
Comprehensive transcriptome analysis of reference genes for fruit development of Euscaphis konishii
- Published
- Accepted
- Received
- Academic Editor
- Rodolfo Aramayo
- Subject Areas
- Agricultural Science, Molecular Biology
- Keywords
- Euscaphis konishii, Fruit development, Reference genes, qRT-PCR
- Copyright
- © 2020 Yang et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2020. Comprehensive transcriptome analysis of reference genes for fruit development of Euscaphis konishii. PeerJ 8:e8474 https://doi.org/10.7717/peerj.8474
Abstract
Background
Quantitativereal-time reverse transcriptase polymerase chain reaction is the common method to quantify relative gene expression. Normalizating using reliable genes is critical in correctly interpreting expression data from qRT-PCR. Euscaphis konishii is a medicinal plant with a long history in China, which has various chemical compounds in fruit. However, there is no report describing the selection of reference genes in fruit development of Euscaphis konishii.
Methods
We selected eight candidate reference genes based on RNA-seq database analysis, and ranked expression stability using statistical algorithms GeNorm, NormFinder, BestKeeper and ReFinder. Finally, The nine genes related to the anthocyanin synthesis pathway of Euscaphis konishii were used to verify the suitability of reference gene.
Results
The results showed that the stability of EkUBC23, EkCYP38 and EkGAPDH2 was better, and the low expression reference genes (EkUBC23 and EkCYP38) were favourable for quantifying low expression target genes, while the high expression reference gene (EkGAPDH2) was beneficial for quantifying high expression genes. In this study, we present the suitable reference genes for fruit development of Euscaphis konishii based on transcriptome data, our study will contribute to further studies in molecular biology and gene function on Euscaphis konishii and other closely related species.
Introduction
As an important economic crop, fruit trees play an important role in the agricultural production of various countries. Fruit development research is an important part of fruit science. Screening out stable reference genes in the fruit developmental stage can lay the foundation for the study of functional genes in neighbouring species and accelerate the process of molecular breeding.
The analysis of gene expression, or more correctly tanscript abundance, is essential in all aspects of molecular biology research to understand gene expression patterns in different biological processes (Dussert et al., 2013). So far, the commonly used methods for the detection of gene expression are northern blotting, gene chip and quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR). Among them, qRT-PCR has become one of the most powerful tools for studying gene expression due to its high sensitivity, accuracy and specificity (Bustin, 2002; Nolan, Hands & Bustin, 2006). qRT-PCR is wonderful for fast and accurate gene expression analysis. However, this technology requires suitable reference genes to normalize expression data and control the quantity of cDNA (Derveaux, Vandesompele & Hellemans, 2010). In previous stages, housekeeping genes were selected as stable reference genes. However, lots of housekeeping genes have significantly different levels of expression in different experimental conditions or related species (Dheda et al., 2004). The stability of reference gene is influenced by experimental conditions and plant species. It is necessary to find additional reference genes for most plants.Therefore, according to different experimental conditions, the key is to accurately quantify target genes and screen one or more reference genes with stable expression from multiple candidate reference genes.
Currently, Gene Expression Omnibus was used as a new tool to screen reference genes due to its accuracy and comprehensiveness. GeneChip research of Arabidopsis thaliana revealed that the protein phosphatase 2A was favourable for quantifying low expression target genes (Czechowski et al., 2005); Coker and Davies’ research showed that the stability of TUA (Tubulin alpha), CYP (Cyclophilin) and GAPDH (Glyceraldehyde-3-phosphate dehydrogenase) is better by using dbEST (Coker & Davies, 2004). The best reference genes of Elaeis guineensis (Xia et al., 2014), Populus trichocarpa (Pang et al., 2017) and Populus trichocarpa (Su et al., 2013) were screened out based on RNA-seq technology.
Euscaphis konishii is a member of the family Staphyleaceae, which is used as an ornamental and medicinal source material in China. The fruit of E. konishii has various chemical compounds, including flavonoid compounds (Liang et al., 2018a), triterpene compounds (Cheng et al., 2010; Li et al., 2016), and phenolic acid compounds (Huang et al., 2014), which have both anti-inflammatory and anticancer effects according to traditional and modern medical research. Moreover, in order to study the gene regulation mechanisms of medicinal compounds in E. konishii, several reference genes in E. konishii were selected by Liang et al. (2019) based on transcriptome data (Liang et al., 2018b). However, the data being studied from Liang et al. lack the transcriptome of the fruit at different developmental stages. Yuan et al. (2018) identified a large number of differentially expressed genes associated with endocarp colouration based on the transcriptome database, but it is unknown which genes are suitable as reference genes related to fruit development. Huang et al. (2019) revealed that the total triterpenes of Euscaphis konishii pericarp showed the immune boosting effect and hepatic protective activity against inflammation, oxidative stress and apoptosis in BCG/LPS-induced liver injury. These findings suggest that pericarp of E. konishii might be a promising natural food for immunological hepatic injury. Because of the information on gene expression in E. konishii is still unclear, transcriptome technology would play an important role in mining the reference gene. Our study provided two available internal control genes for qRT-PCR data normolizetion in E. konishii. It will contribute to analyze the expression patterns of compound related gene, and further to reveal the breeding mechanism.
