IS elements were plotted using R v4.1.2 (R Core Team, 2021) with ggplot.
Phylogeny of alk-gene cluster-carrying genomes
To build the reference phylogenetic tree for this analysis, bcgTree v1.2.0 (Ankenbrand & Keller, 2016) was performed. The bcgTree pipeline builds phylogenetic trees from bacterial core genomes, these were 107 essential genes. Briefly, gene prediction was performed by prodigal v2.6.3 (Hyatt et al., 2010), then the core genes were identified using hmmsearch in HMMER v3.2.1 (Eddy, 1998; Eddy, 2009) and aligned using muscle v3.8.31 (Edgar, 2004). The unreliable alignment regions were removed by using Gblocks v0.91b (Castresana, 2000). The resulting alignment was used to construct a maximum likelihood phylogenetic tree using the RAxML v8.2.12 (Stamatakis, 2014) with rapid bootstrap 1000. The final tree was midpoint rooted and visualized using iTOL (Letunic & Bork, 2021).
Phylogeny of the alkB genes
To infer the phylogenetic structure of the alkB gene, we manually downloaded nucleotide sequences coding alkane 1-monooxygenase from KEGG (http://www.kegg.jp) with ortholog number K00496, along with corresponding taxonomy information for each sequence. The alkB genes with identical nucleotide sequences and taxonomy compared with alkB genes in alk-gene clusters were eliminated.
The retained alkB gene sequences from KEGG (http://www.kegg.jp) and the alkB gene sequences retrieved from alk-fragment were combined for phylogenetic analysis. All the alkB gene sequences were aligned using mafft v7.407 (Katoh & Standley, 2013). And then maximum likelihood phylogenetic tree was constructed using the RAxML v8.2.12 (Stamatakis, 2014) with rapid bootstrap 1000. The xylM, which encodes the xylene monooxygenase subunit, was used as the outgroup, as described in the studies by Nie et al. (2014) and Karthikeyan et al. (2021).
Organization structure of alk-genes
The sequence fragments were extracted from the genome sequence for each alk-gene cluster, from 10kbp upstream of the alkB gene to 28kbp downstream, using inhouse perl scripts. These sequence fragments were defined as alk-fragments. The corresponding gene prediction and annotation information were extracted at the same time. The organization structure was plotted using ggplot2 and gggenes packages in R v4.1.2.
Results
Phylogenetic diversity of genomes carrying alk-gene clusters
To capture as much diversity as possible, >390 000 genomes which include both MAGs and isolate genomes, were screened for the presence of alk-gene clusters. Totally 117 genomes (Table S1) carrying at least one set of alk-gene clusters were observed, which formed 2 distinct clades (Fig. 1). Clade A was composed of 56 strains, all in α-proteobacteria at class level, representing for 6 orders and at least 26 genera. While clade B was composed of 61 strains, all in γ-proteobacteria at class level, representing for at least 3 orders and 6 genera. Strains in genus Pseudomonas, located in clade B, had consistent and complete alk-genes sets. While all the other species had incomplete alk-genes set, more or less. Longer branch length reflected distant phylogenetic relationship between species in clade A, suggesting wider distribution of alk-gene clusters compared with those in clade B.
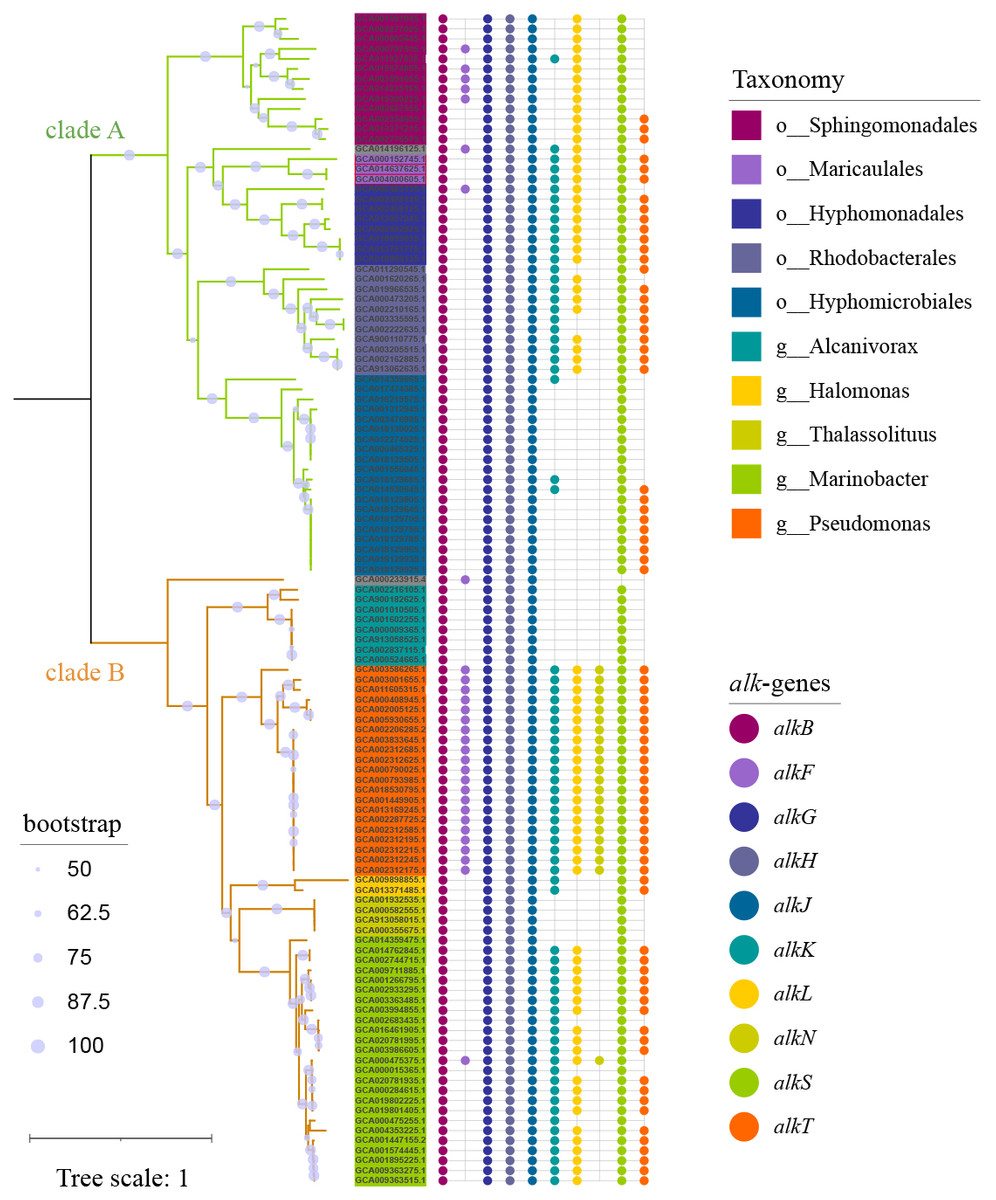
Figure 1: Phylogenetic tree of strains carrying alk-gene clusters.
