A hybrid-hierarchical genome assembly strategy to sequence the invasive golden mussel Limnoperna fortunei
- Published
- Accepted
- Subject Areas
- Bioinformatics, Computational Biology, Conservation Biology, Genetics, Genomics
- Keywords
- invasive, bivalve genomics, golden mussel, amazon, river basin
- Copyright
- © 2017 Uliano da Silva et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Preprints) and either DOI or URL of the article must be cited.
- Cite this article
- 2017. A hybrid-hierarchical genome assembly strategy to sequence the invasive golden mussel Limnoperna fortunei. PeerJ Preprints 5:e2995v1 https://doi.org/10.7287/peerj.preprints.2995v1
Abstract
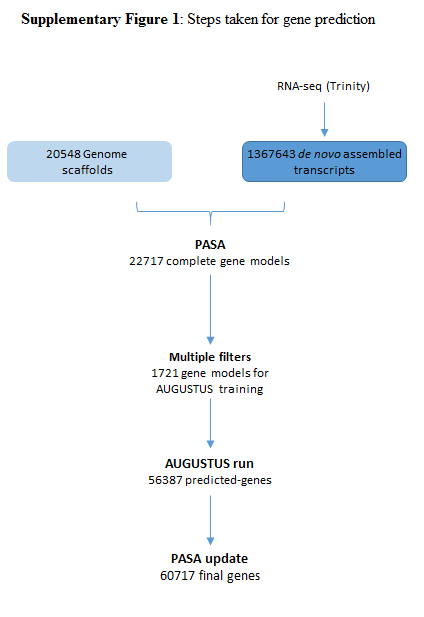
Background: For more than 25 years the golden mussel Limnoperna fortunei has been an aggressive invader in South America freshwaters, having travelled more than 5,000 km upstream across 5 countries. The mussel has outcompeted native species in the invaded environments and since chemicals in the water have been unable to control infestation in closed environments, it has economically harmed aquaculture, hydroelectric generation and ship transit. We sequenced the complete genome of the golden mussel to try to understand the molecular basis of its invasiveness and search for ways to control it. Findings: We have assembled the 1.6 Gb genome into 20548 scaffolds with a N50 length of 312 Kb using a hybrid and hierarchical assembly strategy from short and long DNA reads and transcriptomes. A total of 60717 coding genes were inferred from a customized transcriptome-trained AUGUSTUS run. We also compared predicted protein sets with those of complete molluscan genomes, revealing an exacerbation of protein-binding domains in L. fortunei. Conclusions: We assembled one of the best bivalve genomes available using a cost-effective approach using Illumina pair-end, mate pair and PacBio long reads. We expect the continuous and careful annotation of L. fortunei’s genome to aid to the investigation of bivalve genetics, evolution and invasiveness, as well as the development of biotechnological tools for aquatic pest control.
Author Comment
This is a preprint submission to PeerJ Preprints.
{kind=link}