
DOSCHEDA: a web application for interactive chemoproteomics data analysis
- Published
- Accepted
- Received
- Academic Editor
- Robert Winkler
- Subject Areas
- Bioinformatics
- Keywords
- Quantitative Chemoproteomics, Statistical Models, Web interface, Shiny, TMT, iTRAQ, Proteomics, Quantitative Chemical biology, Drug dose-response, Protein drug profiling
- Copyright
- © 2017 Contrino et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Computer Science) and either DOI or URL of the article must be cited.
- Cite this article
- 2017. DOSCHEDA: a web application for interactive chemoproteomics data analysis. PeerJ Computer Science 3:e129 https://doi.org/10.7717/peerj-cs.129
Abstract
Background
Mass Spectrometry (MS) based chemoproteomics has recently become a main tool to identify and quantify cellular target protein interactions with ligands/drugs in drug discovery. The complexity associated with these new types of data requires scientists with a limited computational background to perform systematic data quality controls as well as to visualize the results derived from the analysis to enable rapid decision making. To date, there are no readily accessible platforms specifically designed for chemoproteomics data analysis.
Results
We developed a Shiny-based web application named DOSCHEDA (Down Stream Chemoproteomics Data Analysis) to assess the quality of chemoproteomics experiments, to filter peptide intensities based on linear correlations between replicates, and to perform statistical analysis based on the experimental design. In order to increase its accessibility, DOSCHEDA is designed to be used with minimal user input and it does not require programming knowledge. Typical inputs can be protein fold changes or peptide intensities obtained from Proteome Discover, MaxQuant or other similar software. DOSCHEDA aggregates results from bioinformatics analyses performed on the input dataset into a dynamic interface, it encompasses interactive graphics and enables customized output reports.
Conclusions
DOSCHEDA is implemented entirely in R language. It can be launched by any system with R installed, including Windows, Mac OS and Linux distributions. DOSCHEDA is hosted on a shiny-server at https://doscheda.shinyapps.io/doscheda and is also available as a Bioconductor package (http://www.bioconductor.org/).
Background
Many drugs fail in the clinic for lack of efficacy or for toxicity and as such, some of the most important steps in drug discovery are evaluation of target engagement and off-targets liabilities. Next Generation Sequencing (NGS) and mass spectrometry proteomics-based drug discovery (Jones & Neubert, 2017) approaches offer unique opportunities to deeply characterize drug-targets biology and pharmaceutical agents (Bantscheff et al., 2007; Bantscheff et al., 2008).
Quantitative chemical proteomics (Bantscheff & Drewes, 2012) and MS-Cellular Thermal Shift Assay (MS-CETSA) (Martinez & Nordlund, 2016) have been recently deployed in drug discovery to aid target deconvolution in the context of phenotypic screens, to elucidate drugs’ mechanisms of action and evaluation of drug repurposing (Schirle, Bantscheff & Kuster, 2012).
A generous number of computational tools has been developed for MS-spectral analysis and protein quantification, such as Proteome Discoverer (https://www.thermofisher.com/), MaxQuant (Cox & Mann, 2008) or PEAKS (http://www1.bioinfor.com) for de novo peptide sequencing. However, the increasing variety of MS based approaches for drug target deconvolution can produce data that need dedicated downstream analysis platforms for facilitating the biological interpretation of results.
Here, we focus on quantitative chemoproteomics used to determine protein interaction profiles of small molecules from whole cell or tissue lysates (Manning, 2002). Chemoproteomics includes reverse competition-based experiments that, in combination with quantitative MS (e.g., tandem mass tags (TMT) or isobaric tag for relative quantification (iTRAQ)), are used for rank-ordering of small molecule-protein interactions by binding affinity (Bantscheff & Drewes, 2012).