The Maximum Likelihood phylogenetic tree was constructed from core genes with seqboot 1000. The green branch represented for clade A, which was composed of strains from α-proteobacteria. The yellow branch represented for clade B, which was composed of strains from γ-proteobacteria. The corresponding taxonomy was represented by the background colors of leaf labels. The Bubble diagram on the right represents the presence of alk-genes.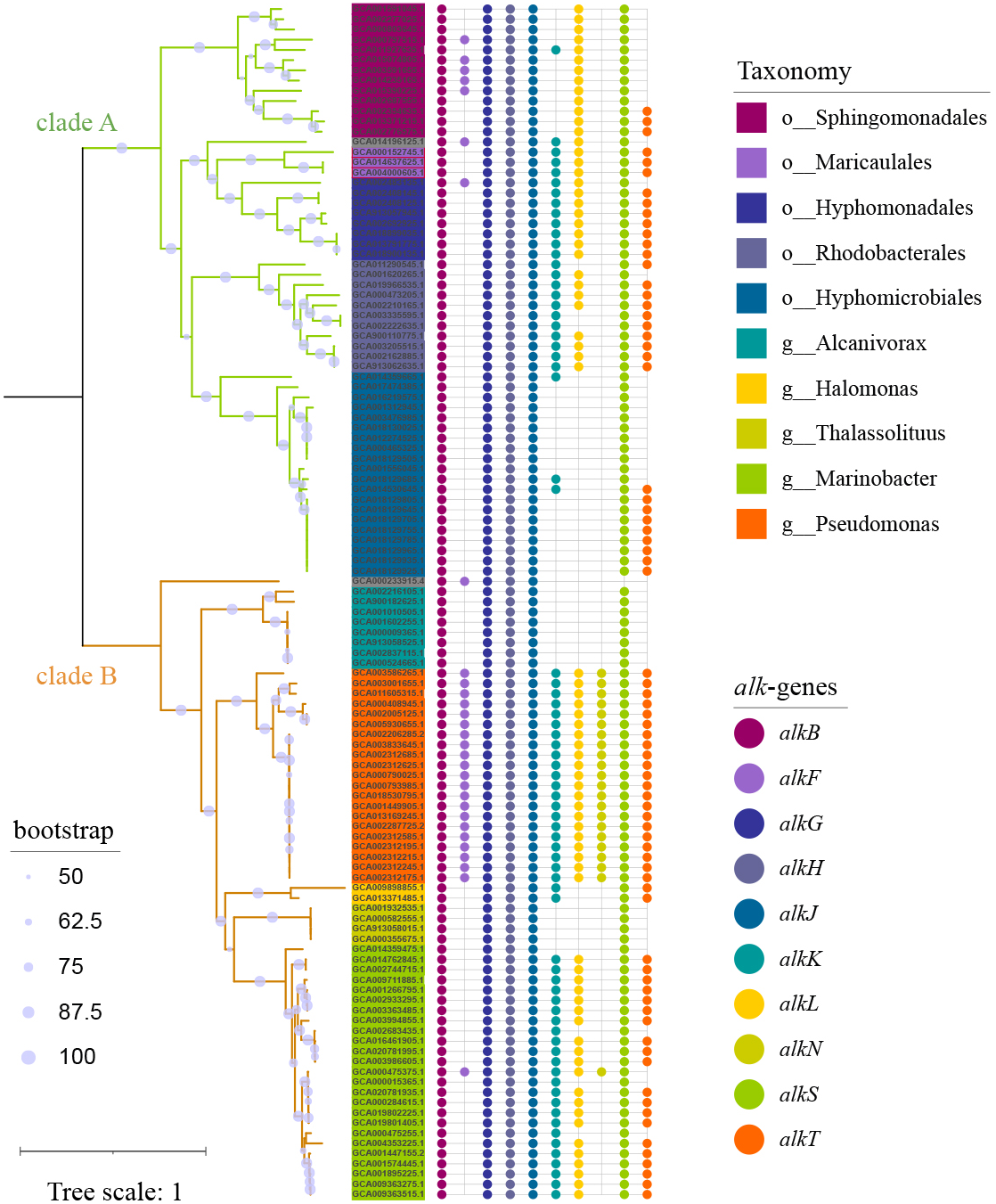{kind=link}
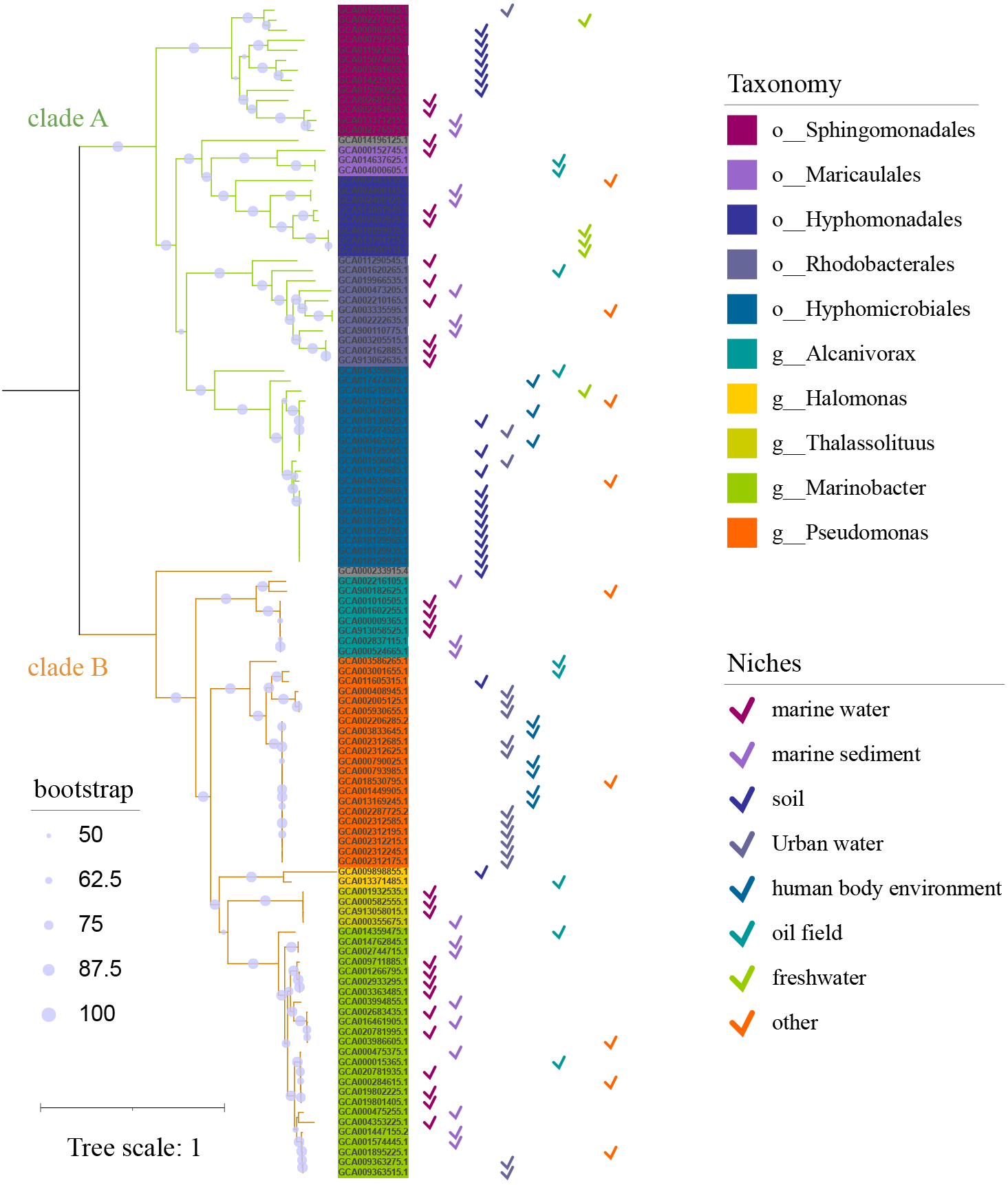
Niches distribution of strains carrying alk-gene clusters
These alk-gene clusters carrying strains had extensive niches distribution, including marine, soil, water, oil field and human as inferred in Fig. S1. But the niches distribution has a strong correlation with taxonomy, rather than alk-genes. Soil niched strains were enriched in α-proteobacteria, accounting for the major of Sphingomonadales and Hyphomicrobiales in clade A. Freshwater niched strains were limited in α-proteobacteria. While urban water niched strains enriched in γ-proteobacteria, account for almost half of Pseudomonas. It is speculated that alk-gene clusters had less impact on niches adaptation. On the other hand, they had the potential for dissemination in a wide range of environments.
