
Comparative and phylogenomic analyses of mitochondrial genomes in Coccinellidae (Coleoptera: Coccinelloidea)
- Published
- Accepted
- Received
- Academic Editor
- Jasmine Janes
- Subject Areas
- Entomology, Molecular Biology, Taxonomy, Zoology
- Keywords
- Phylogenetic, Coccinellidae, Mitochondrial genome
- Copyright
- © 2021 Xinghao et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2021. Comparative and phylogenomic analyses of mitochondrial genomes in Coccinellidae (Coleoptera: Coccinelloidea) PeerJ 9:e12169 https://doi.org/10.7717/peerj.12169
Abstract
The Coccinellidae are one of the most familiar beetle families, the ladybirds. Despite the great ecological and economic significance, the phylogenetic relationships of Coccinellidae remain poorly understood. One of the reasons is that the sequenced mitogenomes available for this family are very limited. We sequenced complete or nearly complete mitogenomes from seven species of the tribe Coccinellini with next-generation sequencing. All species have the same gene content and gene order as the putatively ancestral insect mitogenome. A large intergenic spacer region (> 890 bp) was found located between trnI and trnQ. The potential for using secondary structures of the large and small ribosomal subunits for phylogenetic reconstruction was predicted. The phylogenetic relationships were explored through comparative analyses across more than 30 coccinellid species. We performed phylogenetic analyses with both concatenation methods (Maximum Likelihood and Bayesian Inference) and multispecies coalescent method (ASTRAL). Phylogenetic results strongly supported the monophyly of Coccinellidae. Within Coccinellidae, the Epilachnini and the Coccinellini including Halyziini were monophyletic, while the Scymnini and Coccidulini were non-monophyletic.
Introduction
The family Coccinellidae comprises a group of insects often called ladybirds or ladybugs, a familiar family of beetles, for example, the seven-spot ladybird beetle Coccinella septempunctata Linnaeus. Currently, the family is subdivided into about 360 genera, with approximately 6,000 species worldwide (Vandenberg, 2002). Many of coccinellid species are beneficial due to their predaceous nature, and are well known for roles in biological control. The predatory group is often referred to as aphidophagous, despite sometimes also feeding on other hemipteran species, for example, scales and whiteflies (Hodek & Honek, 2009; Obrycki et al., 2009). Additionally, other ladybirds are phytophagous, including the genus Bulaea Mulsant and the whole subfamily Epilachninae (Giorgi et al., 2009). Some phytophagous ladybird beetles are serious pests and cause damage to important crops, such as Epilachna varivestris Mulsant and Henosepilachna vigintioctopunctata (Fabricius) (Szawaryn et al., 2015).
Despite the monophyly of the whole Coccinellidae is well supported (Giorgi et al., 2009; Magro et al., 2010; Robertson et al., 2015; Seago et al., 2011; Slipinski, 2007), the phylogenetic relationships within the family are uncertain. Redtenbacher (1844) classified the Coccinellidae into two groups: the phytophagous group and the aphidophagous group. The two groups were further classified into six or seven subfamilies. Sasaji (1968; 1971a; 1971b), based on larval and adult morphological characters, proposed a six subfamilies’ classification system (Sticholotidinae, Coccidulinae, Scymninae, Chilocorinae, Coccinellinae, and Epilachninae). This scheme received widespread acceptance (Giorgi et al., 2009). Kovář (1996) added the Ortaliinae into the scheme of six subfamilies to construct the seven subfamilies scheme. The phytophagous group corresponding to the subfamily Epilachninae is often considered to monophyletic (Giorgi et al., 2009; Seago et al., 2011; Song et al., 2020). The aphidophagous group spans the remaining subfamilies (Giorgi et al., 2009). Within the aphidophagous group, the Coccinellinae is monophyletic, while the other subfamilies are non-monophyletic (Giorgi et al., 2009; Magro et al., 2010).
Previous authors have attempted to reconstruct the phylogenetic relationships among the subfamilies based on morphological (Yu, 1994) and/or molecular (Giorgi et al., 2009; Magro et al., 2010; Robertson et al., 2015; Seago et al., 2011) data. However, most of the subfamilies proposed by Sasaji (1968; 1971a; 1971b) were not supported by recent molecular studies (Giorgi et al., 2009; Magro et al., 2010; Seago et al., 2011; Che et al., 2021). Seago et al. (2011) conducted the simultaneous analysis of morphological and multi-locus molecular data to investigate the phylogenetic relationships among major clades of Coccinellidae. Their results found no statistical support for the classification of several subfamilies, except for Coccinellinae and Microweisinae. Che et al. (2021) recognized three subfamilies in Coccinellidae, namely Microweiseinae, Monocoryninae stat. nov., and Coccinellinae, based on multiple nuclear protein-coding gene sequences. In this study, our taxon sampling was focused on the tribe Coccinellini (24 exemplars out of 40 analyzed coccinellid species). The increased taxon sampling allowed us to investigate the relationships below the subfamily level.
Rapid radiations pose one of the most difficult challenges for the phylogenetic estimation of subfamily relationships (Robertson et al., 2015). For other beetle groups (e.g., Galerucinae and Chrysomelidae) with similar divergence time, some authors have successfully used mitogenomes to resolve the subfamily (Nie et al., 2018), family (Nie et al., 2020) and above the family level relationships (Timmermans et al., 2016). Song et al. (2020) and Yuan et al. (2020) provided the first mitogenome phylogenetic analyses of Coccinellidae. The results confirmed the monophyly of Coccinellinae and Epilachninae. But both studies only considered DNA sequence variation in their phylogenetic reconstructions (Song et al., 2020; Yuan et al., 2020).
The previous research work often used the super matrix to construct the phylogenetic tree of Coccinellidae (e.g., Robertson et al., 2015; Song et al., 2020). Mitochondrial DNA as a phylogenetic marker may suffer from phylogenomic biases as associated with incomplete lineage sorting and gene tree heterogeneity. Multispecies coalescent methods have been developed to tackle these problems. A recent study has applied multispecies coalescent analysis to phylogenetic reconstruction within Lepidoptera based on mitogenome sequence data (Kim, Kim & Cameron, 2020). Their results demonstrated that multispecies coalescent analysis can be a reliable inference method for mitogenomic data in resolving insect phylogenetic relationships. The Coccinellidae have experienced rapid radiation (Robertson et al., 2015; Seago et al., 2011; Tomaszewska et al., 2021). Here, we attempted to use the super tree generated by ASTRAL analysis of mitogenome data to investigate the phylogenetic relationships in Coccinellidae.
Besides applying the alignments of mitochondrial DNA sequences to phylogenetic reconstruction, the mitogenome organization (Shao, Campbell & Barker, 2001), and the secondary structures of mitochondrial large subunit (rrnL) and small subunit (rrnS) may provide potential information for phylogenetic studies (Buckley et al., 2000; Page, 2000; Page, Cruickshank & Johnson, 2002; Yoshizawa & Johnson, 2003). The use of both rrnL and rrnS gene sequences in phylogenetic reconstructions is undercut by alignment difficulties. Both rRNA genes contain a number of possibly uninformative conserved regions interspersed with highly compositionally heterogeneous variable regions that are difficult to align. Particularly, it is difficult to align these highly variable regions accurately using the current algorithmic alignment methods (Song et al., 2020). The rRNA molecule forms distinct secondary structures that play an important role in the functioning of ribosomes (Noller, 1984). Secondary structure information can thus be used to identify homologous positions, with higher phylogenetic informativeness.
In this study, we sequenced seven complete or nearly complete mitogenomes of the subfamily Coccinellinae, with three main aims: (1) to compare mitogenome organization and gene content across coccinellid lineages; (2) to identify conserved sequence motifs and the associated secondary structure elements in rrnL and rrnS genes to provide the potential phylogenetic information; (3) to add more mitogenome data to uncover the evolutionary relationships of ladybirds.