Although several comprehensive analysis pipelines, such as OCAP (Wang, Yang & Yang, 2012), ProSightPC (http://proteinaceous.net/software/), TopPIC (Kou, Xun & Liu, 2016), MSstats (Choi et al., 2014), Skyline (MacLean et al., 2010), MaxQuant & Perseus (Tyanova et al., 2016) and DAPAR & ProStaR (Wieczorek et al., 2017), have been developed for the downstream data analysis, to the best of our knowledge there are no tools specifically designed to facilitate chemoproteomics data analysis for scientists with a limited computational background and available as a public server application.
Based on this, we have developed a Shiny-based web application named DOSCHEDA (Down Stream Chemoproteomics Data Analysis), which includes: (i) an open-source code available on Bioconductor for R-users; (ii) a user-friendly Graphical User Interface (GUI) with no programming knowledge required (iii) a traffic-light system to enable the user to rapidly identify and address data incongruences; (iv) an OS-independent implementation which generates a comprehensive final report in addition to analysis results; (v) a flexible data-input routine which enables the user to import different file types (.txt, .xlsx, .csv), typically exported from MS-software such as Proteome Discover or MaxQuant; (vi) the CRAPome flagged proteins based on the contaminant repository database (Mellacheruvu et al., 2013)
DOSCHEDA addresses the need to perform fit for purpose statistical analysis of chemoproteomics experiments, including linear and non-linear models, to provide a ranking of the protein(s) most competed by the investigational compound (the competitor) as well as to generate standardized results that can be further used for downstream analysis or for different experiment comparisons.
Implementation
DOSCHEDA is implemented using R (R Core Team, 2014), an open-source software for statistical computing and coded in Shiny (Chang et al., 2015).
DOSCHEDA processes the data based on a series of pipelines developed and integrated into the application. The user selects what pipeline to be executed based on the experimental design and data input.
Data input
The application is designed to take three different types of data:
-
Peptide Intensities. These are obtained from Proteome Discoverer, MaxQuant or similar software. The same procedure applied in Proteome Discoverer 2.1 has been implemented in DOSCHEDA for summing the reporter ions to protein relative quantification. The protein fold changes [Ctrl]/[treated] are then converted to log2 scale and then passed into the pipeline.
-
Fold Changes. These are the protein fold changes, [Ctrl]/[treated].
-
Log2 Fold Changes. These are the log2 protein fold changes.
DOSCHEDA has been optimized for Proteome Discoverer 2.1 (PD 2.1), but it can also use data from other software given that the input file contains specific columns as described in Table 1.
| Input data from PD2.1 | Input data not from PD2.1 | |
|---|---|---|
| Peptide intensities | Peptide qvality score, protein accessions, peptide names, intensities | Peptide qvality score, protein accessions, peptide names, intensities |
| Fold changes | Protein fold changes | Protein accessions, protein fold changes, unique peptides |
| Log-fold changes | Protein log-fold changes | Protein accessions, protein log-fold changes, unique peptides |
Statistical analyses
The experimental design as shown in Table 2 will determine the type of analysis that can be performed in DOSCHEDA. The standard pipeline utilizes a linear model with a quadratic form where a0 is the intercept, a1 the slope and a2 the quadratic coefficient. This pipeline is suitable for experimental designs with less than 5 channels (4-plex) and more than 1 replicate (Table 2). Increasing free drug concentrations and generation of a dose-response relationship with the protein target(s) provides information about specific drug-target interactions. The linear model analysis implemented in DOSCHEDA by using the limma R package (Smyth, 2004) will identify proteins with a significant p value (slope, intercept, quadratic) as the protein drug-target(s).