Materials & Methods
Plant material
The fruits of E. konishii have three developmental stages, including the green stage, turning stage, and red stage. These were collected from Qingliu County, Fujian Province, China, from June to September. The samples were provided by Sanming yisheng agricultural and forestry co., LTD. All pericarp samples were separated from the fruit, wrapped in tin foil and then frozen in liquid nitrogen and immediately stored at −80 °C until they were qualified for further analysis. Three biological replicates for each sample were used for RNA extraction.
Establishment of the RNA-Seq database
We sequenced the transcriptome of the E. konishi i pericarp in three developmental stages. The library produced 67.78 G of clean data, and the average clean data in the nine samples were 6.78 G. In this study, high-quality libraries with mapping rate were higher than 79.74% and Q30 values highter than 92.70%. In total, 86,120 unigenes with a mean length of 893.34 bp and N50 length of 1,642 nt were assembled by using Trinity (Grabherr et al., 2011) with min_kmer_cov set to 2 by default and all other parameters set default. The length distribution of all unigenes were shown in Fig. 1. The raw data has been submitted to NCBI, Sequence Read Archive (SRA) submission: SRR9267648 to SRR9267656. We used BLAST software to compare unigene sequences to NR (https://blast.ncbi.nlm.nih.gov/Blast.cgi), Pfam (http://pfam.xfam.org/), GO (http://www.geneontology.org/), KEGG (http://genome.jp/kegg/), Pwiss-Prot (http://uniprot.org/), KOG (http://www.ncbi.nlm.nih.gov/KOG), eggNog (http://eggongdb.embl.de/), and COG (http://www.ncbi.nlm.nih.gov/COG). Transcriptome sequences were used as references to analyse the expression profiles of each sample. The sequencing results of each sample were compared to the reference sequence, and the expression amount of each unigene in different samples was obtained.
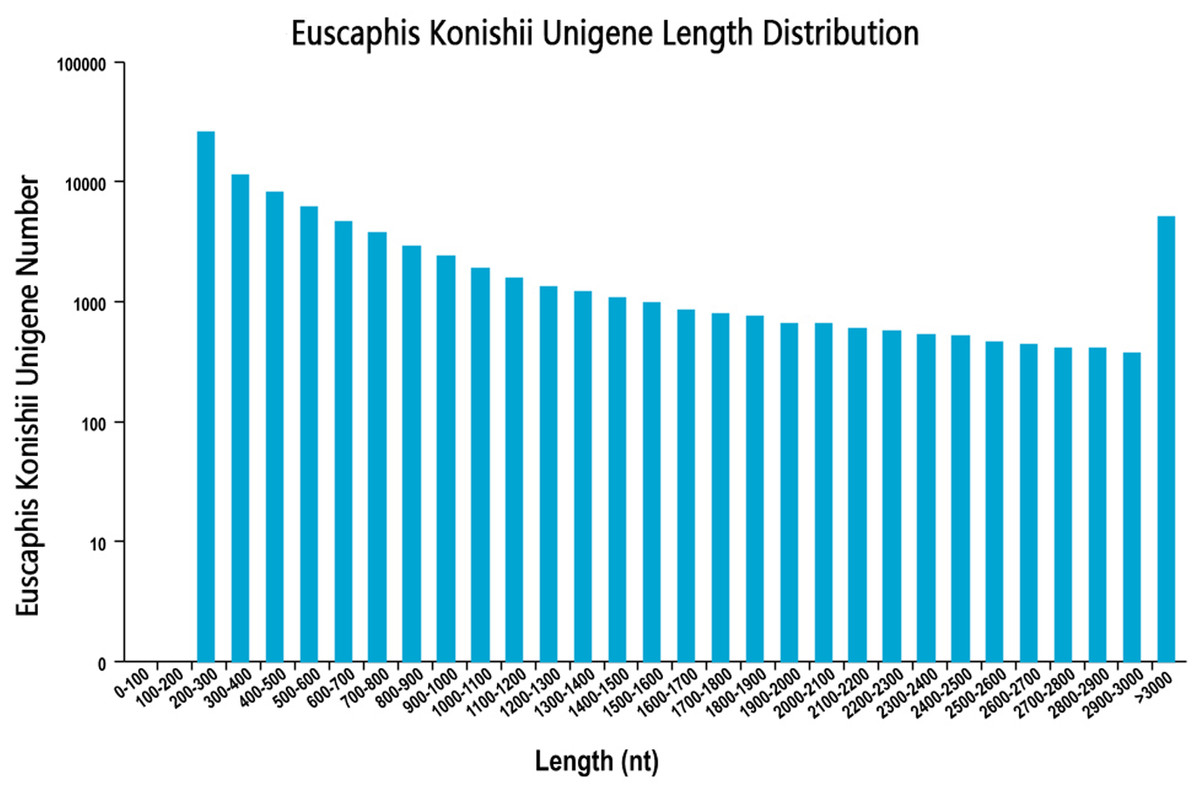
Figure 1: Unigenes length distribution.