Materials and Methods
Taxon sampling
A total of 46 mitogenomes were analyzed in this study: 40 species of ladybird beetles and other six species of Cucujifamia outgroups (Table 1). Of which, seven mitogenomes of the tribe Coccinellini were newly sequenced in this study. We extracted genomic DNA from seven specimens, Coccinella lama Kapur, Hippodamia variegata (Goeze), Coccinella transversoguttata Faldermann, Adalia bipunctata (Linnaeus), and Oenopia dracoguttata Jing were collected from Lhasa, Tibet, China in June 28, 2019; Harmonia axyridis (Pallas) and Harmonia eucharis (Mulsant) were collected from Linzhi, Tibet, China in August 3, 2019. Thoracic muscle was used for DNA extraction, using the TIANamp Genomic DNA Kit (TIANGEN BIOTECH CO., LTD), according to the manufacturer’s protocol. Extracted DNA was stored at −20 °C.
| Item | Family | Subfamily | Tribe | Species | Accession number | Reference |
|---|---|---|---|---|---|---|
| Outgroup | Bothrideridae | Bothriderinae | Bothriderini | Dastarcus helophoroides | NC_024271 | Zhang et al., 2015 |
| Corylophidae | Corylophinae | Rypobiini | Gloeosoma sp. | JX412843 | Unpublished | |
| Discolomatidae | Discolomatinae | – | Discolomatinae sp. | JX412748 | Unpublished | |
| Endomychidae | Endomychinae | – | Endomychus coccineus | JX313667 | Unpublished | |
| Erotylidae | Xenoscelinae | Loberonotha olivascens | JX412784 | Unpublished | ||
| Latridiidae | Latridiinae | – | Enicmus brevicornis | JX313681 | Unpublished | |
| Ingroup | Coccinellidae | Scymninae | Scymnini | Nephaspis sp. DPP-2018 | MG253275 | Unpublished |
| Coccinellidae | Scymninae | Scymnini | Scymninae sp. 2 ACP-2013 | MH940166 | Crampton-Platt et al., 2015 | |
| Coccinellidae | Scymninae | Scymnini | Nephus includens | MN164642 | Magro et al., 2020 | |
| Coccinellidae | Scymninae | Scymnini | Nephus reunioni | MN164643 | Magro et al., 2020 | |
| Coccinellidae | Scymninae | Scymnini | Nephus sp. 1 EL-2020 | MN164644 | Magro et al., 2020 | |
| Coccinellidae | Scymninae | Scymnini | Nephus voeltzkowi | MN164645 | Magro et al., 2020 | |
| Coccinellidae | Scymninae | – | Coccinellidae sp. 1 EF-2015 | KT780638 | Unpublished | |
| Coccinellidae | Coccidulinae | Coccidulini | Cryptolaemus montrouzieri | KT878320 | Unpublished | |
| Coccinellidae | Coccidulinae | Coccidulini | Coccidula rufa | JX412767 | Unpublished | |
| Coccinellidae | Ortaliinae | Noviini | Rodolia quadrimaculata | MN053055 | Song et al., 2019 | |
| Coccinellidae | Epilachninae | Epilachnini | Afissula sp. XL-2019 | MN053057 | Song et al., 2019 | |
| Coccinellidae | Epilachninae | Epilachnini | Henosepilachna pusillanima | KJ131489 | Behere et al., 2014 | |
| Coccinellidae | Epilachninae | Epilachnini | Henosepilachna vigintioctopunctata | MG584727 | Unpublished | |
| Coccinellidae | Epilachninae | Epilachnini | Subcoccinella vigintiquattuorpunctata | KT780695 | Unpublished | |
| Coccinellidae | Epilachninae | Epilachnini | Epilachna admirabilis | MN053053 | Song et al., 2019 | |
| Coccinellidae | Chilocorinae | Chilocorini | Chilocorus bipustulatus | MN053054 | Song et al., 2019 | |
| Coccinellidae | Coccinellinae | Halyziini | Halyzia sedecimguttata | KT780652 | Unpublished | |
| Coccinellidae | Coccinellinae | Halyziini | Halyziini sp. HA | MG584728 | Zhang et al., 2017 | |
| Coccinellidae | Coccinellinae | Coccinellini | Adalia bipunctata | MW029465 | This study | |
| Coccinellidae | Coccinellinae | Coccinellini | Aiolocaria hexaspilota | MK583344 | Seo et al., 2019 | |
| Coccinellidae | Coccinellinae | Coccinellini | Anatis ocellata | KX035143 | Unpublished | |
| Coccinellidae | Coccinellinae | Coccinellini | Anisosticta novemdecimpunctata | KT876880 | Linard et al., 2016 | |
| Coccinellidae | Coccinellinae | Coccinellini | Calvia championorum | KX132085 | Unpublished | |
| Coccinellidae | Coccinellinae | Coccinellini | Calvia decemguttata | KX087252 | Unpublished | |
| Coccinellidae | Coccinellinae | Coccinellini | Cheilomenes sexmaculata | KM244706 | Tang et al., 2014 | |
| Coccinellidae | Coccinellinae | Coccinellini | Coccinella septempunctata | JQ321839 | Kim et al., 2011 | |
| Coccinellidae | Coccinellinae | Coccinellini | Coccinella lama | MW029464 | This study | |
| Coccinellidae | Coccinellinae | Coccinellini | Coccinella transversoguttata | MW029466 | This study | |
| Coccinellidae | Coccinellinae | Coccinellini | Coelophora saucia | MN053056 | Song et al., 2019 | |
| Coccinellidae | Coccinellinae | Coccinellini | Cycloneda sanguinea | KU877170 | Unpublished | |
| Coccinellidae | Coccinellinae | Coccinellini | Eriopis connexa | MG253268 | Unpublished | |
| Coccinellidae | Coccinellinae | Coccinellini | Harmonia quadripunctata | KX087296 | Unpublished | |
| Coccinellidae | Coccinellinae | Coccinellini | Harmonia axyridis | MW029463 | This study | |
| Coccinellidae | Coccinellinae | Coccinellini | Harmonia eucharis | MW029462 | This study | |
| Coccinellidae | Coccinellinae | Coccinellini | Hippodamia convergens | KX755332 | Unpublished | |
| Coccinellidae | Coccinellinae | Coccinellini | Hippodamia undecimnotata | KX087298 | Unpublished | |
| Coccinellidae | Coccinellinae | Coccinellini | Hippodamia variegata | MW029468 | This study | |
| Coccinellidae | Coccinellinae | Coccinellini | Oenopia dracoguttata | MW029467 | This study | |
| Coccinellidae | Coccinellinae | Coccinellini | Propylea japonica | KM244660 | Tang et al., 2014 | |
| Coccinellidae | Coccinellinae | Coccinellini | Propylea sp. HSL-2016 | KX132084 | Unpublished |
Notes:
Bold denotes the newly sequenced species.
Library construction and next-generation sequencing
About 500 ng genomic DNA of the individual species were used for library preparation using the Illumina TruSeq TM DNA Sample Prep Kit (Illumina, San Diego, CA, USA), according to manufacturer’s instructions. DNA were sheared into 350-bp fragments in a Covaris M220 instrument (Covaris Inc.). Libraries were sequenced on the Illumina HiSeq 2500 platform with 150-bp paired-end reads, at Shanghai OE Biotech. Co., Ltd, China.
Mitogenome assembly and annotation
Statistics for next-generation sequencing are presented in Table 2. Adapters and low-quality reads were trimmed from raw data by using NGS Toolkit (Patel & Jain, 2012). High-quality reads (Q20 ≥ 97.71%, and Q30 ≥ 93.43%) were used in subsequent genome assemblies in Geneious R11, with the following parameters: iterate up to 100 times (slow), maximum gaps per read 5%, maximum gap size 20 bp, minimum overlap 50 bp, and minimum overlap identity 95%. The mtDNA sequence of Cycloneda sanguinea (Linnaeus) (accession number: KU877170) was used as reference for assembly.
Preliminary mitogenome annotations were conducted in MITOS web (Bernt et al., 2013), under the default settings and the invertebrate genetic code for mitochondria. The gene boundaries of protein-coding and ribosomal RNA were refined by alignment against published Coccinellini mitogenome sequences. Transfer RNAs (tRNA) were annotated in MITOS web (Bernt et al., 2013), with secondary structures inferred. Secondary structures for rrnL and rrnS genes were predicted by reference to the darkling beetle Gonocephalum outreyi Chatanay (Song et al., 2018), and figured manually in Adobe Illustrator CS6. Annotated mitogenome sequences have been submitted to GenBank with the accession numbers of MW029462–MW029468.
| Species name | Total number of sequenced raw paired reads | Raw Q30 | Total number of clean paired reads | Clean Q30 |
|---|---|---|---|---|
| Coccinella lama | 18.52 Mbp | 84.02% | 15.57 Mbp | 90.41% |
| Hippodamia variegata | 20.07 Mbp | 85.28% | 17.14 Mbp | 91.12% |
| Coccinella transversoguttata | 21.61 Mbp | 85.15% | 18.45 Mbp | 91.00% |
| Adalia bipunctata | 19.47 Mbp | 84.58% | 16.53 Mbp | 90.72% |
| Oenopia dracoguttata | 22.48 Mbp | 87.47% | 19.24 Mbp | 91.98% |
| Harmonia axyridis | 17.24 Mbp | 83.73% | 14.47 Mbp | 90.28% |
| Harmonia eucharis | 21.28 Mbp | 86.22% | 18.57 Mbp | 91.52% |
Phylogenetic analyses
Before sequence alignment, we added the mitochondrial genes of the seven newly sequenced coccinellid species into the dataset constructed from the mitognome sequences downloaded from GenBank. For the phylogenetic analyses, we concatenated nucleotide alignments from all 37 mitochondrial genes. Protein-coding genes were individually aligned using MAFFT (Katoh & Standley, 2013) in the TranslatorX (Abascal, Zardoya & Telford, 2010) server. Each tRNA, and rRNA was aligned in the MAFFT (E-INS-I algorithm) alignment server and adjusted following reference to secondary structural models. Ambiguously aligned sites in each alignment were removed with Gblocks (Talavera & Castresana, 2007), using the less stringent selection option. Alignments were concatenated using FASconCAT_v1.0 (Kueck & Meusemann, 2010). The sequence alignment used in the phylogenetic analyses is provided in the Supplementary File 1.