Alk-fragments distribution on genome
Totally 120 alk-fragments were extracted from 117 genomes, as two-copies of alk-fragments exist in three genomes. All of the three two-copies alk-fragments carrying strains are located in clade B (γ-proteobacteria). Marinobacter nauticus VT8, a species of Marinobacter isolated from oil producing well, had both alk-fragments on a single chromosome, separated by ∼150 kb distance. The other two strains were Marinobacter salarius strain HL2708#2, a species of Marinobacter isolated from Lava and Alcanivorax sp. N3-2A, a species of Alcanivorax isolated from coastal sediments, had one alk-fragment on chromosome and the other on plasmid.
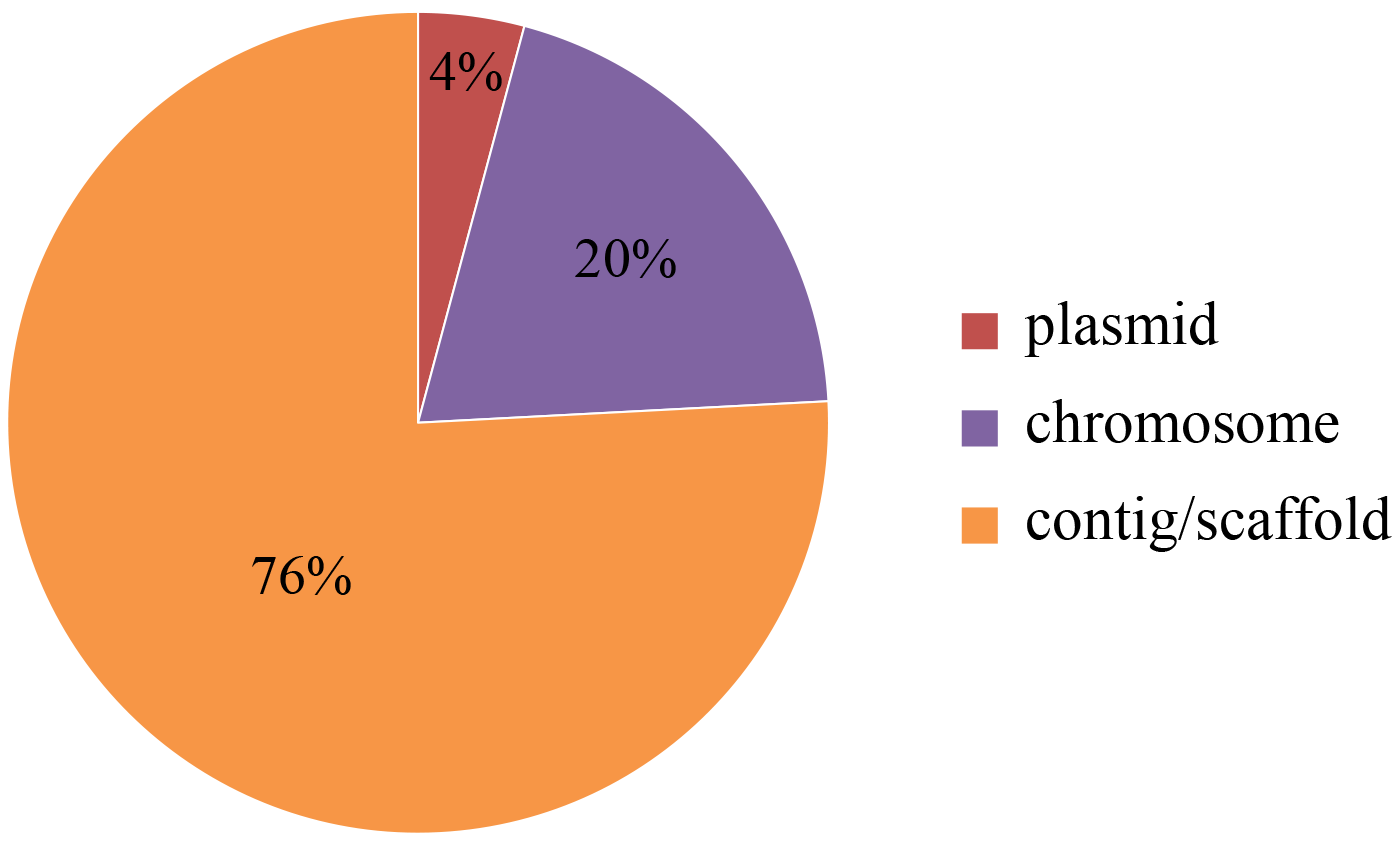
Among all of the 120 alk-fragments observed, there were 24 cases located on chromosome, while only 5 cases were on plasmid. And the remaining 91 cases were located on contig or scaffold, owning to assemble level. Although alk-gene clusters were initially found on plasmids, they were also observed on chromosomes (Fig. S2).