| 1 Replicate | More than 1 replicate | |
|---|---|---|
| Less than 5 channels | Not enough data | Linear model |
| 5 or more channels | Sigmoidal | Linear model |
Alternatively, in cases where biological input material is not a limiting factor, chemoproteomics experiments consist of a full-scale dose-response, in which 5 or more concentrations are tested (e.g., 6-plex or 10-plex). In this case, the user can run the sigmoidal pipeline where the four-parameter log- logistic (Ritz et al., 2015) model is applied (Table 2). Here, the p values of the model parameters (min, max, slope, RB50) will be computed to rank proteins based on their selectivity profile (Smyth & Collins, 2009). The half maximal residual binding (RB50) is a measure of the effectiveness of a drug in binding to a protein. Thus, this quantitative measure indicates how much of a drug or small molecule is needed to saturate binding to a protein by half, and can be used to compare drug-protein profiles. The RB50 values are typically expressed as molar concentration and are computed in the sigmoidal pipeline for each protein within DOSCHEDA. Furthermore, the corrected RB50, according to Daub (2015) corresponds to the ratio (r) of the proteins enriched in the second incubation (supernatant) versus those retained in the first incubation (DMSO or blank) with the affinity matrix. This pulldown of pulldown or depletion factor (r) enables the calculation of a Kd for each protein and it will be part of DOSCHEDA’s outputs.
Peptide removal process using Pearson correlation
When peptide intensities and two replicates are available, DOSCHEDA implements a prior step to the peptide aggregation process to consider of potential technical experimental errors.
The peptide removal function is tailored having in mind the typical chemoproteomics experimental design and aims to leverage the empirical peptides information of experimental replicates avoiding data imputation or using spectra features of peptides (Fischer & Renard, 2016) as they might not be necessarily available information to the final user.
The peptide removal algorithm is described in Fig. 1, in this procedure peptides that are inconsistent (e.g., anti-correlate) between replicates are excluded. This implementation is not intended to address the ratio compression (Savitski et al., 2013) but to leverage the information available on the same peptide between the two replicates.
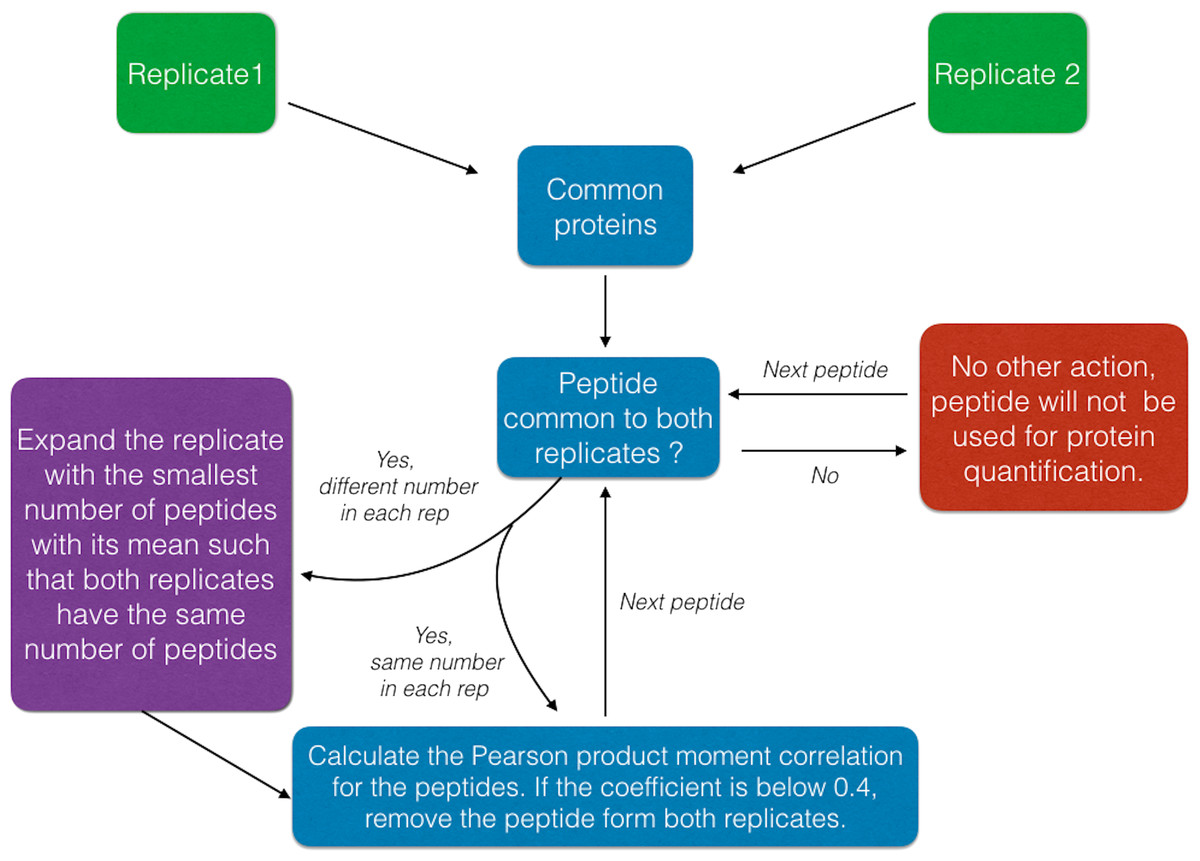
Figure 1: Schematic of the Peptide Removal process algorithm.