Phylogenetic relationships were inferred using Maximum Likelihood (ML) and Bayesian Inference (BI). PartitionFinder 2 (Lanfear et al., 2017) was used to select best-fitting partition schemes and corresponding substitution models for the concatenated alignment (Table S1). Data blocks were defined by codon position and by gene for protein-coding genes. All 22 tRNA genes were included in a single partition, while each of rRNA genes was defined as a separate partition. PartitionFinder analysis was conducted with the CIPRES web portal (Miller, Pfeiffer & Schwartz, 2010), using the corrected Akaike information criterion (AICc). ML analysis was performed using IQ-TREE (Lam-Tung et al., 2015; Nguyen et al., 2015; Trifinopoulos et al., 2016) at the CIPRES web portal (Miller, Pfeiffer & Schwartz, 2010). Nodal supports were estimated with 10,000 ultrafast bootstrap replicates (Hoang et al., 2018). The SH-aLRT branch test (Guindon et al., 2010) was conducted with 1,000 replicates. The command -spp was employed to consider the FreeRate heterogeneity model.
BI analysis was performed with PhyloBayes MPI (Lartillot et al., 2013) implemented on the CIPRES web portal. The site-heterogeneous CAT-GTR model (Lartillot & Philippe, 2004) was employed, with constant sites removed. Two independent Markov chain Monte Carlo (MCMC) chains starting from a random tree were run for 20,000 generations. The initial 20% cycles in each MCMC chain were discarded as burn-in. A consensus tree was computed from the remaining trees. Convergence of the two chains was indicated by a “maxdiff” value of 0.1.
We used ASTRAL v 5.7.1 (Mirarab et al., 2014; Zhang et al., 2018) to estimate a species tree. ML tree searches were conducted for individual gene alignments (13 protein-coding genes, two rRNA genes as single genes plus the 22 tRNA genes combined as a single alignment), with IQ-TREE. Gene trees were then used as input for ASTRAL, using bootstrap replicates from the IQ-TREE estimated gene trees for branch support values estimation.
Results
Genome sequencing and mitogenome assembling
For the newly sequenced species, the total number of raw reads varied between 17.24 Mbp (H. axyridis) and 22.48 Mbp (O. dracoguttata) (Table 2). The proportion of raw reads with phred scores equal to or greater than Q30 ranged between 83.73–87.47%. After filtering low-quality data, the total number of clean reads ranged from 14.47 Mbp to 19.24 Mbp. The proportion of cleaned reads with phred scores equal to or greater than Q30 ranged between 90.28–91.98%.
Of the clean reads, 0.75% (O. dracoguttata) to 2.19% (C. magnifica) corresponded to mitochondrial reads. Length of assembled mitogenomes varied between 17,963 bp (C. transversoguttata) and 21,391 bp (H. eucharis) (Table 3). The average values for sequencing depth of mitogenomes ranged from 501-fold (H. eucharis) to 1,251-fold (C. magnifica). Sequencing depth was not closely correlated with the mitogenome length. The current sequencing depth was sufficient to cover the entire mitogenome.
General characteristics of mitogenome
All seven newly-sequenced mitogenomes contained the typical 37 mitochondrial genes and a complete control region (Fig. S1). All seven coccinelid mitogenomes had the same arrangement of protein-coding genes, tRNA genes, and rRNA genes as the putatively ancestral insect mitogenome (Cameron, 2014a; Cameron, 2014b).
Nucleotide composition was strongly biased toward A and T. The average A+T content for the whole mitogenomes was 77.9%, made up of 76.5% in the protein-coding genes, 78.9% in the tRNA genes, 78.1% in the rRNA genes and 82.5% in the control region. Twelve of the 13 protein-coding genes started with the typical codon ATN (ATT, ATG, and ATA). However, for cox1, the putative initiation codon was TCG(Ser) in all newly sequenced mitogenomes. Canonical stop codons (TAG and TAA) were present for 11/12 protein-coding genes depending on species. Incomplete stop codons (T or TA) were inferred for cox2 (H. variegata and H. eucharis), cox3 (C. lama and C. transversoguttata), nad1 (O. dracoguttata and H. eucharis), and nad2 (H. axyridis).
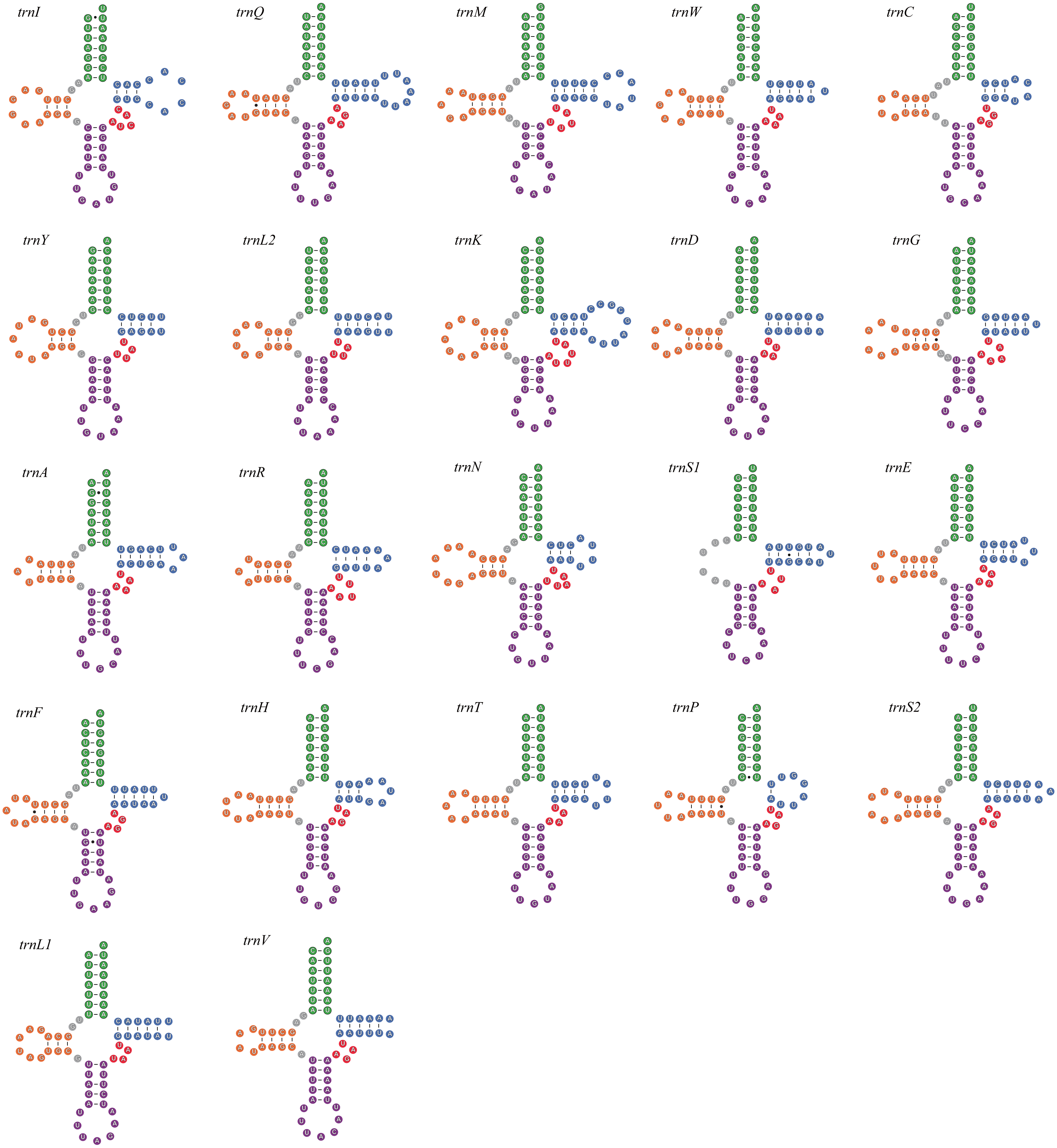
All seven newly-sequenced mitogenomes had a full set of 22 mitochondrial tRNA genes, which ranged from 50 bp (trnH, A. bipunctata) to 71 bp (trnK, A. bipunctata, H. axyridis and H. eucharis) in size. All of mitochondrial tRNA genes had secondary structures commonly seen in other insects, with the exception for trnS1, which lacked a complete dihydrouridine (DHU) arm (e.g., C. lama in Fig. 1).