{kind=link}
RNA-Seq database analysis and primer design
Genes with expression levels lower than 5 FPKM in tanscriptomes would make poor qPCR references due to the difficulties in detecting and quantifying their expression. (Kimmy, Patrick & Joshua, 2017). After their removal, a total of 75,048 genes in E. konishii were evaluated. We calculated the mean expression value, standard deviation, and coefficients of variation based on the raw RNA-seq data. Base on the requirements CV ≤ 0.2 and FC ≤ 0.2, 1,131 genes (Table S1) were obtained by removing overabundant genes with low expression levels. Finally, we selected the eight candidate genes according to genome annotation of each, which was assigned base on the best mach of the alignments using Blast to NR, TrEMBL and KEGG databases. The selecting steps of candidate reference genes were shown in Fig. S1. In order to ensure the accuracy of the reference gene predictions, the coding sequences of eight selected genes was used as queries for BLAST orderly through the TAIR database (http://www.arabidopsis.org/). The sequences with the highest homology with Arabidopsis were shown in Table 1.
| #ID | Arabidopsis ortholog | Gene | E-value | FPKM Mean | SD | CV | FC | ||
|---|---|---|---|---|---|---|---|---|---|
| FC-1 | FC-2 | FC-3 | |||||||
| c71660.graph_c1 | AT1G13440.1 | GAPDH2 | 4e−34 | 1,418.24 | 145.33 | 0.10 | 0.13 | 0.08 | 0.05 |
| c67439.graph_c2 | AT5G19770.1 | TUA3 | 3e−68 | 165.32 | 18.28 | 0.11 | 0.12 | 0.05 | 0.08 |
| c67539.graph_c0 | AT3G01480.1 | CYP38 | 1e−59 | 19.10 | 3.07 | 0.16 | 0.08 | 0.08 | 0.01 |
| c67010.graph_c0 | AT2G16920.1 | UBC23 | 2e−158 | 18.30 | 2.05 | 0.11 | 0.19 | 0.13 | 0.04 |
| c62586.graph_c0 | AT3G52590.1 | UBQ1 | 3e−10 | 230.36 | 21.98 | 0.10 | 0.19 | 0.00 | 0.18 |
| c65728.graph_c0 | AT3G15020.2 | mMDH2 | 5e−35 | 187.30 | 18.88 | 0.10 | 0.11 | 0.20 | 0.08 |
| c63030.graph_c0 | AT3G47520.1 | MDH | 2e−19 | 19.87 | 1.95 | 0.10 | 0.16 | 0.14 | 0.03 |
| c63658.graph_c0 | AT5G09810.1 | ACT7 | 2e−38 | 407.67 | 35.20 | 0.09 | 0.09 | 0.17 | 0.08 |
Notes:
- FPKM
-
Fragments Per Kilobase of transcript per Million fragments mapped; SD
- CV
-
coefficient variation
- FC
-
flod change
- GAPDH2
-
Glyceraldehyde-3-phosphate dehydrogenase c-2
- TUA3
-
Tubulin alpha-3
- CYP38
-
cyclophilin 38
- UBC23
-
Ubiquitin-conjugating enzyme 23
- UBQ1
-
Ubiquitin extension protein 1
- mMDH2
-
Lactate/malate dehydrogenase family protein
- MDH
-
malate dehydrogenase
- ACT7
-
actin 7
Here we obtained the nucleotide sequences of reference genes from our laboratory. The nucleotide sequences of candidatate reference genes were appended in Table S2. According to the nucleotide sequence of candidate reference genes and the design principle of qRT-PCR, forward and reverse primers of all candidate reference genes were designed using Primer Premier 6.0 software and were synthesized by Jinweizhi Biotechnology Co., LTD (Suzhou, China) and purified by PAGE. The primer sequences, amplicon size, anneal temperature were shown in Table 2.