Phylogenetic diversity of alkB from alk-fragments
As previously reported by Nie, alkB genes were clustered into eight clusters (Nie et al., 2014). The cluster IV had more taxonomy diversity, which were from α-, β- and γ-proteobacteria. Consistent phylogenetic structure of alkB genes was obtained in this study, as shown in Fig. 2. Interestingly, the 120 alkB genes located in alk-gene clusters were limited to a mono-clade, which formed a subclade of cluster IV.
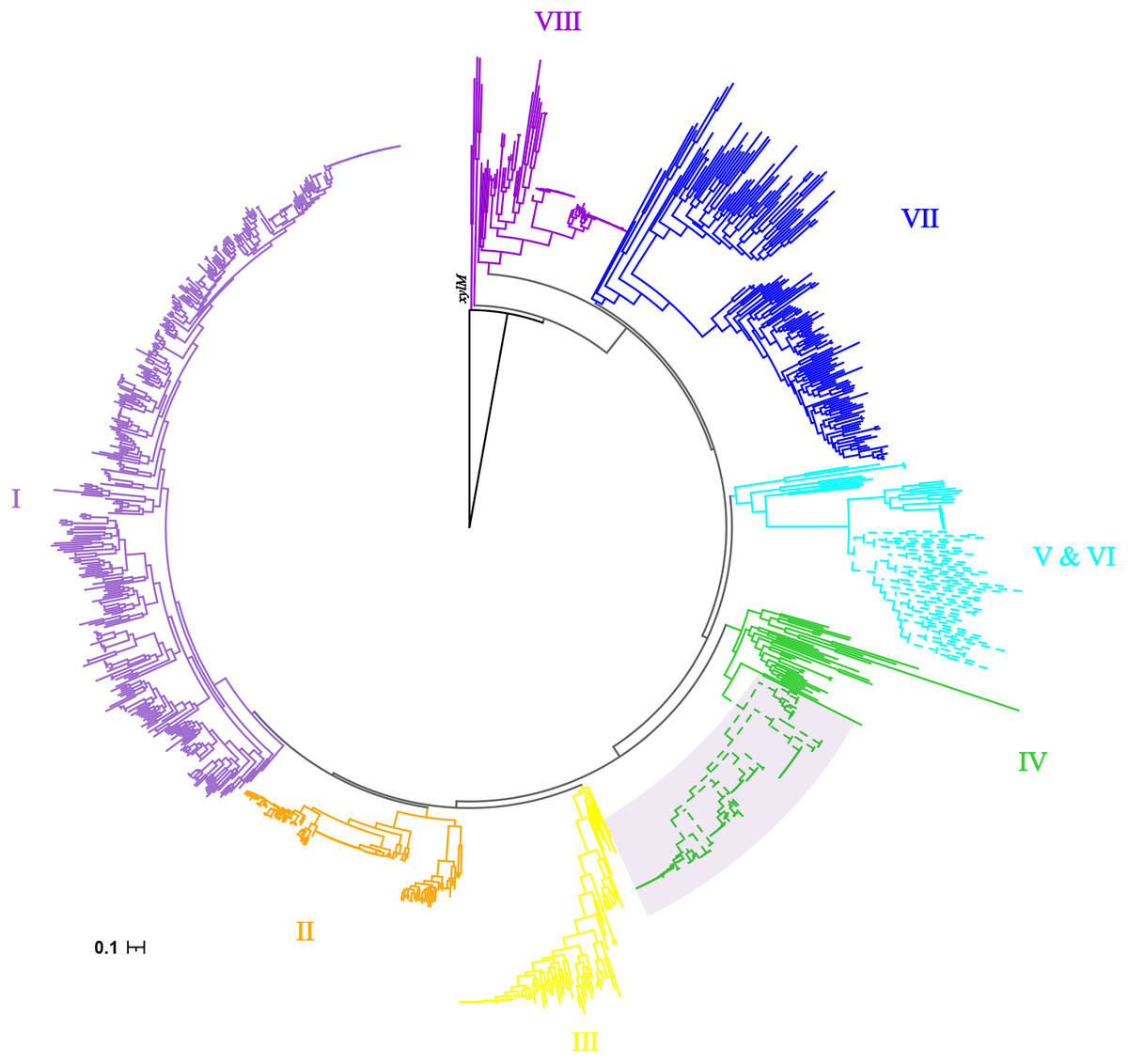
Figure 2: Phylogenetic tree of alkB genes.
The Maximum Likelihood phylogenetic tree was constructed from the nucleotide sequence of alkB genes with seqboot 1000. The xylM gene was used as outgroup. The cluster IDs were consistent with those reported by Nie, with branches colored by solid color. The alkB genes extracted from alk-fragments were marked with a fan-shaped shadow background.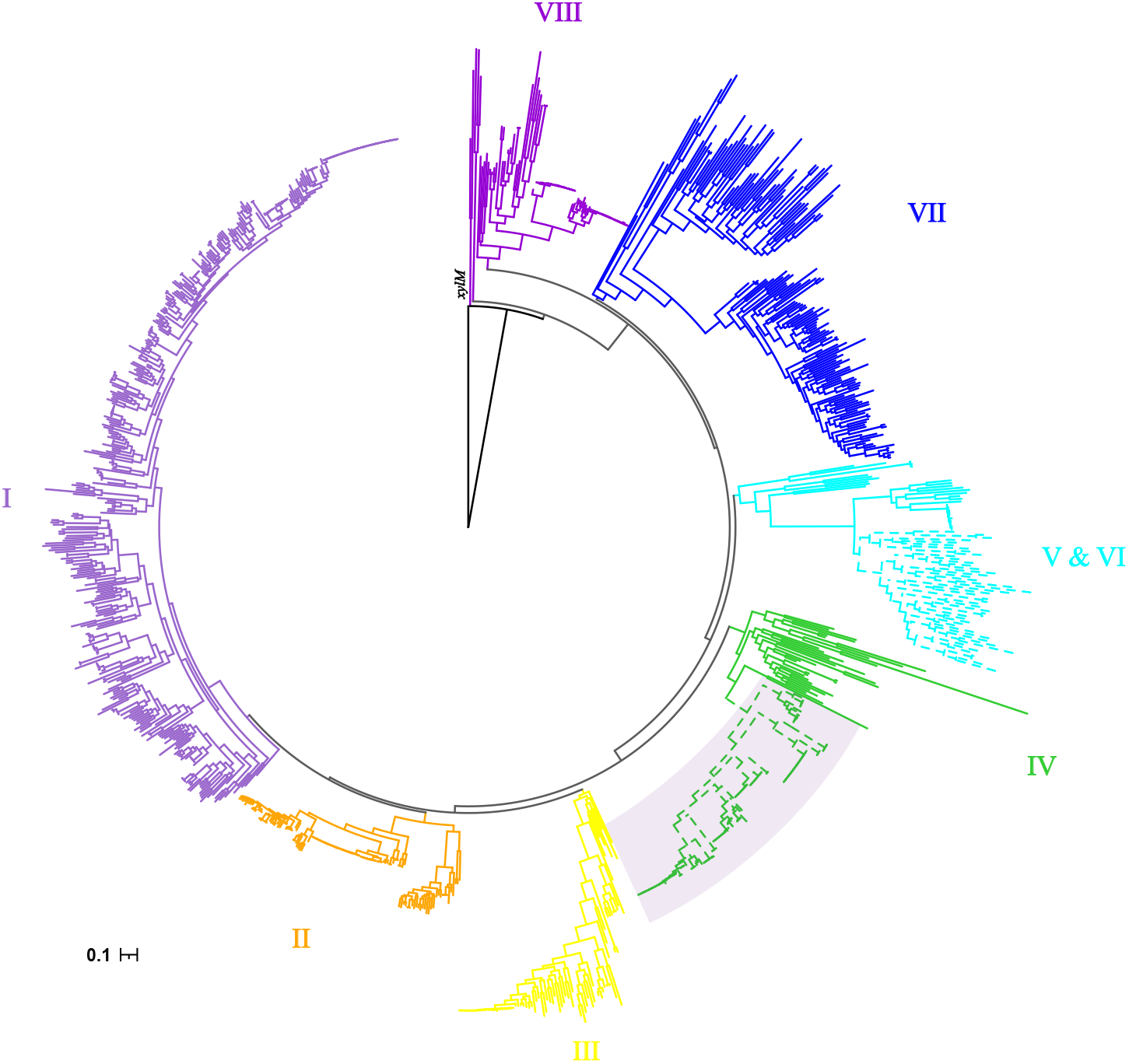{kind=link}
As mentioned above, alk-gene clusters contained a complete gene set for alkane degradation, representing for lower adaptability requirements of the recipient in the transfer process between species, suggesting its potential for wider dissemination. But the results here showed that the spread of alk-gene clusters was limited. Their phylogenetic relationship