Compositional and functional analysis of the gut microbiota of Radix auricularia (Linnaeus) via high-throughput Illumina sequencing
- Published
- Accepted
- Subject Areas
- Biodiversity, Microbiology, Freshwater Biology
- Keywords
- Radix auricularia, Intestinal bacterial communities, 16S rRNA gene, Illumina Miseq sequencing.
- Copyright
- © 2018 Hu et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Preprints) and either DOI or URL of the article must be cited.
- Cite this article
- 2018. Compositional and functional analysis of the gut microbiota of Radix auricularia (Linnaeus) via high-throughput Illumina sequencing. PeerJ Preprints 6:e26512v1 https://doi.org/10.7287/peerj.preprints.26512v1
Abstract
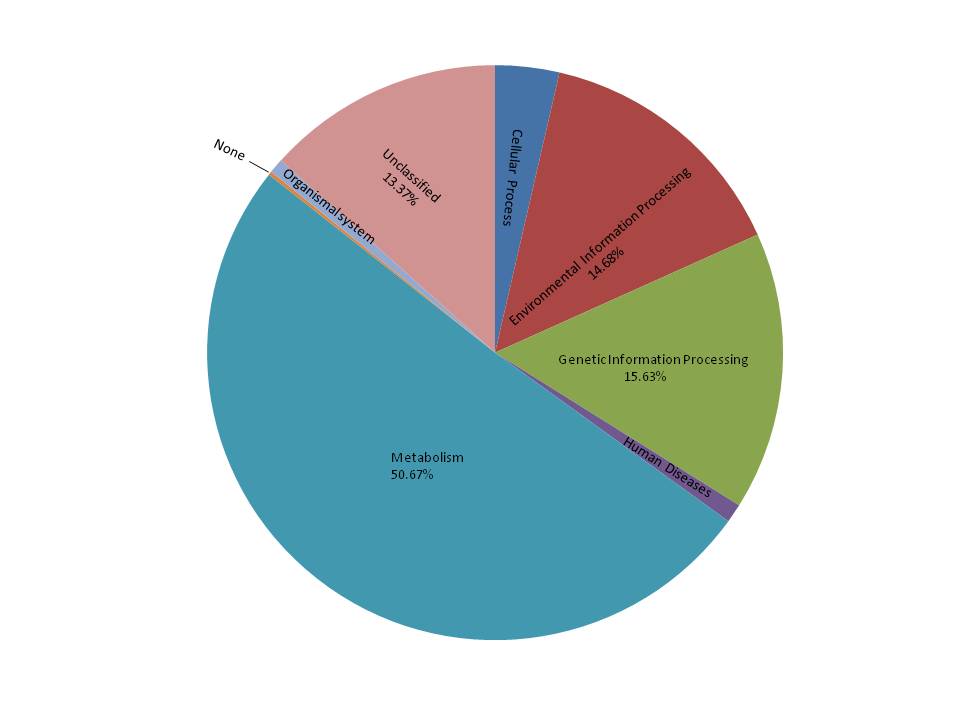
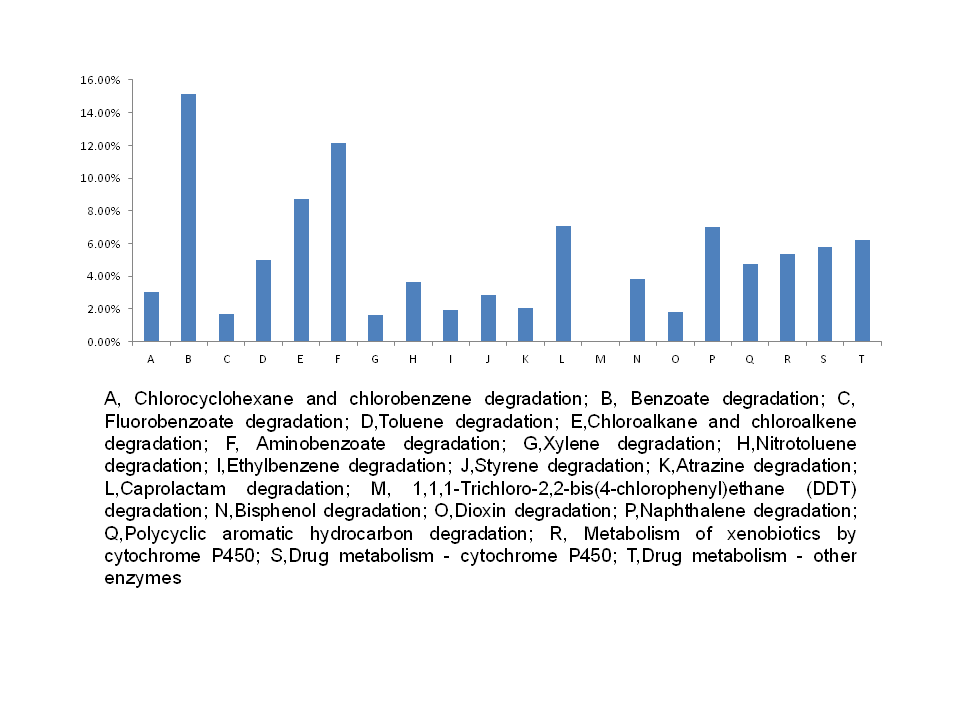
Widely distributed across the world, the freshwater snail Radix auricularia plays an important role in freshwater systems. In this study, gut bacterial communities of R. auricularia were characterized using 16S rRNA amplicon sequencing, then intestinal bacteria were compared at different growth stages: adult snails (AS) (with complete gonadal development) and juvenile snails (JS) (with incomplete gonadal development). We obtained 251,072 high quality sequences which were clustered into 1,196 operational taxonomic units (OTUs) with 97% sequence identity. The predominant phyla were Proteobacteria and Cyanobacteria, followed by Chloroflexi, Firmicutes, and Actinobacteria. Other bacterial species such as Tenericutes, Bacteroidetes, Fusobacteria and Verrucomicrobia were present to a lesser extent. 52 bacterial families and 55 genera were found in > 1% of each sample. A large number of species could not be successfully identified. 469 core OTUs were found to make up 39.38% of all OTUs and 88.38% of all sequences. Samples obtained from juvenile organisms possessed higher ratios of Ruminococcaceae, Subdoligranulum, and Faecalibacterium than adult species. Furthermore, 16S rRNA gene data was used to predict function, showing that genes related to metabolism and environmental information processing were rich in snail samples.
Author Comment
This is a submission to PeerJ for review.
Supplemental Information
The sequence data of all snail samples
A_1, A_2, A_3, A_4 represented adult snail samples, J_1, J_2, J_3, J_4 represented juvenile snails samples.
Taxa of core bacteria shared by all R. auricularia samples and their relative abundance
the table showed the core bacteria with abundance >0.5%.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}