| Specis | Mitogenome (bp) | Mapped reads | Minimum sequencing depth | Average sequencing depth | Mitochondrial reads (%) |
|---|---|---|---|---|---|
| Coccinella lama | 19,337 | 170,237 | 169 | 1,251 | 2.19 |
| Hippodamia variegata | 18,347 | 127,665 | 136 | 993 | 1.49 |
| Coccinella transversoguttata | 17,963 | 79,131 | 121 | 642 | 0.86 |
| Adalia bipunctata | 18,750 | 117,955 | 167 | 941 | 1.43 |
| Oenopia dracoguttata | 19,359 | 72,567 | 55 | 550 | 0.75 |
| Harmonia axyridis | 18,737 | 93,440 | 549 | 729 | 1.29 |
| Harmonia eucharis | 21,391 | 71,886 | 68 | 501 | 0.77 |
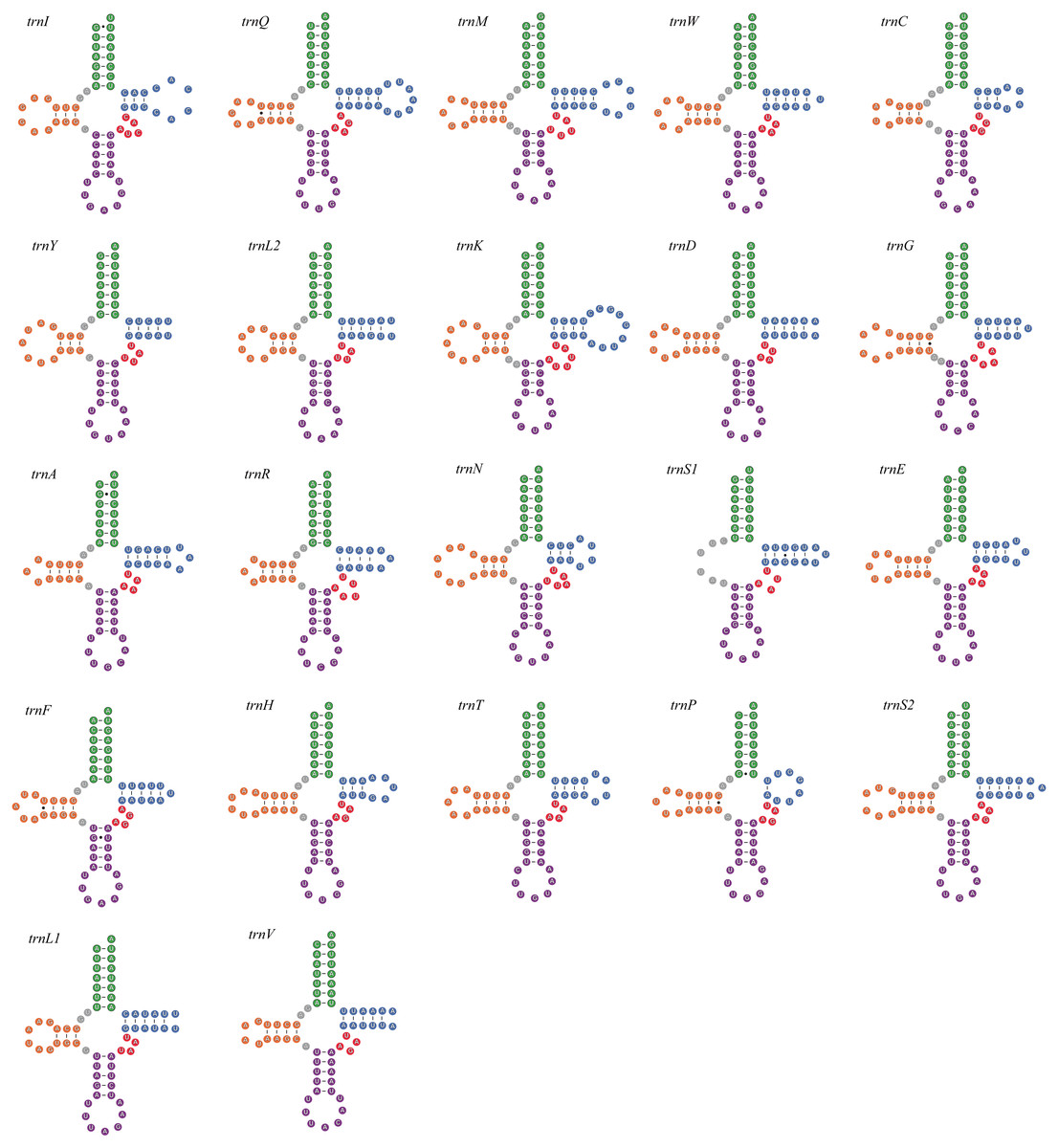
Figure 1: The secondary structures of tRNA genes inferred for Coccinella lama.
{kind=link}
The rrnL and rrnS genes were 1,200–1,299 bp and 746–762 bp, respectively. The secondary structures inferred for the rrnL and rrnS genes were similar to the secondary structure models proposed for other beetles (e.g., G. outreyi, Song et al., 2018) (Figs. 2–3 and Fig. S2). In the rrnL genes, there were differences in the number of helices. H. variegate, A. bipunctata, O. dracoguttata and H. axyridis had 44 helices. C. lama and C. transversoguttata had 45 helices, while H. eucharis had 43 helices. Base mismatches and sequence length variation resulted in observed differences. Each rrnL gene contained five domains (I-II, IV-VI), and lacked domain III. rrnS genes had three domains (I, II, III) composed of 26 helices.
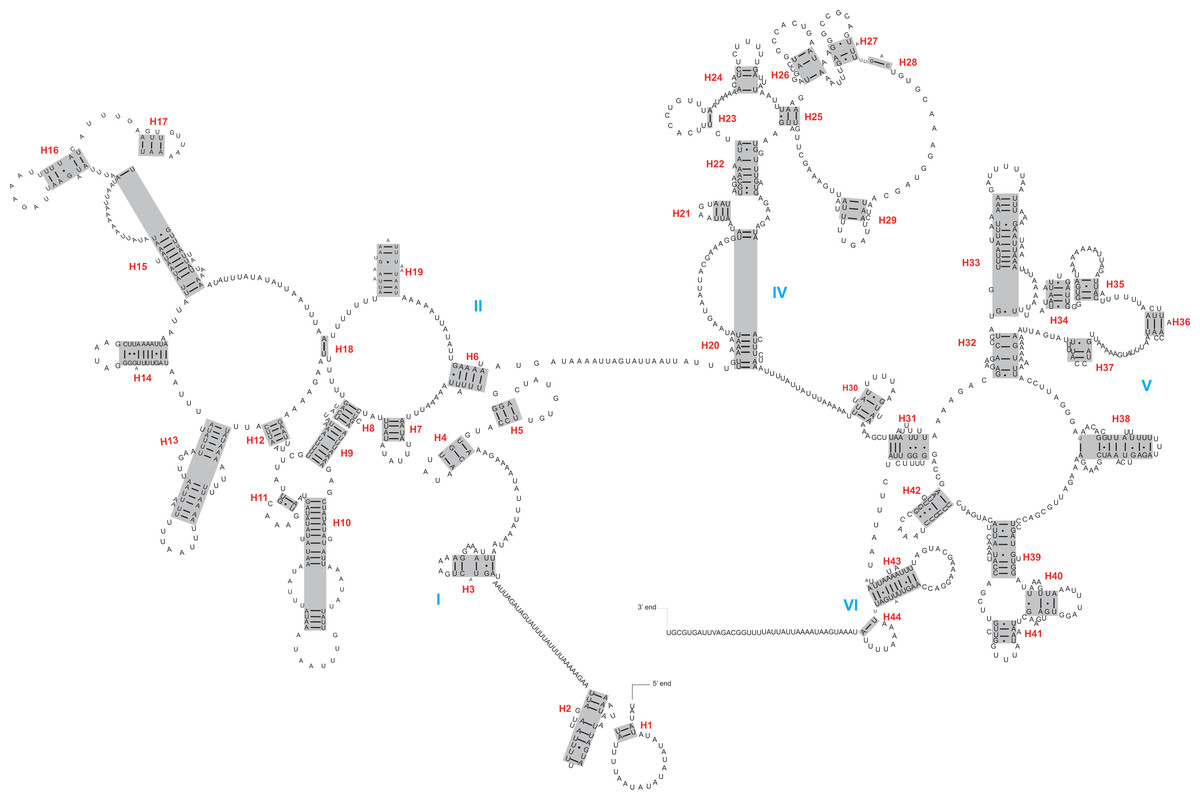
Figure 2: The secondary structure of rrnL inferred for Harmonia axyridis.