{kind=link}
In fact, the peptide removal process is based on the calculation of the Pearson correlation coefficient between the same peptide abundances of two replicates as a pre-filtering step before summing the remaining peptide intensities to finally infer back the protein abundance.
Initially, the peptides are filtered by their Peptide Quality score (Qs < 0.05) column, a mandatory input column in this case. Next, data are filtered to have a minimum of 2 peptides (shared or unique) per protein and per replicate such that the Pearson correlation coefficient (R2) can be calculated between matched peptides. Peptides with R2 < 0.4 are removed and summed intensities are computed from the remaining peptides; although this cut-off can be modified by the user within DOSCHEDA, we observed that 0.4 is a reasonable default threshold that removes extreme peptide quantifications (e.g., low correlated and anti-correlated peptides) and keeps almost unchanged the proteome coverage, as shown in Fig. 2 R2 < 0.4. Intuitively, larger R2 values will reflect into a larger peptide removal and consequently proteins loss as shown by Figs. 2A–2D.

Figure 2: Effect of different Pearson coefficient cut offs.
X- and Y-axis are Log2 fold changes. The total number of proteins 1,360 (A, 100%) reduces to 1,084 with Pearson coefficient R2 = 0.2 (B, ∼80%), 1,044 with R2 = 0.4 (C,∼77%), and 382 with R2 = 0.8 (D, 28%).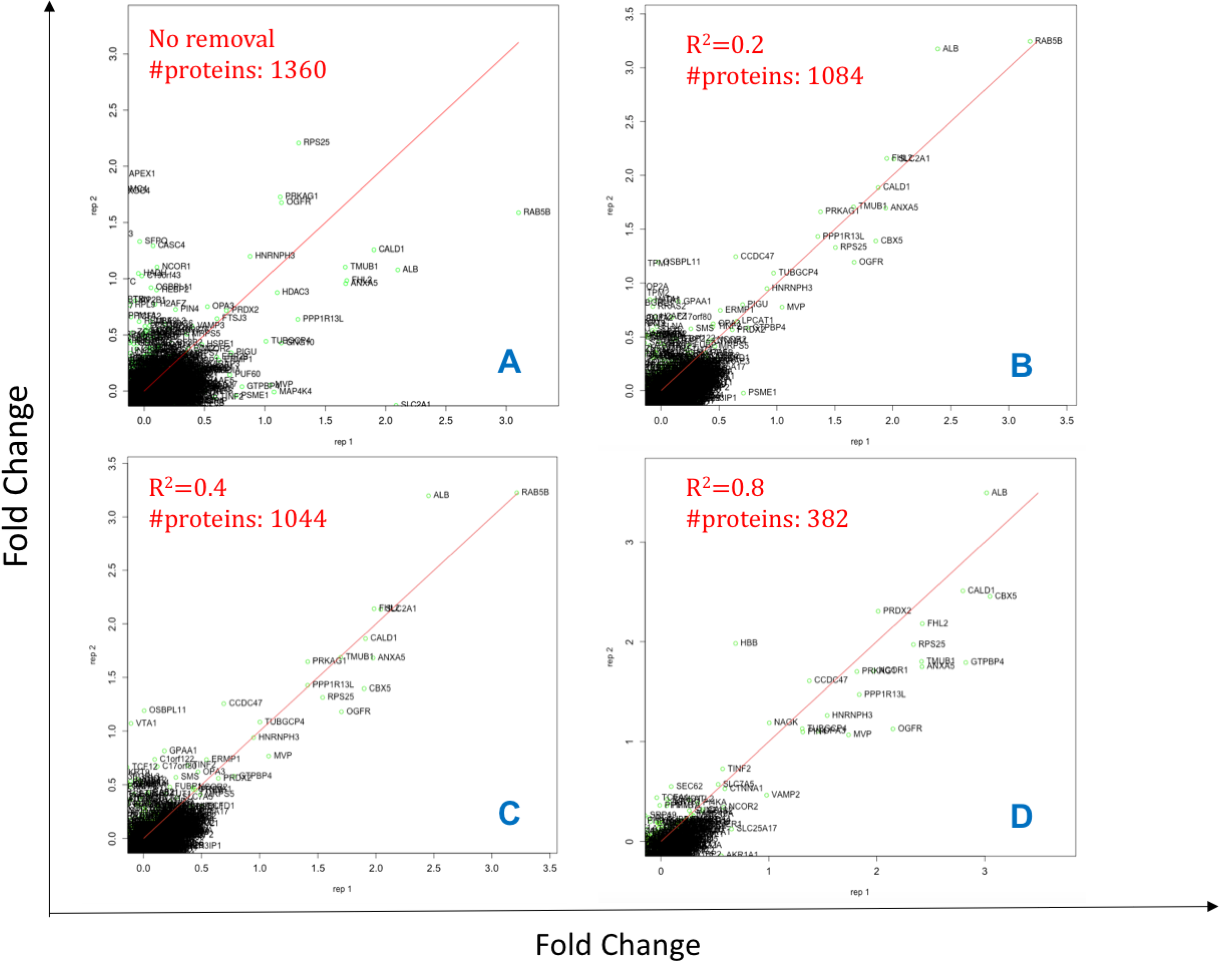{kind=link}
The R2 can be adjusted by the user within DOSCHEDA allowing quick results comparison in a similar fashion of Fig. 2 and, empowering the analyst to fine tuning this threshold based on project aims.
Finally, only proteins with more than one unique peptide are retained for the downstream analysis. Table 3 summarizes all the possible scenarios handled by the peptide removal function.