was very conservative. Together with its limitation within a certain class of species as results described above, it was suggested that there might be restrictive factors which limit its dissemination.
Organization diversity of alk-genes and IS elements distribution
The organization diversity of alk-genes was related to taxonomy lineage, as inferred from Fig. 3. Species with similar genetic relationships showed similar organization types of alk-genes. The alk-genes in Pseudomonas had the highest integrity, including alkBFGHJKL, alkN and alkST. As a distinctive feature, almost all alkF and alkN were distributed in the species of Pseudomonas. The species in genus of Marinobacter located in clade B and the species in orders of Rhodobacterales, Hyphomonadales and Maricaulales located in clade A had relatively lower alk-genes integrity, these were alkST and alkBGHJKL. Most species in order of Sphingomonadales further lost alkK, and alkT. The alkS and alkBGJH were dominated in the genus of Alcanivorax and in order of Hyphomicrobiales. This phenomenon suggested that species barrier played an essential role in alk-genes dissemination, which needs to be broken in the transmission process.
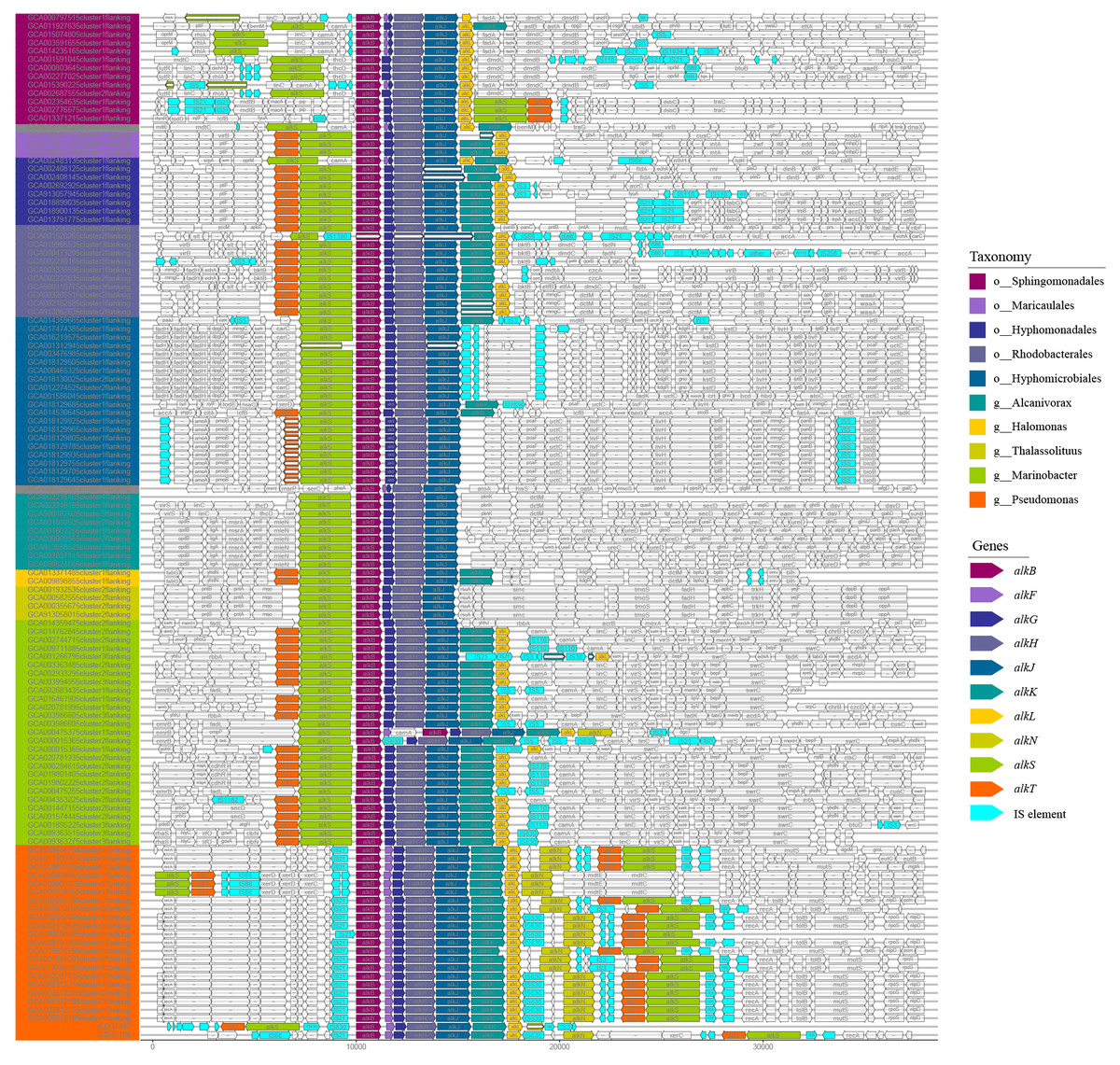
Figure 3: Comparative analysis of alk-genes organization structure.