Red numbers denote the helices and blue Roman numerals denote the domains.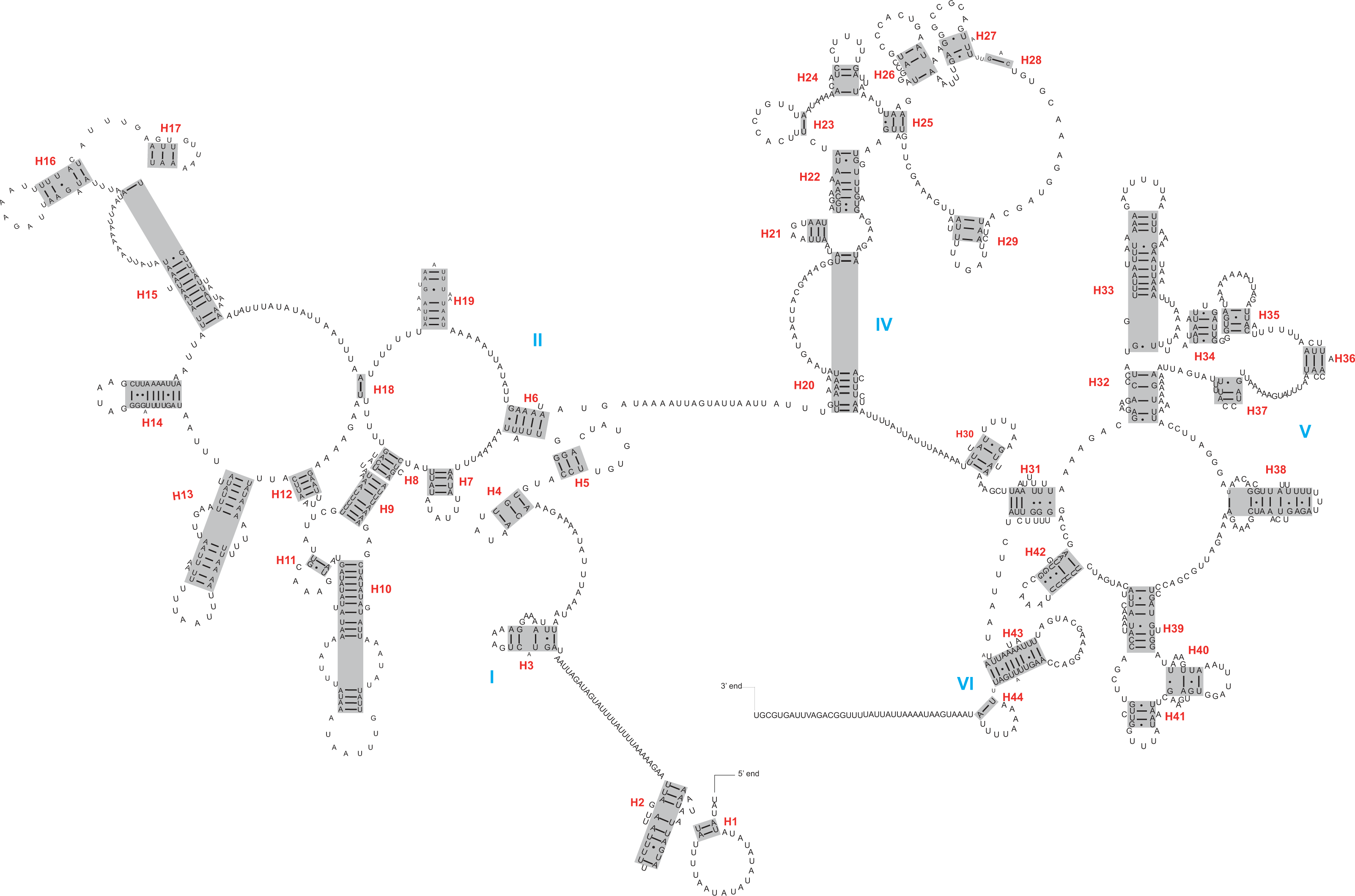{kind=link}
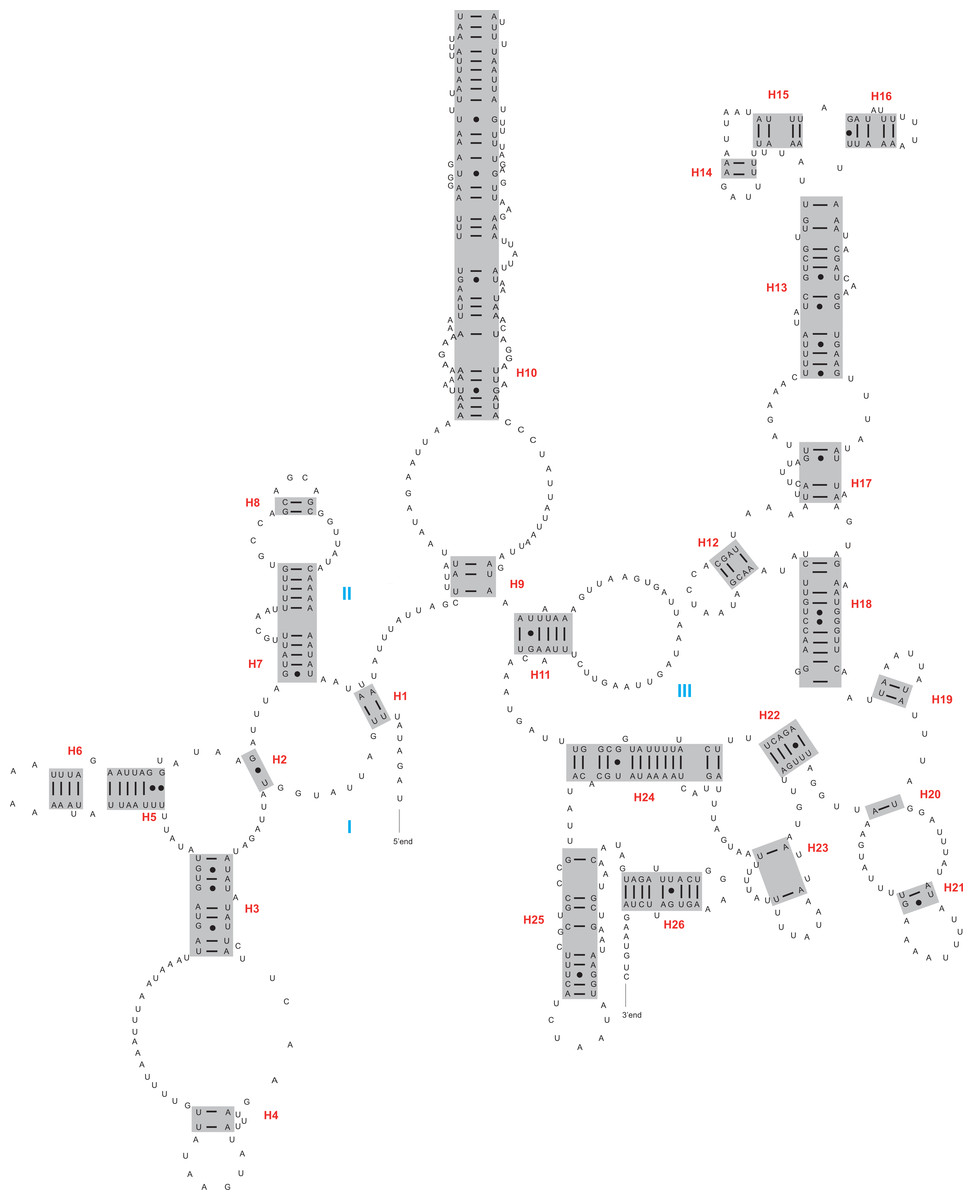
Figure 3: The secondary structure of rrnS inferred for Harmonia axyridis.
