LuxS quorum sensing system, its protein modeling and active-binding sites and phylogenetic analysis from Aeromonas hydrophila
- Published
- Accepted
- Subject Areas
- Bioinformatics, Microbiology
- Keywords
- Aeromonas hydrophila, LuxS, Quorum sensing, 3D-structure, In silico
- Copyright
- © 2017 Ali et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Preprints) and either DOI or URL of the article must be cited.
- Cite this article
- 2017. LuxS quorum sensing system, its protein modeling and active-binding sites and phylogenetic analysis from Aeromonas hydrophila . PeerJ Preprints 5:e3109v1 https://doi.org/10.7287/peerj.preprints.3109v1
Abstract
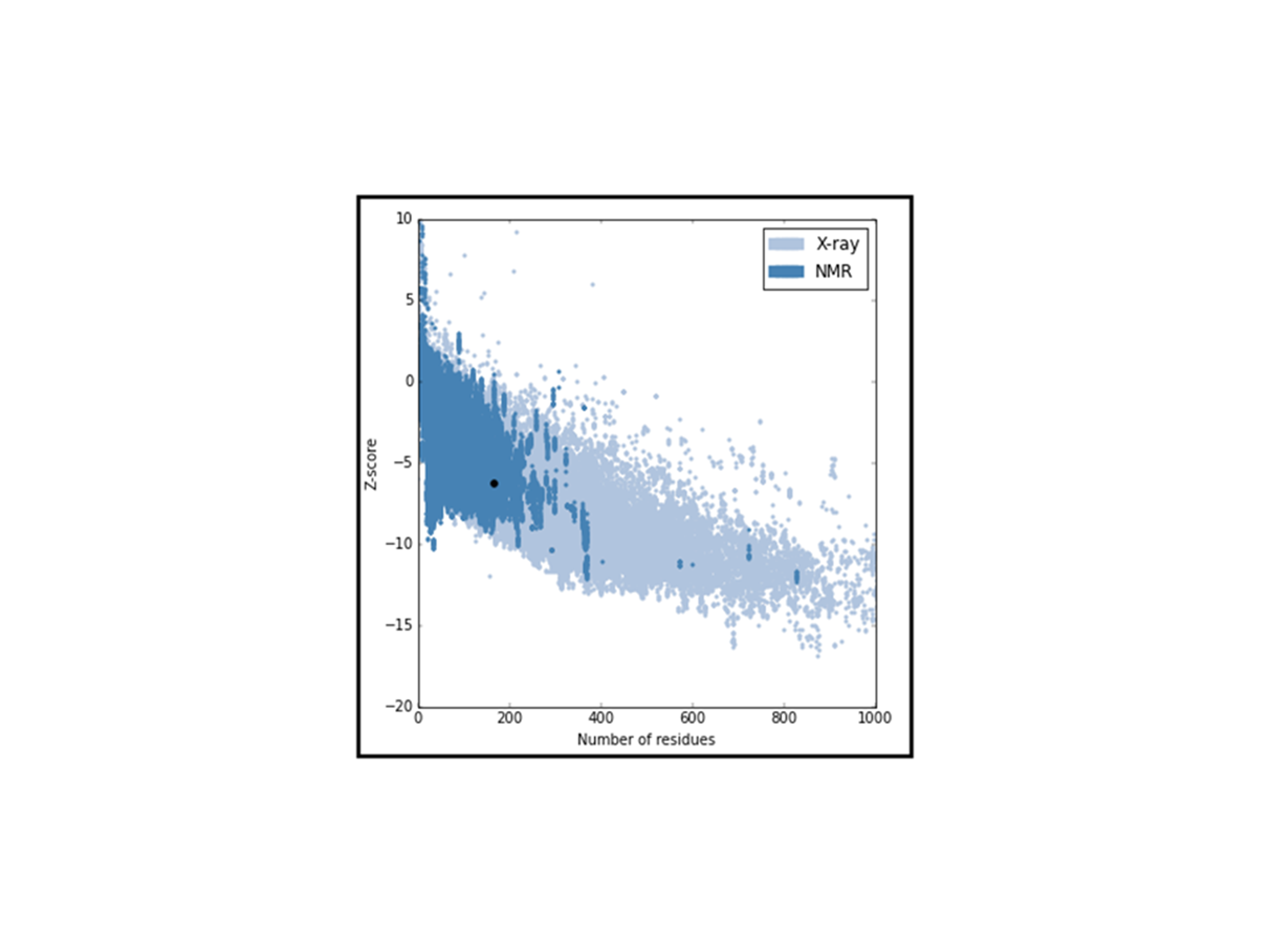
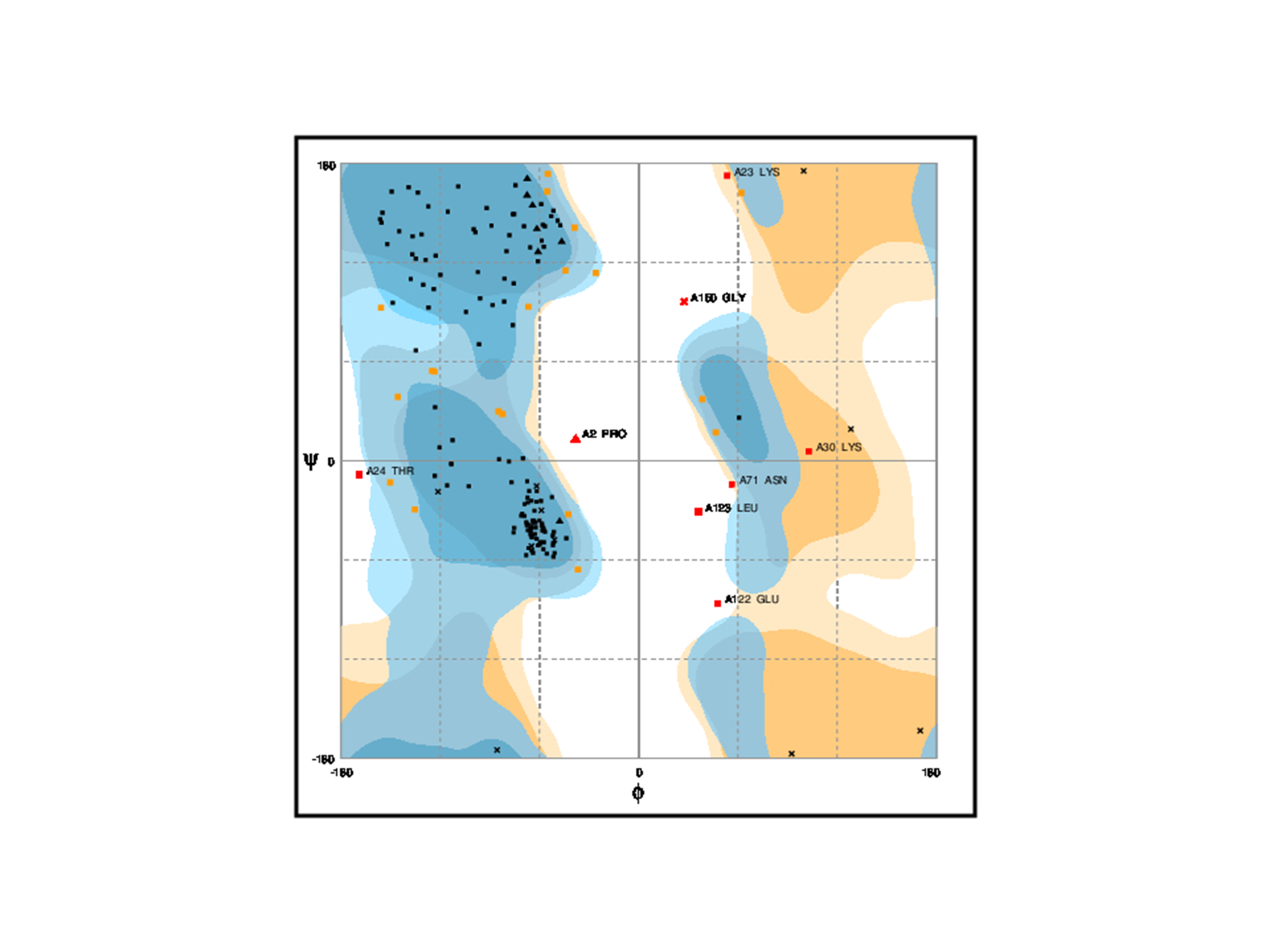
LuxS is commonly found in various bacterial species, like A. hydrophila which causes infection in fish, shrimps, and prawns and is a great threat to aquaculture industry as well as public health. It is an essential enzyme and highly conserved in various bacterial species, and has a wide range of functions such as involved in quorum sensing (QS), sporulation, virulence and synthesis of biofilm. This study focused on the prediction of 3D-sturcture of LuxS by template similarity and its ligand binding sites analysis to define its structure-function relationship. Primary structure analysis of LuxS examined that about 42% of residues content are alpha-helix, which makes it stable for three-dimensional structure homology. For the con struction of homology modeling of LuxS, crystal structure (5e68.1.A) has been used as a template and Swiss model as a work space. The validation of model by ProSA, SAVES, PROCHECK, PROSAII and RMSD. All results analysis shows that refined model is reliable and it has78.11% amino acids sequence similarity with the template,0.4Åas RMSD, and Z-score is -6.21 and Ramachandran plot analysis shows that 83.4% of residues found in the most favored regions where only 0.4% falls into the disallowed regions. Zinc ion ligand was predicted with highest MAMMOTH score and its binding residues His-54, His-58 and Cys-128 were analyzed by COACH-Meta server. LuxS phylogeny was constructed by sequences and structures of the most similar sequences were analyzed. In silico, the information has been generated in this work expects to be the first step towards the structure determination of LuxS in A. hydrophila.