| Prot. name | Peptides Rep1 | Peptides Rep2 | Pearson correlation coefficient | Action |
|---|---|---|---|---|
| Protein 1 | A, B | A, B | RA>0.4 RB>0.4 | No removal, computed the summed intensities as in PD2.1 |
| Protein 2 | A, B | A | NULL | Lack of peptide measurements; Protein 2 will be removed |
| Protein 3 | A, B, C | A, B, C | RA>0.4 RB>0.4 RC <0.4 | A, B considered and summed. C is removed, reason of noise in the data |
Notes:
A,B,C in the real case scenario are peptide sequences.
The user should initially run the exploratory data analysis in DOSCHEDA up to the Principal Component Analysis (PCA) and the correlogram plot to evaluate the correlation between replicates. Only at this point the user can make an informed decision whether to apply the peptide removal function or continue to analyze the data without applying the removal process. Yet, being an open source tool the peptide removal process could be replaced by any other similar function that might be available in literature (see the DOSCHEDA Bioconductor vignette).
Additional Features. The server version of DOSCHEDA comprises additional uploads that are useful when researchers are comparing datasets, including: (i) intersections of the enriched proteins with a user uploaded GeneID list (e.g., protein kinesis); (ii) default mapping from Uniprot accession to GeneIDs using InterMine or in case the organism of interest is not present in the drop-down list, DOSCHEDA allows the user to specify the mapping file.
Furthermore, DOSCHEDA allows: (i) two normalization/scaling options (see DOSCHEDA manual); (ii) interactive 2D plots with user-defined x- and y- axes; (iii) quality control (QC) traffic lights flags; (iv) downloadable results and detailed report of the analyses.