Red numbers denote the helices and blue Roman numerals denote the domains.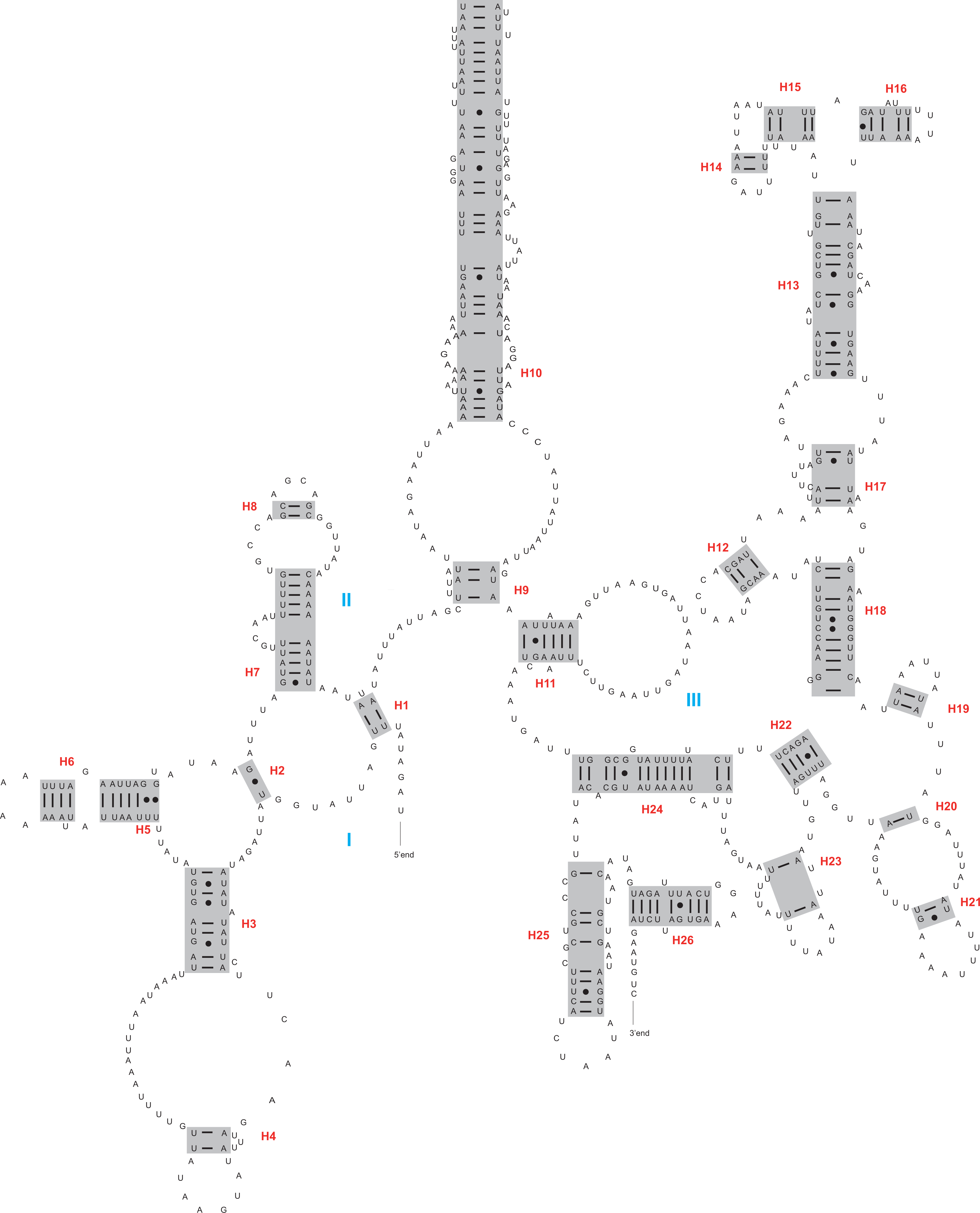{kind=link}
The complete control region between rrnS and trnI was identified within assembled mitogenomes, demonstrating circulation of the molecule. Besides the control region, a large non-coding region (or intergenic spacer region) was present between trnI and trnQ. The sequence lengths of this region varied between 895 bp (C. transversoguttata) and 2,745 bp (C. lama).
Mining phylogenetic information from mitochondrial ribosomal RNA secondary structures
Through comparison of 34 mitochondrial rrnL and 31 rrnS secondary structures of coccinellid species, we found that species in the same genus shared conserved motifs. For rrnL domain V, the loop located in the tip of helix 35 had largely identical nucleotide composition within a single genus (Fig. 4 middle and Fig. S3), but it was distinguisable between genera. Within Coccinellinae, two species from the tribe Halyziini shared the secondary structure character of rrnS helix 7 (Fig. 3). Additionally, they did not have the rrnS helix 8, that was distinct from all other Coccinellidae. Although the structures of rrnS helix 8 of Coccinellini are basically similar to Epilachnini/Scymnini, there is a discrepancy of two nucleotides (AGUU vs AGCA) between them (Fig. 4). In addition to the secondary structures illustrated in Fig. 4, we found that rrnS helix 4 also contained potential phylogenetic information (Fig. S4). The Coccinellini shared an identical nucleotide composition of helix 4, while it was distinguishable from other lineages.
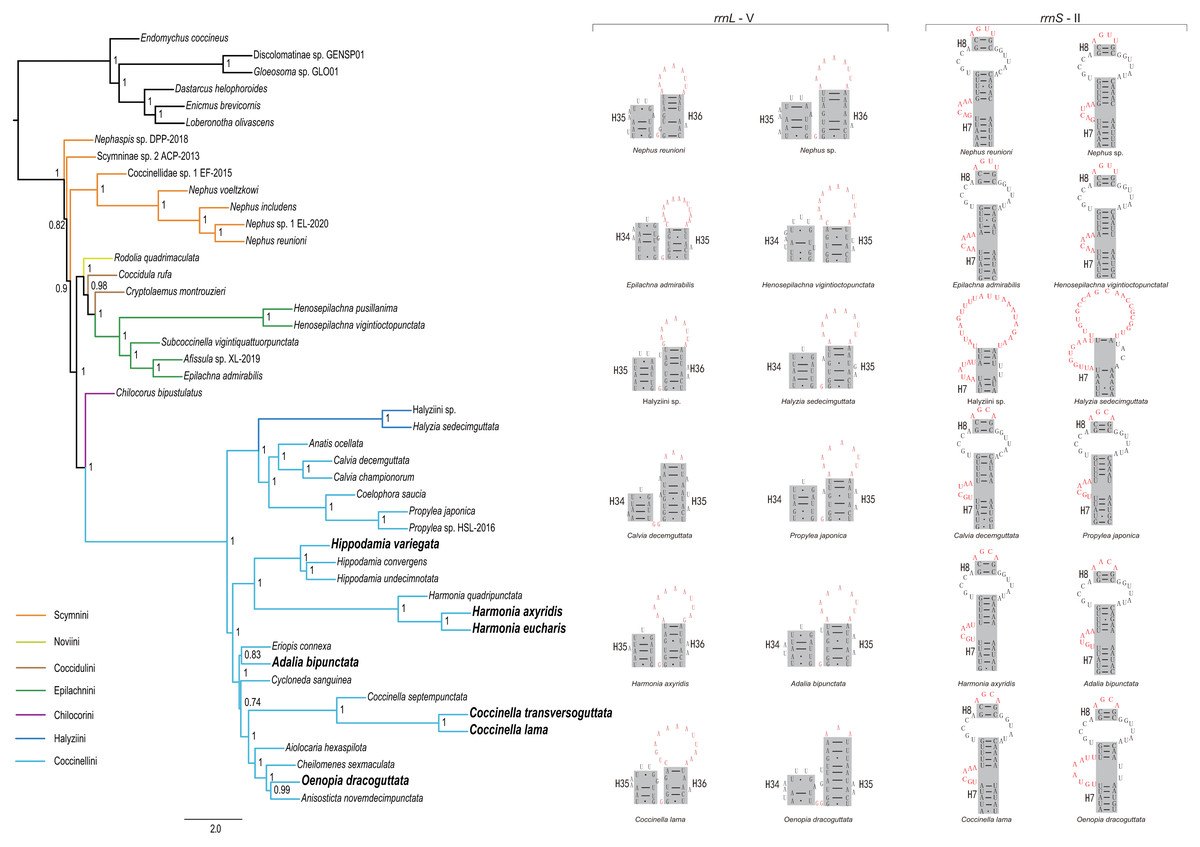
Figure 4: Phylogenetic relationships within Coccinellidae inferred using ASTRAL analysis (left), and comparisons of the secondary structures of rrnL (middle) and rrnS (right).