Author Comment
This is a submission to PeerJ for review.
Supplemental Information
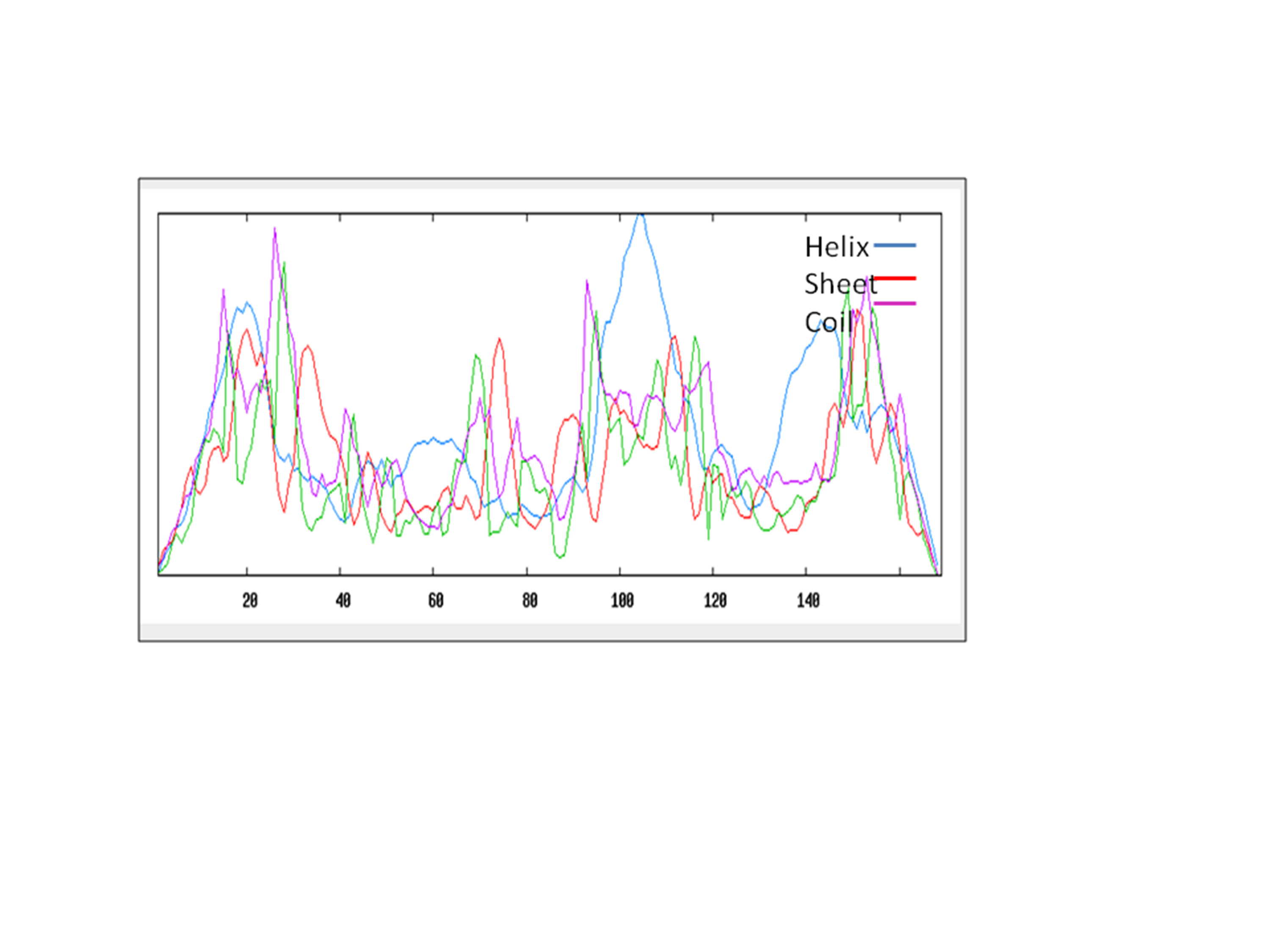
Secondary structure analyses of LuxS
(C) Stand for coil, (e) extended strand, (h) alpha-helix and (t) beta turn
{kind=link}
Distribution of secondary structure elements in LuxS
{kind=link}
{kind=link}
Ramachandran plot classifies the residues falls int o specific regions is shown in quadrangle
{kind=link}
Primary structure analysis of LuxS
Total number of negatively charged residues (Asp + Glu) = 23; Total number of positively charged residues (Arg + Lys) = 15; Grand average of hydropathicity (GRAVY) = -0.179; Number of amino acids = 169; Molecular weight = 18777.45; Theoretical pI: 5.30
Identification of functional regions/domain in LuxS
Sequence alignment between template and the target
E-value:4.9e-118, Bit-Score:880 Ident.:78.11% Positives :100.0% Query Length:169 Match Length:169