| #ID | Gene name | Primer sequence | Length | Distance from 3′ | Anneal temp |
|---|---|---|---|---|---|
| c71660.graph_c1 | GAPDH2 | F:CCGTGTTCCTACTGTTGATGT | 95 | 1,244 | 62.0 |
| R:CCTCCTTGATAGCAGCCTTAAT | 1,338 | 61.9 | |||
| c67439.graph_c2 | TUA3 | F:GGGTGGTAGCAAACCCTATTAC | 103 | 203 | 62.4 |
| R:CCGAAGGTGCAGATGATGAA | 305 | 62.2 | |||
| c67539.graph_c0 | CYP38 | F:ATCTGTTGGAACTCCTCCATTC | 114 | 2,251 | 62.0 |
| R:AGCCCTGAAGCAAGGTAAAG | 2,364 | 62.2 | |||
| c67010.graph_c0 | UBC23 | F:AGCCACATAATCTCCGTGTAAG | 105 | 4,161 | 62.0 |
| R:GCTGACCATGTTCGAGTAGTT | 4,265 | 62.0 | |||
| c62586.graph_c0 | UBQ1 | F:ACGAGCCAAAGCCATCAA | 105 | 1,435 | 62.0 |
| R:GGCCGAACTCTTGCTGATTA | 1,539 | 62.2 | |||
| c65728.graph_c0 | mMDH2 | F:CATCGTAAGTCCCTGCTTTCT | 104 | 956 | 61.9 |
| R:TGCCAAGTACTGCCCTAATG | 1,059 | 62.0 | |||
| c63030.graph_c0 | MDH | F:ATGAAGAAGTCCACGAGCTAAC | 97 | 995 | 62.1 |
| R:GCCATAGACAGAGTAGCAGAAC | 1,091 | 62.0 | |||
| c63658.graph_c0 | ACT7 | F:GATCTGGCATCACACCTTCTAC | 112 | 394 | 62.3 |
| R:CTGAGTCATCTTCTCCCTGTTG | 505 | 62.0 |
Notes:
- F
-
forward primer
- R
-
reverse primer
RNA Extraction and cDNA Synthesis
Total RNA was extracted and purified by using an RNAprep Pure Plant Kit (Polysaccharides and Polyphenolics-Rich, Tiangen, Beijing, China), according to the manufacturer’s instructions. Two-hundred nanograms of total RNA of each sample was used as the template. In addition, the cDNA synthesis strand was performed by using a Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher, Foster City, CA, USA), according to the manufacturer’s instructions and was stored at −80 °C for further experiment.
Candidate reference genes for RT-PCR amplification
The RT-PCR mixture contained 25 µL L of 2 ×EasyTaq®PCR SuperMix, 0.4 µL L of forward primer (10 µM), 0.4 µL L of reverse primer (10 µM), and 5 µL L of diluted cDNA in a final volume of 50 µL L. PCR conditions: 40 cycles of 3 min at 94 ° C, 94 °C at 30 s, 56 °C at 10 s, 72 °C at 30 s, and 72 °C at 7 min. RNA quality was determined by 1.2% agarose gel electrophoresis.
Candidate reference genes for qRT-PCR amplification
The qRT-PCR mixture contained 3 µL L of diluted cDNA, 5 µL of 2 × SYBR Green Master, 0.4 µL L of forward primer (10 µM), and 0.4 µL L of reverse primer (10 µM), with ddH2O added to achieve a final volume of 10 µL L. All PCRs were performed using the QuantStudioTM Real-Time PCR System under the following conditions: 40 cycles of 2 min at 95 °C, 5 s at 95 °C and 30 s at 60 °C. The procedure ended with a melt-curve ramping from 60–95 °C. The melting curve was analysed to determine primer specificity. For further confirmation, PCR products were cloned into pGXT vectors respectively and then sequenced by Biosune Biotech (Fuzhou, China).
Statistical analysis
The instrument calculated the Cq value of each sample based on the qRT-PCR experiment. We can analyse the stability of candidate reference genes using GeNorm, NormFinder, and BestKeeper software. Finally, ReFinder was used to calculate the comprehensive ranking of the stability of candidate reference genes based on analysis results.
Validation of the candidate reference genes
Anthocyanin is the key factor affecting fruit colouration as an important plant pigment. In general, fruit colouration is closely related to the content and proportion of anthocyanin. To verify the results of our experiments, the expressions of nine genes related to the anthocyanin synthesis pathway were calculated with the reference genes selected (Fig. S5). Moreover, the EkSS3 gene related to glycometabolism pathway was also used to validate the reliability of reference genes. The details of primer were shown in Table 3. The qRT-PCR experimental method was the same as described above, and the relative expression level was calculated by the 2−ΔΔct method. Experimental data from three biological replicates were analysed using analysis of variance (ANOVA), followed by Student’s t-test (P < 0.05).
| Locus | Primer sequence (5′–3′) |
|---|---|
| c50541.graph_c0 (CHS) | F:GGAACTCGCTGTTCTGGATAG/R:CCTTGTGGCCCTTAACTTCT |
| c59825.graph_c0 (CHS) | F:GCATGTGTTGTGCGAGTATG/R:CCTTCCCTTCTTCCAGAGATTT |
| c60763.graph_c0 (ANS) | F:CAGCTTGAGTGGGAAGACTATT/R:TACTCGCTTGTTGCCTCTATG |
| c64532.graph_c1 (F3H) | F:GGTTCAAGATTGGCGTGAAATAG/R:CATCAGCTTCCCACTGTACTC |
| c69442.graph_c0 (CHI) | F:TCTTGCTGAGGATGATGACTTT/R:TCTCTAGCTGCACTCCATACT |
| c69862.graph_c1 (UFGT) | F:ACCGCTAATCCCAACTCTTTC/R:GTGGTTCGGTGTGCCTATT |
| c71357.graph_c2 (FLS) | F:CGACAATCGCTCCATCTTCT/R:ATGGCCTCCTTCCTGTATTAAC |
| c72659.graph_c0 (CHS) | F:TTGGTGACGCCGAAGATAAA/R:GAGGTCCAGCTACAGTTCTTG |
| c72737.graph_c0 (CHI) | F:CTCTTGTCCAGCAGCATTCTA/R:CAGAGTTTGGCTGCAAGAATATC |
| c58939.graph_c0(SS3) | F: TGAATGGATGCAGGTGACTGGAAC/R: CCACACTTGCTGAGTTGCTCTTTC |
Notes:
- F
-
forward primer
- R
-
reverse primer
Results
Differentially expressed genes between fruit with varied development
Based on the theree comparisons of green vs. turning, green vs. red, and turning vs. red, we identified a total of 4,804 differentially expressed genes (DEGs). Among them, 2,175 DEGs between green vs. turning, 3,935 DEGs between green vs. red, and 936 DEGs between turning vs. red were detected. Among the 4804 DEGs selected to predict functions by gene ontology (GO) annotation and KEGG pathway analysis. The genes related to the glycometabolism, anthocyanin biosynthesis, flavonoid biosynthesis and carotennoid biosynthesis performed different expression in different stages of fruit ripening.