These fragments were ordered according to the corresponding strains shown in the phylogenetic tree of Fig. 1. These gene clusters were aligned according to the starting code of alkB. The nested white bars inside represented for pseudogenes.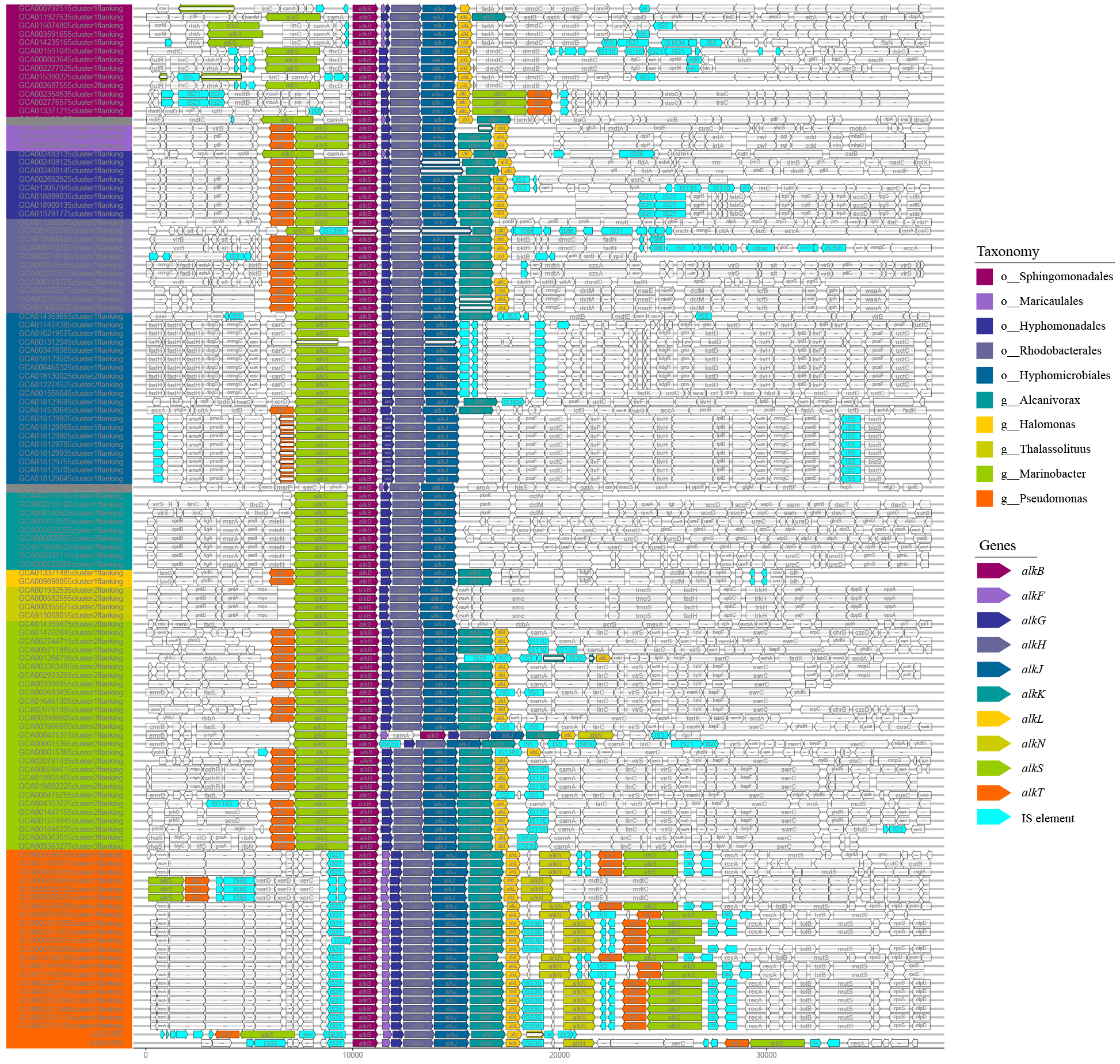{kind=link}
At the same time, many IS elements existed near alk-genes, as shown in Fig. 3. These IS elements belong to 11 families. Previous reports suggested that IS elements might play a role in mobilizing the alk-gene clusters (Van Beilen et al., 2001). We further explored the distribution of IS elements among different taxonomy. As shown in Fig. 4, IS elements are distributed unevenly in different taxonomy.
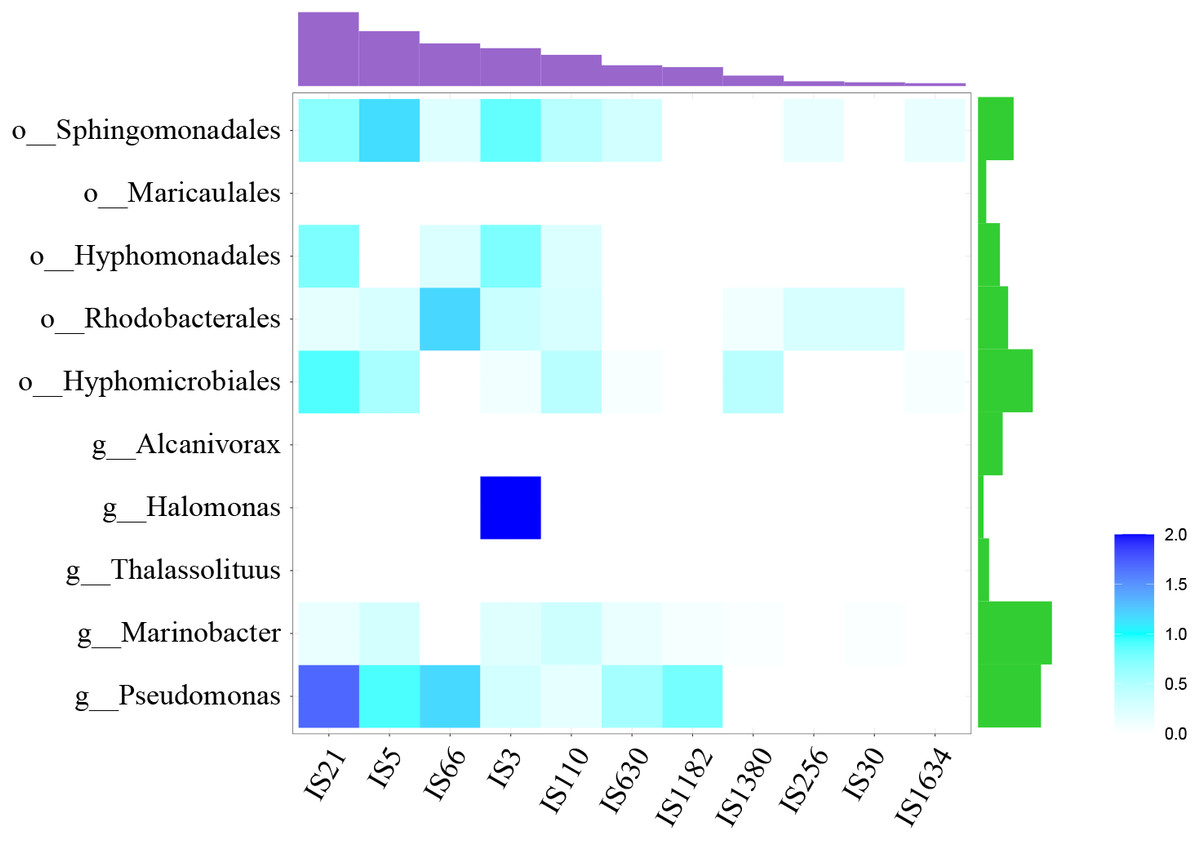
Figure 4: Distribution of IS elements.