In the phylogenetic tree, numbers around nodes indicate the local posterior probabilities. Bold denotes the newly sequenced species.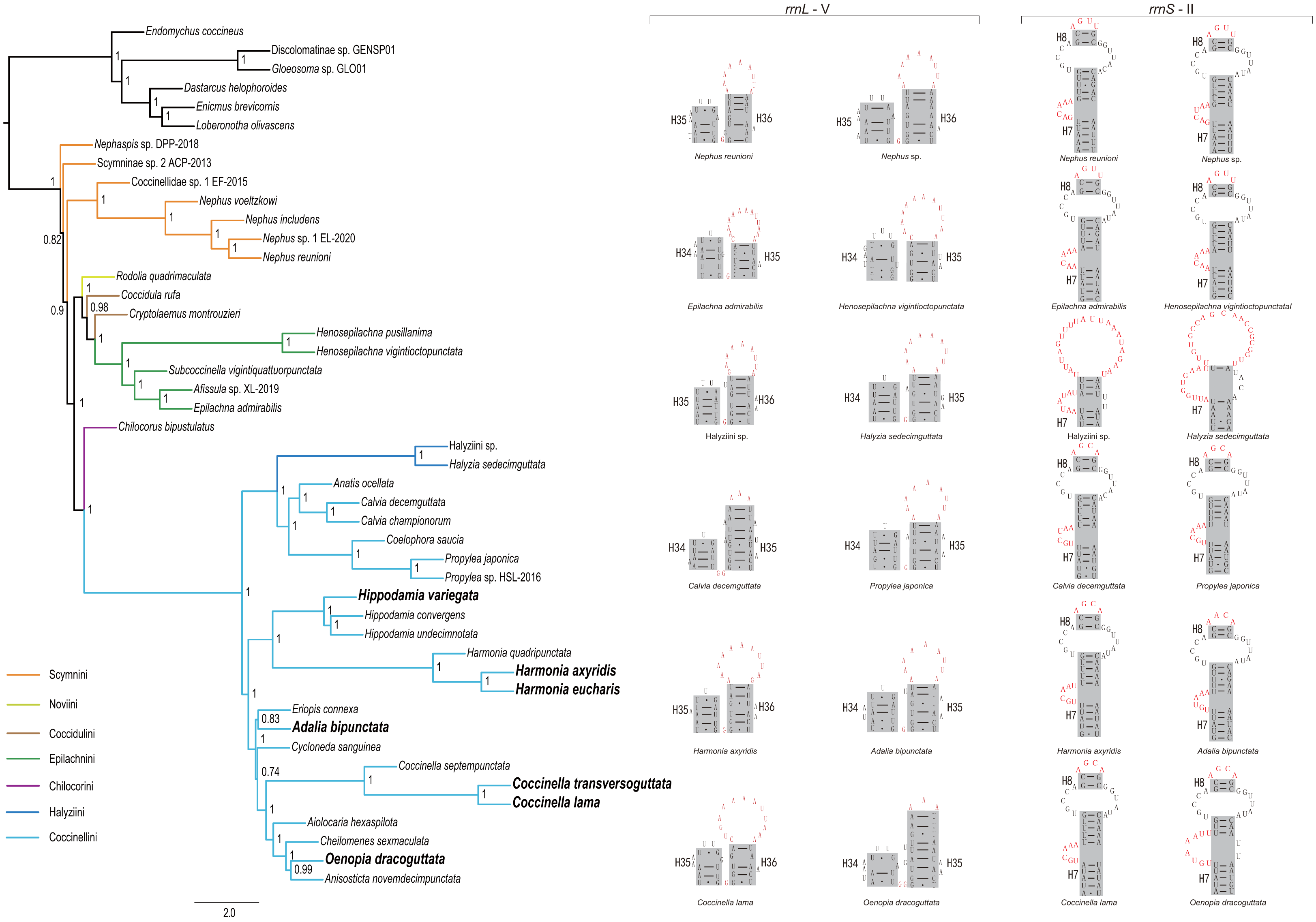{kind=link}
Phylogenetic inference
Monophyly of the Coccinellidae was strongly supported in both ML and BI analyses (BS = 100, PP = 1, Fig. 5). Within Coccinellidae, the Chilocorini was recovered as the sister group to a clade comprising Halyziini and Coccinellini. This large clade was sister to Epilachnini. However, the sister-group relationship received no statistical support (BS = 30, PP = 0.71). The Epilachnini was consistently supported as monophyletic (BS = 100, PP = 1). In the ML analysis, the Cryptolaemus was sister to Epilachnini (BS = 38). A clade comprising a part of Scymnini (Nephaspis) and the species representing Noviini (Rodolia quadrimaculata) emerged as sister to the rest of the family. But these relationships received no statistical supports (BS = 47). Similarly, in the BI analysis, the basal relationships within Coccinellidae were ambiguous. In particular, relationships among Scymnini, Noviini and Coccidulini were unresolved. The Scymnini and Coccidulini were non-monophyletic.
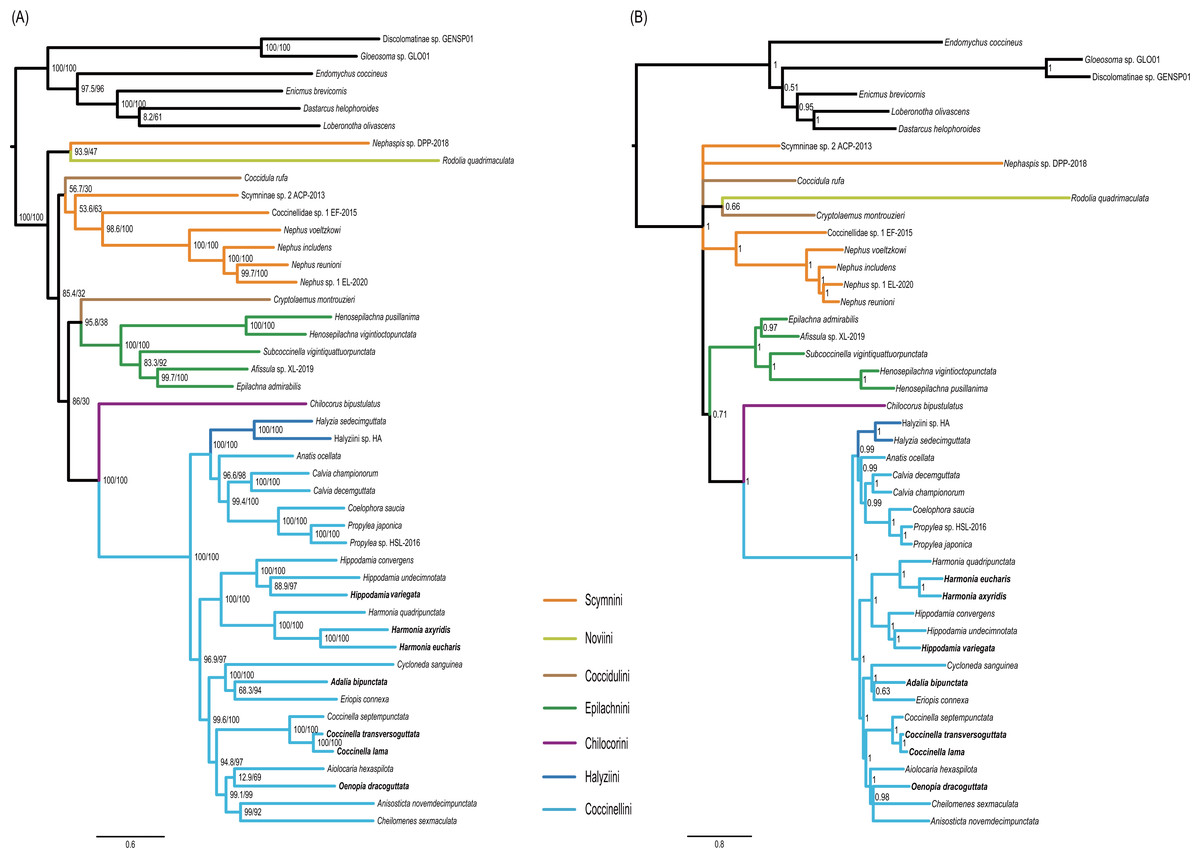
Figure 5: Phylogenetic trees inferred from the super matrix constructed by 37 mitochondrial genes.
(A) Maximum Likelihood analysis was performed using IQ-tree. Numbers around nodes indicate support values from SH-aLRT test (left) and ultrafast bootstrap analysis. (B) Bayesian Inference was performed using PhyloBayes. Numbers around nodes indicate posterior probabilities.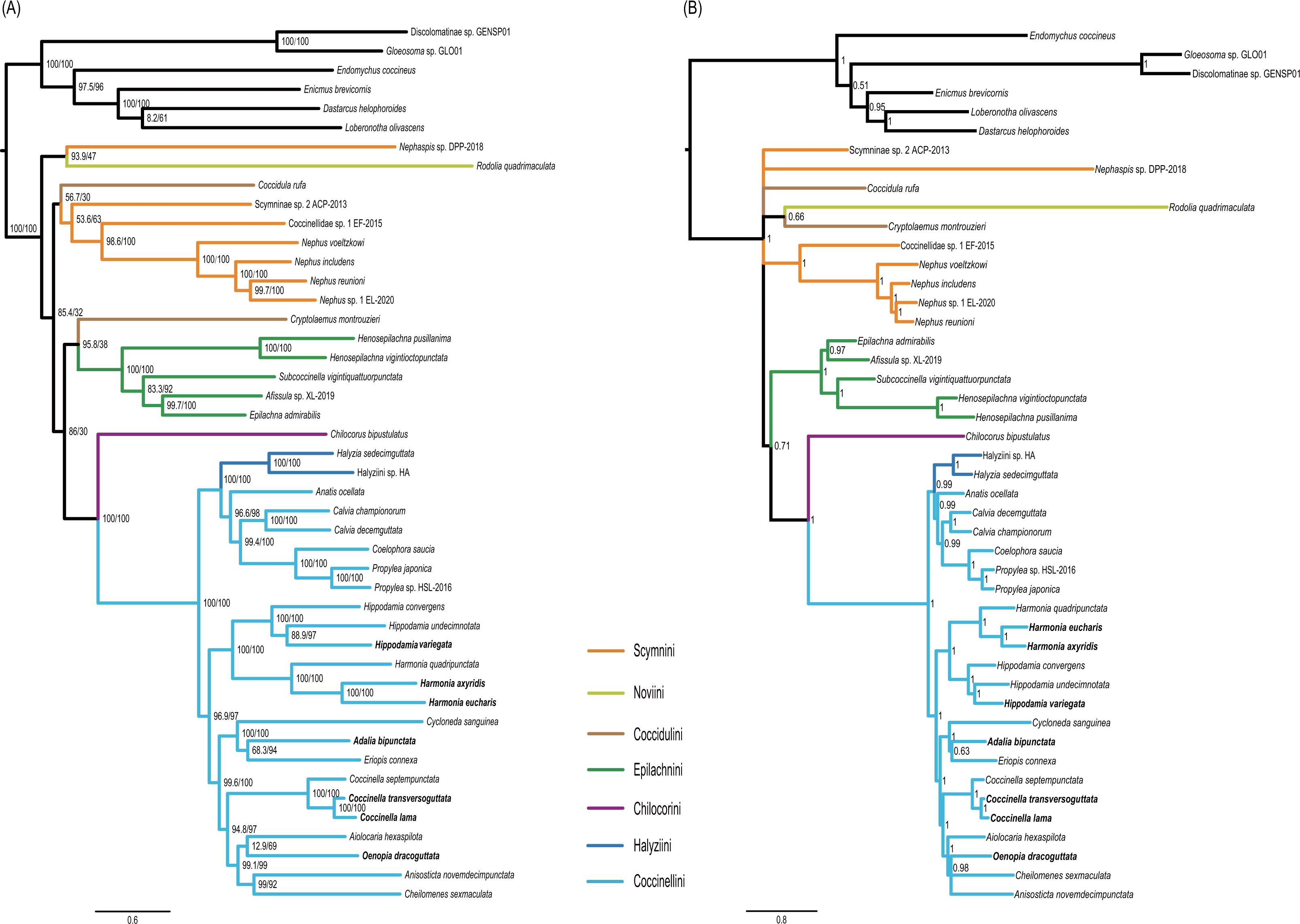{kind=link}
The Coccinellini including Halyziini formed a large clade. In both ML and BI analyses, the Halyziini was sister to a clade comprising Anatis, Calvia, Coelophora and Propylea (BS = 100, PP = 0.99). All genera with more than two species included in this study (i.e., Halyzia, Calvia, Propylea, Harmonia, Hippodamia and Coccinella) were supported as monophyletic (BS = 100, PP =1).