Reference gene selection and primer design
According to selecting process (Fig. S1), eight candidate reference genes were chosen to perform the gene normalization studies. The sequences of these candidate reference gene were used to design the qRT-PCR primers. The details of eight reference genes were lised in Table 1. The gene models of eight candidate reference refered to the homologous genes of Arabidopsis, and the details were shown in Fig. S2.
Candidate reference genes for RT-PCR amplification
PCR results of the 8 candidate internal reference genes are shown in Fig. S4 with a single band, no primer dimer and nonspecific amplification, which could be used for subsequent qRT-PCR analysis. We sequenced the PCR amplification products of eight genes and blast the correctness of the PCR amplified fragments (Table S3).
Cq values of candidate reference genes
The Cq values for all 8 reference genes are shown in Fig. 2. The Cq values varied from 24.65 (EkGAPDH2) to 35.27 (EkACT7). Moreover, as shown in Fig. 2, EkmMDH2, EkMDH and EkACT7 are more variable than other candidate reference genes. The stability of EkmMDH2, EkMDH and EkACT7 expression is poor.
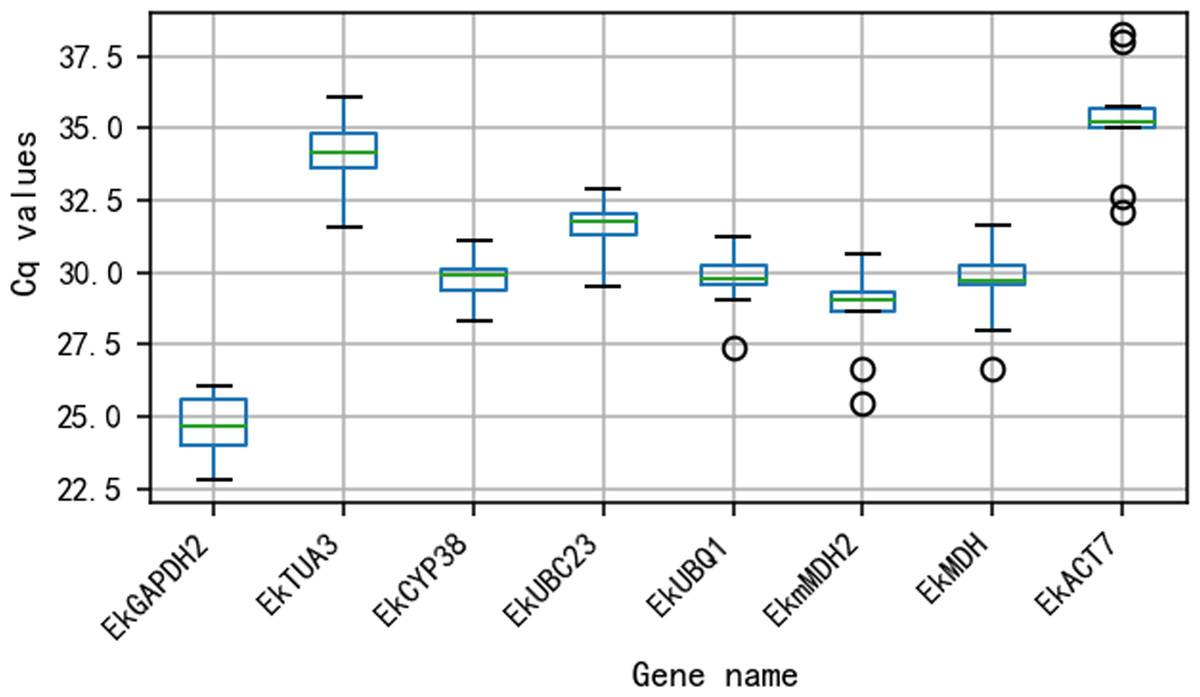
Figure 2: Quantification cycle(Cq) value of eight candidate reference genes across all the experimentalsamples.