Heatmap showed the distribution of IS element, using IS element numbers normalized by alk-fragment number in each taxonomy. Amethyst bars at the top represented for the absolute number of each IS family. Green bars on the right represented for strain number of each taxonomy.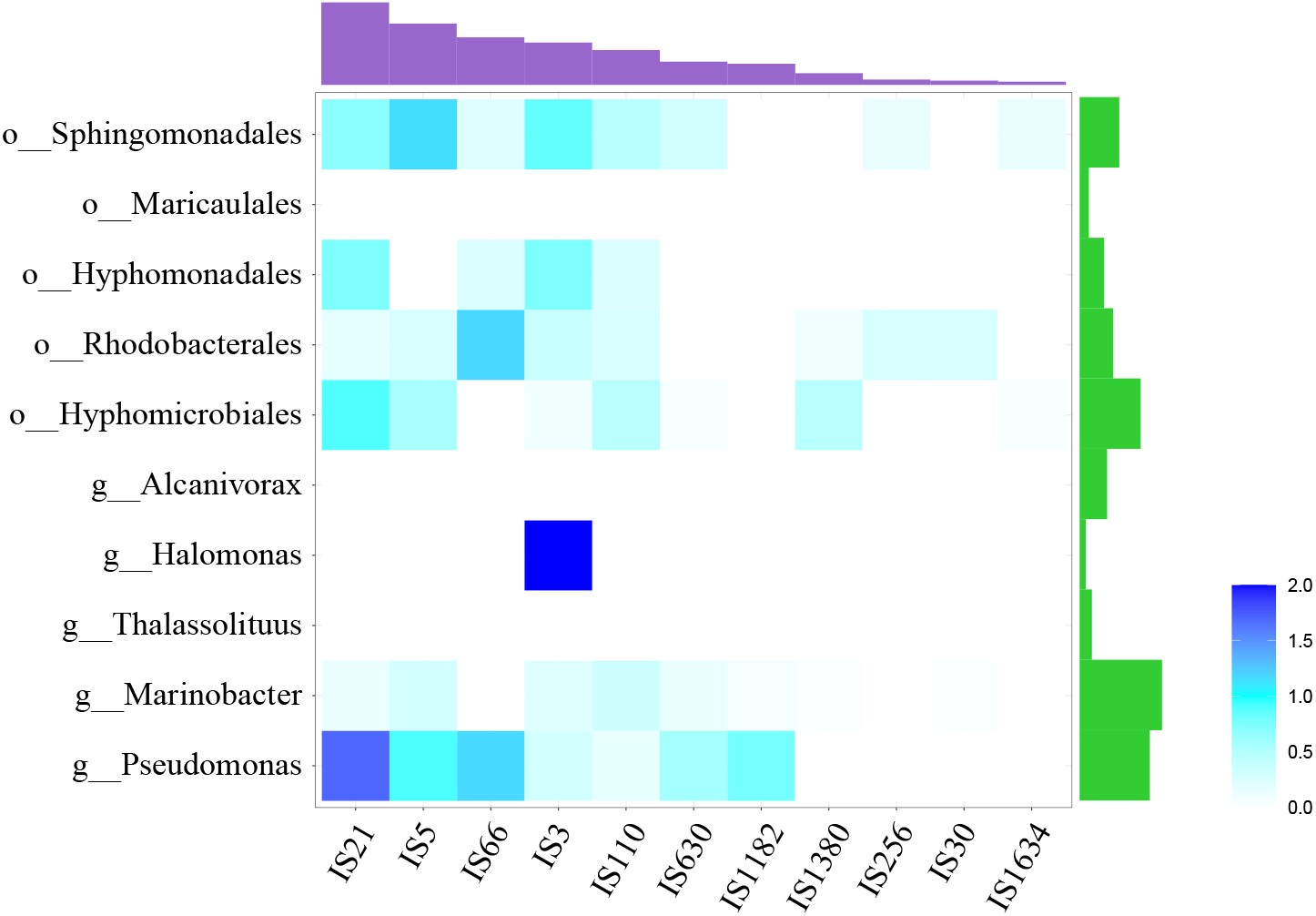{kind=link}
The alk-fragments from Pseudomonas and Marinobacter, Hyphomicrobiales, Rhodobacterales, Hyphomonadales and Sphingomonadales had amount of IS elements belonging to diverse families. The alk-fragments from Halomonas contained IS elements belonging to only one family, while almost no IS element was observed in alk-fragments from Thalassolituus Alcanivorax and Maricaulales. The results showed that in addition to IS elements, there might be other ways to transmit alk-gene clusters. In order to explore these factors for further research, larger scale data may be required.
Discussion
In the present work, we performed a comprehensive investigation of alk-gene clusters focusing on their distribution, organization, phylogenetic relationship and dissemination potential. Using phylogenetic analysis methods, we characterized a novel alkane monooxygenase (alkB) clade, which was extracted from alk-gene clusters. According to our knowledge, this was the first comprehensive report on the survey of alk-gene clusters and their phylogenetic relationship in a mono-clade so far.