Results
DOSCHEDA generates a variety of different plots depending on the user’s data input, as outlined in Fig. 3. Overall there are two sets of output plots, one designed for the quality control (QC), the other one to visualize the results of the analyzed data.
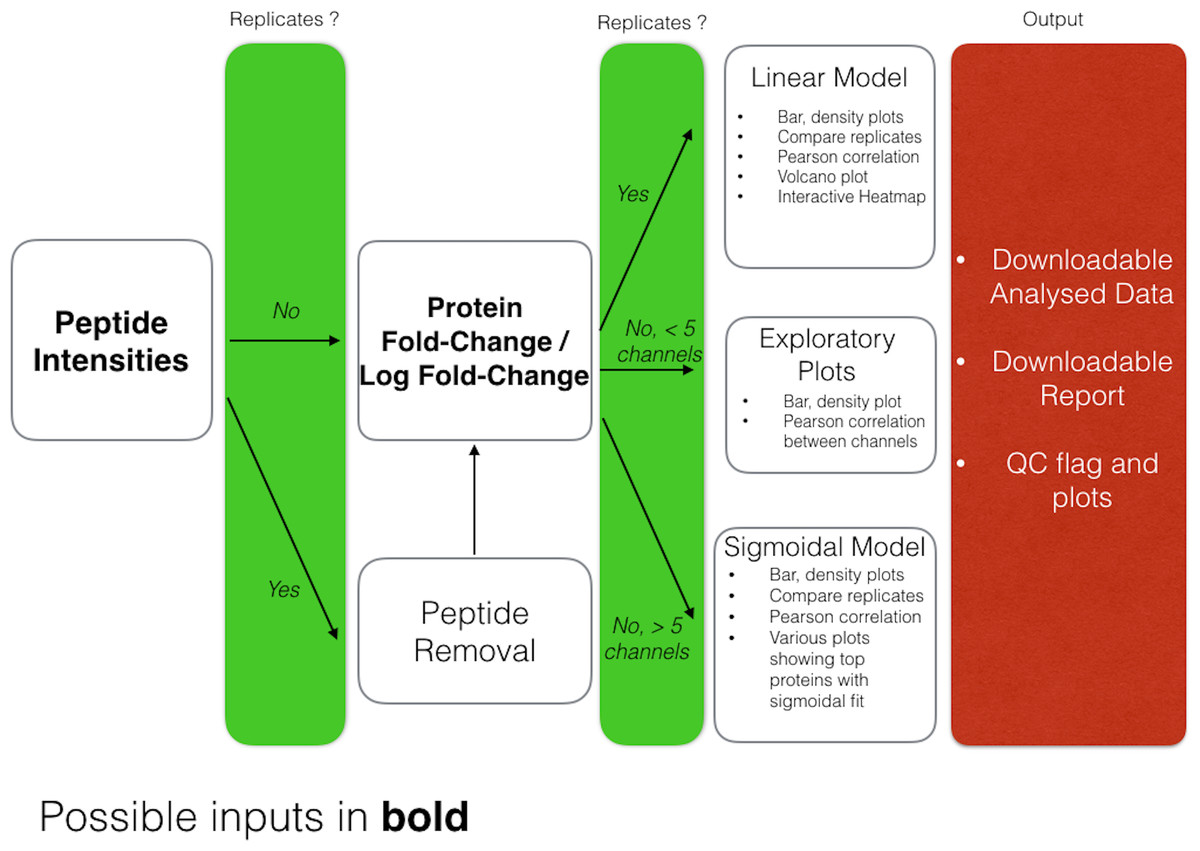
Figure 3: DOSCHEDA workflow.
{kind=link}
The DOSCHEDA application provides standard QC plots such as box, whisker plots, data distributions within each iTRAQ/TMT channel, correlogram plots to quantify the correlation between replicates, a 2D-plot to verify the variance independence from the data mean, and a plot of the first two principal components. Based on the QC outcomes, if data are consistent the user can proceed to the inspection of the statistical analysis results, otherwise, if peptide intensities are available the peptide removal process can be applied.