The green line across the box depicts the median. Whiskers represent the local maximum and minimum values, and points beyond the end of each whisker mark the outliers. The box indicates the 25% and 75% percentiles.{kind=link}
qRT-PCR analysis
As shown in Fig. S3, the melting curve of the 8 candidate reference genes at different developmental stages only had a single main peak, and the amplification curves between the repeated samples had high repeatability.
Expression stability analysis of reference genes
Evaluating the expression stability of candidate reference genes depended on statistical analysis. Four different statistical software programs (GeNorm, NormFinder, BestKeeper and ReFinder) were commonly used to calculate the variability of the candidate genes expression and determine the one which were the most suitable.
Gene expression stability was determined by the M-value in GeNorm. The gene expression stability increases as the M-value decreases (Wu et al., 2017; Vandesompele et al., 2002). It was considered that candidate reference genes can be used as reference genes when the M-value is less than 1.0; the result of GeNorm revealed that EkUBQ1 was the most stable gene with the lowest M-value, followed by EkUBC23, EkCYP38 and EkGAPDH2. However, EkACT7, EkmMDH2, EKMDH and EkTUA3 were unsuitable as reference genes, with a value greater than 1.0. Expression stability values analysed by GeNorm are shown in Fig. 3A.
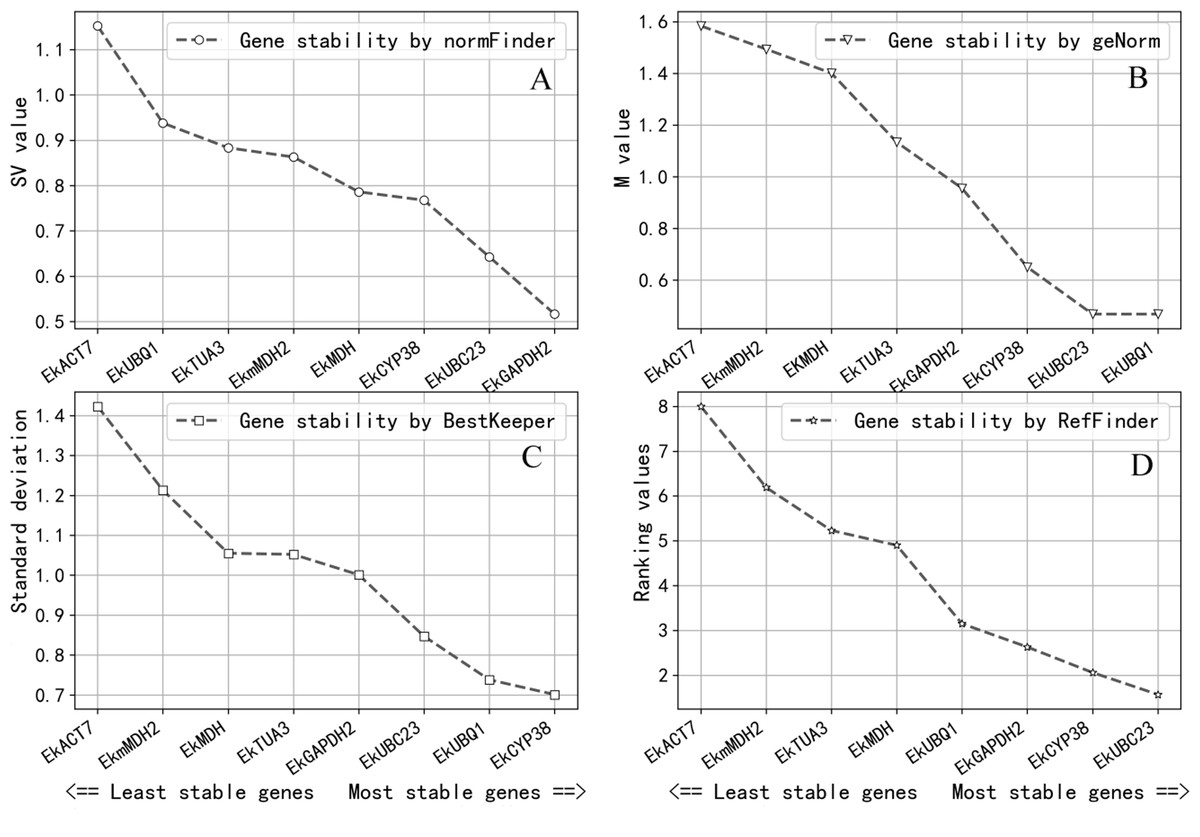
Figure 3: Expression stablility analysis of reference genes by NormFinder (A), geNorm (B), BestKeeper (C) and ReFinder (D).