According to previous reports (Schneiker et al., 2006; Van Beilen et al., 2001), it can be inferred that alk-gene clusters were not limited to plasmids nor in P. putida. But their detailed distribution had not been reported. Results of this study showed that the alk-gene clusters can be observed only in α- and γ-proteobacteria. A mono-clade phylogenetic relationship of alkB extracted from alk-genes clusters was observed as shown in Fig. 2. This phenomenon was quite different from the ubiquitous distribution of alkB genes, from at least α-, β-, γ- and δ-proteobacteria, Actinobacteria, Bacteroidetes, and Spirochaetes (Nie et al., 2014; Wang & Shao, 2013).
The genes from alk-gene clusters performed co-evolution, which can be observed in Fig. 3 as a similar organization within closely related species. The closer the genetic relationship of species, the more similar the arrangement of genes in alk-gene clusters, indicating the restriction of interspecific transmission. Although the alk-gene clusters encode all proteins necessary for converting alkanes into corresponding fatty acids, the operon structures are not necessary for the function of alkane hydroxylase (Tsai et al., 2017).
The alk-gene clusters were observed on chromosome with higher frequency in this study, suggesting its transmission mechanism might not be dominated by plasmid transfer among species. These DNA fragments were relatively large (typically >20kb), resulting in difficulty in horizontal transfer between species. A positive correlation between the abundance of mobile genetic elements and the frequency of HGT was generally observed (Springael & Top, 2004). The horizontal transfer of large DNA fragments depends on large mobile elements. There were abundant IS elements in alk-gene clusters. These IS elements were relatively concentrated and focused on several special families, which might be responsible for the limiting factor of its spread among extensive species. But we observed almost no IS element in flanking regions of alk-gene clusters from Thalassolituus Alcanivorax and Maricaulales, suggesting other ways than IS elements for transmission of alk-gene clusters. Although no evidence is presented here, we suspect that the exchange of large fragments between genomes through homologous recombination is also a factor leading to the spread of ALK fragments.
The alk-gene clusters encode proteins involved in alkane degradation, which might make it possible for the host to obtain more energy sources. We observed extensive niches adaption of alk-gene clusters carrying strains. But the niches adaptions were more likely linked with the taxonomy of the host. Although the importance of host niches adaptability was not observed, we can infer that environment had less impact on the dissemination of alk-gene clusters. It was suspected that they had potential for dissemination in much wider ecological niches.
Conclusion
In this study, a set of 120 alk-gene clusters was identified using a large-scale genome survey (>390,000 genomes). Phylogenetic, taxonomy distribution, niches distribution, organization structure, chromosome location and IS elements analyses were used to provide complete information about the alk-gene clusters. Although the alk-gene clusters were limited in α- and γ-proteobacteria, they had extensive niches distribution. At the same time, a large number of IS elements were observed nearby the alk-gene clusters. The benchmark dataset of alk-gene clusters established using genome-resolved approach in this study can provide base line for further investigation in metagenomic datasets, to explore its evolution and its position in ecology.
Supplemental Information
Niches distribution of strains carrying alk-gene clusters along with the corresponding phylogenetic tree
The phylogenetic tree was the same as in Fig. 1. The niches distribution is represented by the Check Mark on the right.
{kind=link}
{kind=link}
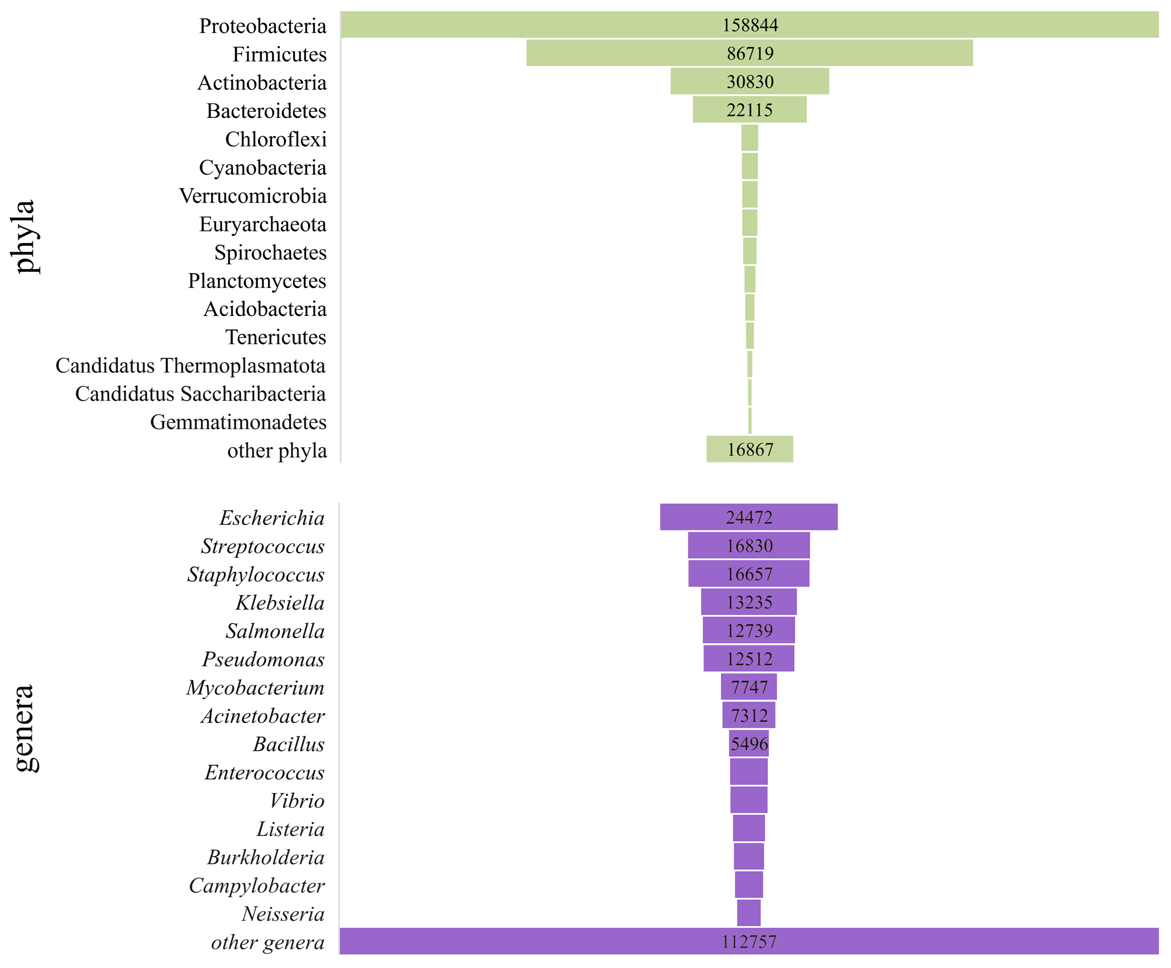
Taxonomic distribution of all available genomes included in this analysis
The number of genomes from the top 15 phyla (A) and top 15 genera (B) was represented by bar size.
{kind=link}