For the analysis, depending on the chosen statistical model, DOSCHEDA will generate different outputs.
In case of a linear model the p values distributions for the three coefficients (a0, a1 and a2) and their corresponding interactive volcano plots are displayed.
In case of a sigmoidal model DOSCHEDA will output three plots: the first showing the sigmoidal curves with a difference higher than 30% between the top and the bottom value; the second and the third showing the proteins that have a significant RB50 (p value <0.05) and a significant slope (p value <0.05), respectively.
Independently of the applied statistical model, the R package d3heatmap is used to generate an interactive heatmap of the input data.
Once the analysis has been completed, the summary section of the application allows the user to visualize the results in a table format, where functions like “search” and “sorting” are also enabled for quick queries.
The summary section contains quality control (QC) traffic light flags with green for data consistency and orange for data incongruences with an accompanying warning message. There are two types of traffic lights flags; see Fig. 4.
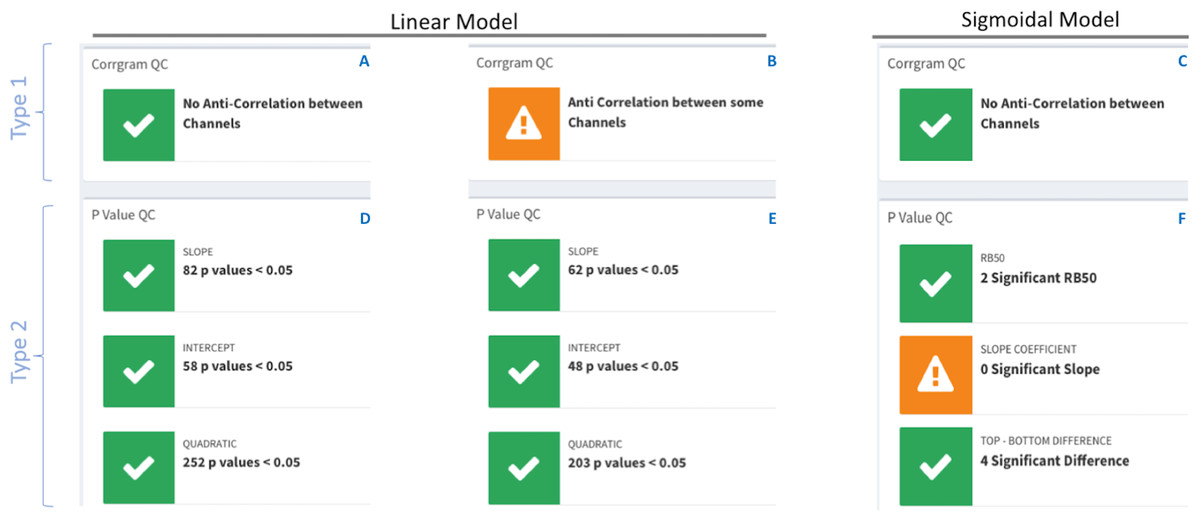
Figure 4: DOSCHEDA traffic lights flags system.