Analysis was performed after pooling Cq data across all three growth conditions. The designation of the analyses were perfomed on all eight candidate reference genes. Gene were ranked from the least stable (on the left) to the most stable (on the right). (A) Genes were ranked according to their NormFinder SV value. (B) Genes were ranked according to their geNorm M value. (C) Genes were ranked according to their BestKeeper SD value. (D) Genes were ranked according to their ReFinder Ranking value.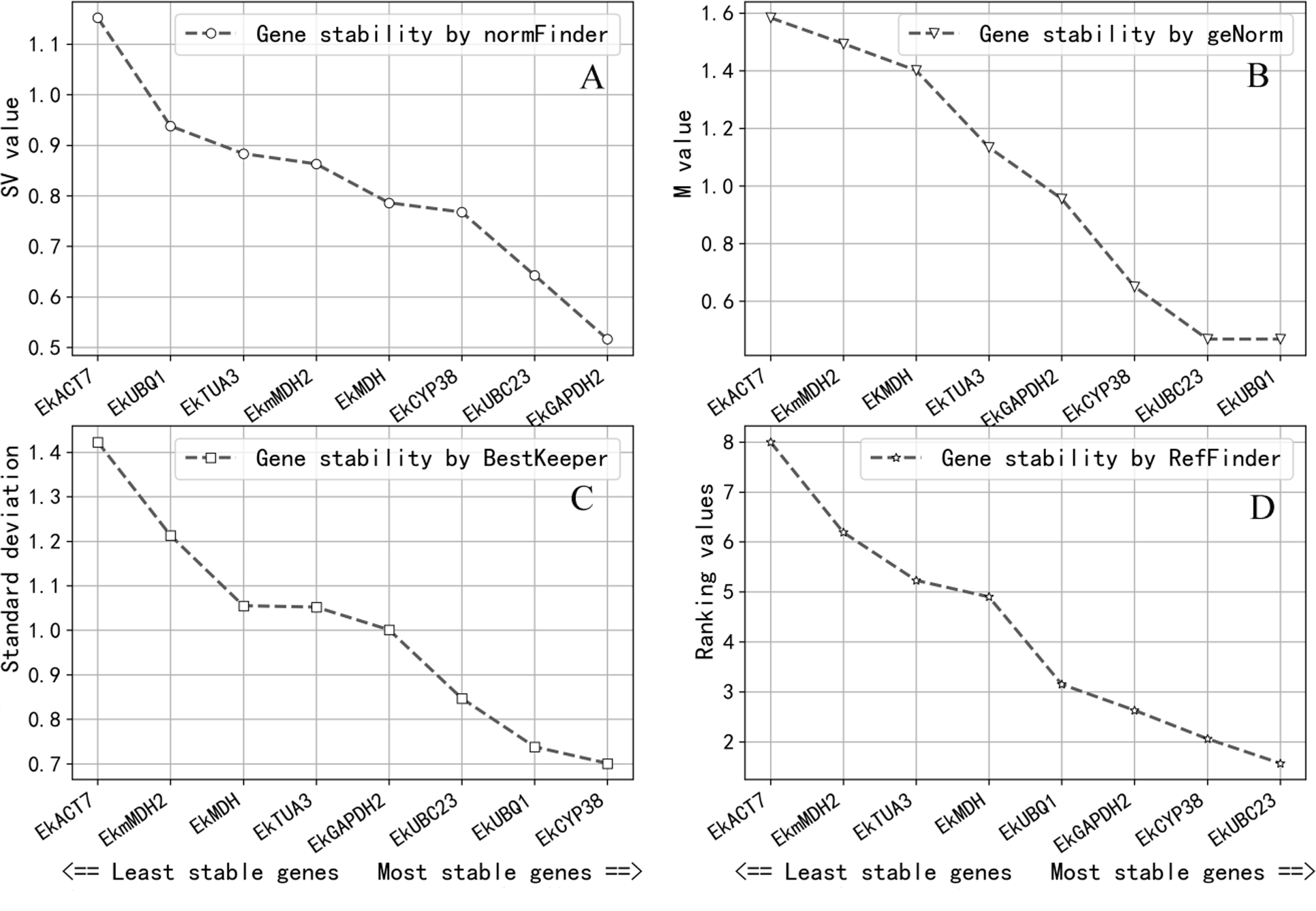{kind=link}
NormFinder software ranks all reference gene candidates based on intra- and inter-group variation and combines results into a stability value for each candidate reference gene. A better stable gene expression is indicated by a lower stability value (Andersen, Jensen & Ørntoft, 2004). The analysis results of NormFinder shown in Fig. 3B revealed that EkGAPDH2 was the most stable gene for all samples.
BestKeeper software determines the most stably expressed genes based on the standard deviation (SD). The lower the SD, the greater the reference gene expression stability will be (Pfaffl et al., 2004). As the analysis results in Fig. 3C show, the SD values of EkACT7, EkmMDH2, EkMDH, EkTUA3 and EkGAPDH2 are greater than 1 and are considered unacceptable as reference genes, according to the selection criteria of BestKeeper software. EkUBC23, EkUBQ1 and EkCYP38 are stable genes with low SD values.