ASTRAL analysis showed better performance in resolving deeper nodes in the Coccinellidae (Fig. 4) with respect to nodal support. Although the Scymnini was still non-monophyletic, the majority of this tribe formed a paraphyletic grade relative to the remaining coccinellid lineages. The two exemplars of Coccidulini formed a paraphyletic grade to Epilachnini. In comparison, members of the Scymnini and Coccidulini were scattered on the trees from both ML and BI analyses. Other difference between multispecies coalescent analysis and the concatenated analyses under ML and BI criteria was the nodal support. The tree from multispecies coalescent analysis typically had higher nodal support values for the internal nodes.
Discussion
Large intergenic spacer region in mitogenome
Large intergenic spacer regions have been frequently found in insect mitogenomes (Du et al., 2017; Song et al., 2020; Wang et al., 2019; Wei et al., 2010). In the present study, all newly sequenced coccinellid species had a large intergenic spacer located between trnI and trnQ. The position of the intergenic spacers accorded with our prior study (Song et al., 2020). Longer sequence lengths in this region contribute to the larger size of the whole mitogenomes of the species in this study. A 54 bp spacer region between trnQ and nad2 was found in the mitogenome of Manduca sexta (Linnaeus) (Lepidoptera: Sphingidae) in a previous study (Cameron & Whiting, 2008). The position of this spacer region is adjacent to the large intergenic spacer region found in coccinellid species sequenced in this study. The phylogenetic utility of this arrangement can be evaluated by sequencing and comparing more mitogenomes from related insect groups in future studies.
Phylogenetic inference
In this study, traditional concatenation methods of analyzing mitogenomes failed to resolve the relationships between the Scymnini and Coccidulini in the BI tree. This result is consistent with other work that found no support for the delimitation of most subfamilies in Coccinellidae (Giorgi et al., 2009; Magro et al., 2010; Seago et al., 2011). Due to mitogenome data availability, the taxon sampling of Scymnini and Coccidulini is still limited. In future studies, we need to sequence more species from both groups to confirm the result. Here we expanded sampling within the Coccinellini, which continued to robustly support the monophyly of the subfamily Coccinellinae and of Coccinellinae genera. This demonstrates that mitogenomic data can be effective in resolving relationships below the subfamily level within the Coccinellidae.
Recent studies recovered members of the former tribe Halyziini within Coccinellini (Nattier et al., 2021; Tomaszewska et al., 2021). Some authors have proposed that Coccinellini, including Halyziini, constitutes a monophyletic group (Che et al., 2021; Nattier et al., 2021; Seago et al., 2011; Tomaszewska et al., 2021). The present study recovered a similar branching pattern based on the mitogenome data, with Halyziini and Coccinellini being placed in a clade. This result is congruent with the results of phylogenetic reconstruction based on combining analyses of nuclear and mitochondrial gene fragments (Nattier et al., 2021; Tomaszewska et al., 2021).
Giorgi et al. (2009) recovered the monophyly of Coccinellinae, based on the 18S and 28S rRNA sequences. Magro et al. (2010), based on multiple gene sequence data (nuclear 18S, 28S rRNA, and mitochondrial rrnS, rrnL and cox1), also supported the Coccinellinae as a monophyletic group. Our result is consistent with the previous studies. The current mitogenome data supported Epilachnini as monophyletic. This was congruent with Che et al. (2021), but contrasted with Magro et al. (2010). The Epilachnini was paraphyletic in the analyses of Magro et al. (2010). The Scymnini was non-monophyletic across our analyses. This arrangement was also retrieved in the previous studies based on nuclear gene (Che et al., 2021; Giorgi et al., 2009; Robertson, Whiting & McHugh, 2008) and combined data of nuclear and mitochondrial gene sequences (Magro et al., 2010). A sister group relationship between the Chilocorus (Chilocorini) and the clade Coccinellini + Halyziini was supported by the present mitogenome analyses. This arrangement is congruent with Escalona et al. (2017), Magro et al. (2010) and Seago et al. (2011), but contrasted with the morphological analyses of Sasaji (1968; 1971a; 1971b).
Multispecies coalescent analysis with ASTRAL showed improved performance in recovering relationships within Coccinellidae, especially for the basal relationships among the lineages of Scymnini and Coccidulini. In addition, the sister group relationship between Chilocorini and Coccinellini was robustly supported. Within Coccinellini, the sister-group relationships between Coelophora and Propylea, between Hippodamia and Harmonia were supported, consistent with the prior studies by Escalona et al. (2017), Nattier et al. (2021) and Tomaszewska et al. (2021).
Implications of mitochondrial rRNA secondary structures for phylogenetic relationships
Exploration of meaningful characters for phylogenetic analysis can be essential to better understand insect evolution. As illustrated for this the secondary structures inferred for this study, two classes of regions are observed in rRNA molecules: the double-stranded stems and single-stranded loops. It has been long debated whether stem characters or loop characters contain useful information for phylogenetic inference (Dixon & Hillis, 1993; Smith, 1989; Wheeler & Honeycutt, 1988). Our analyses showed that loop characters of mitochondrial rRNA genes are phylogenetically informative for the Coccinellidae.
Conclusions
In the present study, we utilized NGS data to reconstruct complete or nearly complete mitogenome of the Coccinellidae. The mitogenomes are very large, ranging from 17,963 bp (C. transversoguttata) to 21,391 bp (H. eucharis), due to a large intergenic spacer (>890 bp) located between trnI and trnQ. The mitochondrial rRNA secondary structures were compared to provide a better phylogenetic alignment. Besides the control region between rrnS and trnI often found in insect mitogenomes, the presence of a large intergenic region in other position is interesting and further studies are needed to investigate the underlying mechanisms creating and preserving such mitochondrial arrangements. The newly sequenced mitogenomes also contribute to a better understanding of the phylogenetic relationships and evolutionary history of ladybirds. The monophyly of Coccinellidae and of several genera within it are recovered, the bootstrap support value has reached 100. Despite this, increased taxon sampling from other species of the coccinellid group other than Coccinellini is needed to comprehensively evaluate the phylogenetic relationships in Coccinellidae.
Supplemental Information
Organizational maps of the seven new mitogenomes sequenced in this study
The secondary structure of rrnL inferred for Coccinella magnifica
Red numbers denote the helices and blue roman numbers denote the domains.
The secondary structure of rrnS inferred for Coccinella magnifica
Red numbers denote the helices and blue roman numbers denote the domains.
The secondary structure of rrnL inferred for Hippodamia variegate
Red numbers denote the helices and blue roman numbers denote the domains.
The secondary structure of rrnS inferred for Hippodamia variegate
Red numbers denote the helices and blue roman numbers denote the domains.
The secondary structure of rrnL inferred for Coccinella transversoguttata
Red numbers denote the helices and blue roman numbers denote the domains.
The secondary structure of rrnS inferred for Coccinella transversoguttata
Red numbers denote the helices and blue roman numbers denote the domains.
The secondary structure of rrnL inferred for Adalia bipunctata
Red numbers denote the helices and blue roman numbers denote the domains.
The secondary structure of rrnS inferred for Adalia bipunctata
Red numbers denote the helices and blue roman numbers denote the domains.
The secondary structure of rrnL inferred for Oenopia dracoguttata
Red numbers denote the helices and blue roman numbers denote the domains.
The secondary structure of rrnS inferred for Oenopia dracoguttata
Red numbers denote the helices and blue roman numbers denote the domains.
The secondary structure of rrnL inferred for Harmonia eucharis
Red numbers denote the helices and blue roman numbers denote the domains.
The secondary structure of rrnS inferred for Harmonia eucharis
Red numbers denote the helices and blue roman numbers denote the domains.