Two types of QCs’ are available, type 1 based on pairwise samples correlation obtained from the correlogram plot (A–C) and, type 2 based on model’s p values coefficients (D–E) obtained by running the linear or sigmoidal statistical analysis. In both types, green flags will be displayed when there is no anti-correlation between any samples and the number of proteins with p value(s) <0.05 is larger than zero. On the contrary orange flags will be displayed when anti-correlation is observed (B) in at least one of the pairwise samples (i.e., Pearson Correlation Coefficient, R2 < 0) and/or there are no proteins that passes the threshold of p value(s) < 0.05 (F).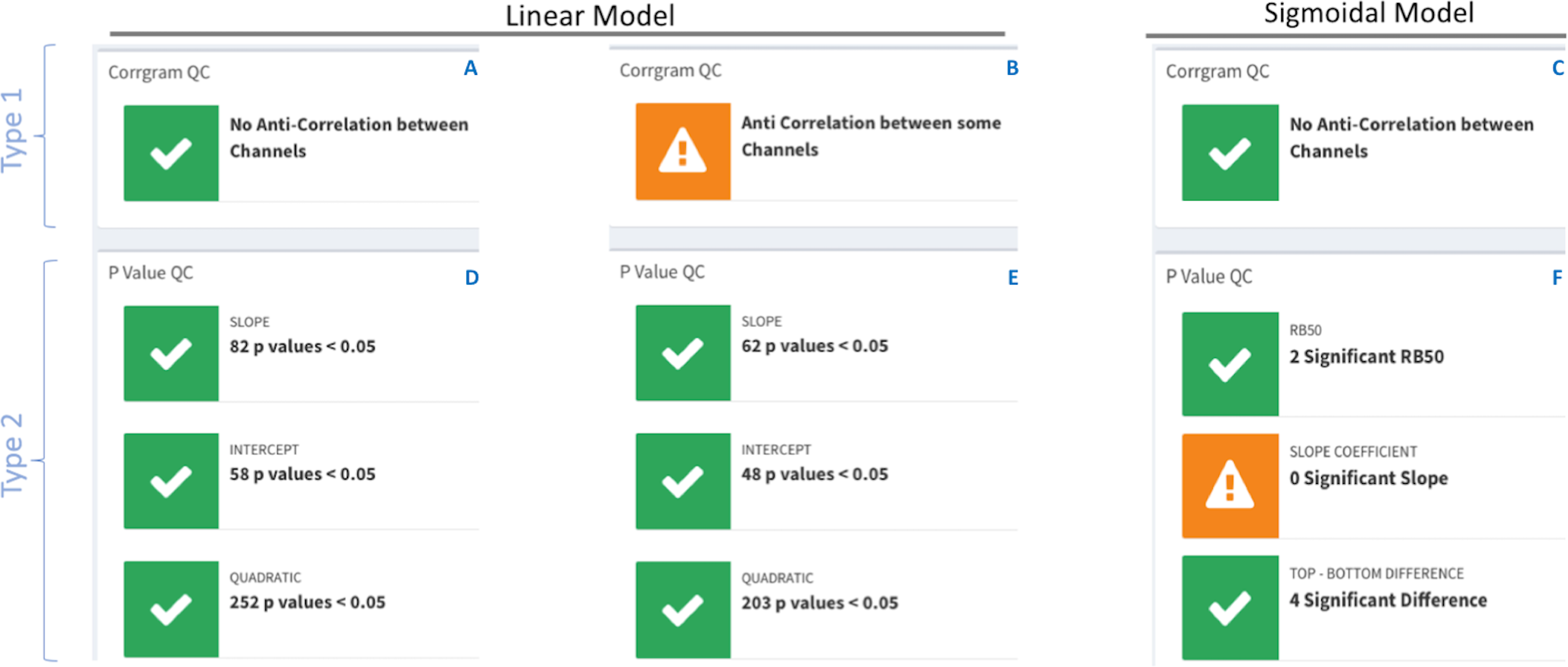{kind=link}
Finally, in the download section of the application the user can download (i) a .csv file containing the results of the analysis which also includes the user-input dataset and (ii) an html report with all the plots produced in the current run, the key tables as well as the session information to facilitate data reproducibility.
Conclusion
DOSCHEDA enables researchers with limited programming experience to perform, evaluate, and interactively visualize chemoproteomics data analysis. DOSCHEDA includes linear and non-linear statistical analysis whose results can be exported in excel spreadsheet format. Also, the user will be able to generate a full report of the executed data analysis, facilitating data reproducibility. Being open source, DOSCHEDA can be easily extended and modified to fit specific additional analyses.
Availability and requirements
DOSCHEDA server lives at https://doscheda.shinyapps.io/doscheda/ and it will be constantly maintained by the authors; the users by following the link above can upload and analyze their own datasets without additional requirements. Finally, to facilitate traceability and reproducibility DOSCHEDA is also available as an open source Bioconductor package.