ReFinder software was used to rank the stability obtained by the analysis of GeNorm, NormFinder and BestKeeper on the comprehensive index. The stability of the reference gene expression is directly related to the size of the index. According to the analysis result of ReFinder shown in Fig. 3D, the stability ranking obtained by ReFinder software is as follows: EkUBC23, EkCYP38, EkGAPDH2, EkUBQ1, EkMDH, EkTUA3, EkmMDH, and EkACT7. The results showed that the stability of EkUBC23, EkCYP38 and EkGAPDH2 was better, and the low expression reference genes (EkUBC23 and EkCYP38) were favourable for quantifying their targets, while the high expression reference gene (EkGAPDH2) was beneficial for quantifying its target.
The suitability of the reference gene
According to the results of the four algorithms (GeNorm, NormFinder, BestKeeper and ReFinder), EkUBC23, EkCYP38 and EkGAPDH2 performed more stably. EkUBC23 and EkCYP38 showed similar expression levels during fruit developmental stages. However, EkUBC23 was used to normalize the expression due to its greater stability. Relative expression levels were normalized using the low expression reference gene (EkUBC23) and high expression reference gene (EkGAPDH2). The results showed that there were some differences between the expression levels of 9 genes related to the anthocyanin synthesis pathway and transcriptome sequencing (Fig. 4). When EkUBC23 was used as the reference gene, the results had no significant difference. When EkGAPDH2 was used for normalizing low expression target genes [c57877.graph_c0( DFR), c59825.graph_c0(CHS) and c69862.graph_c1(UFGT) ], the results were different from the expression of transcriptome. However, EkGAPDH2 has better accuracy than EkUBC23 when it was used for normalizing high expression target genes [c72659.graph_c0(CHS) and c72737.graph_c0(CHI) ]. Therefore, we suggest that the stability and expression of reference genes should be considered as important selection conditions.
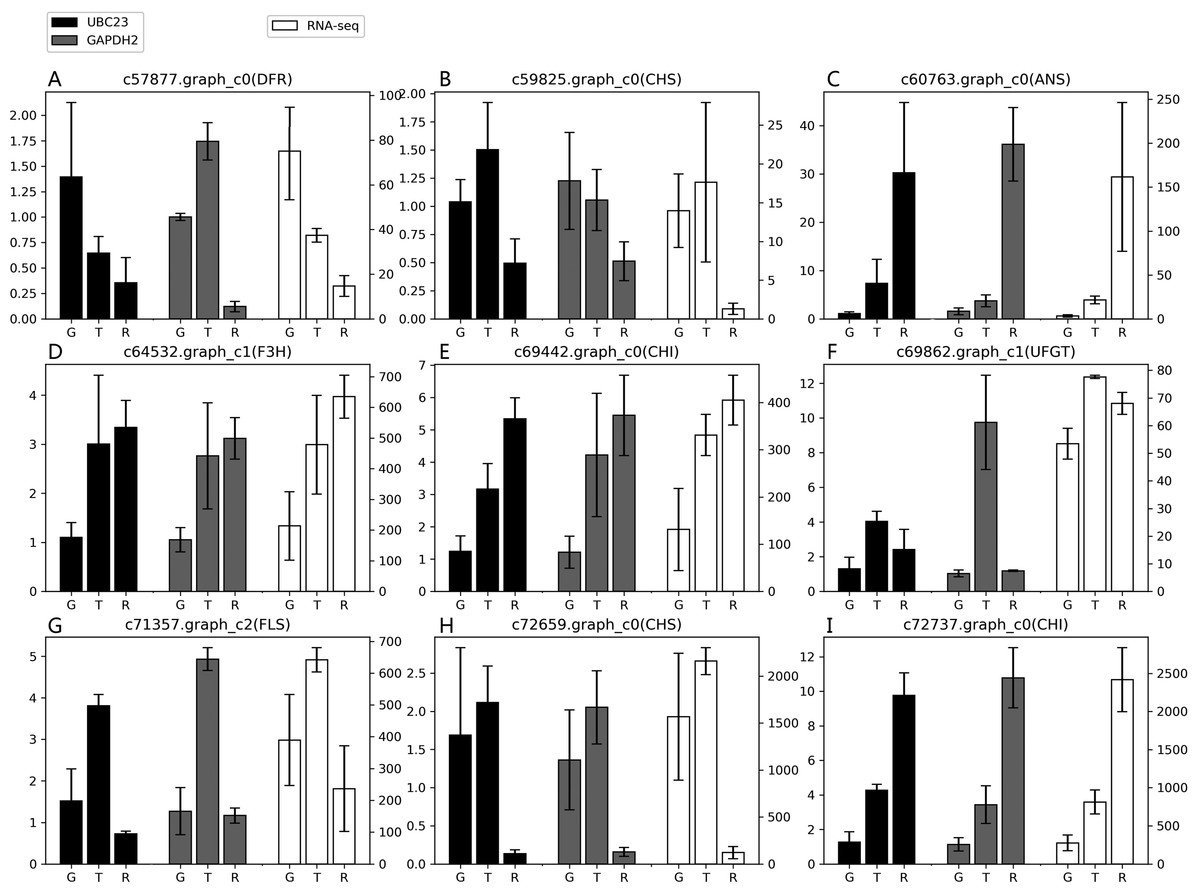
Figure 4: The suitability of reference gene.
(A–I) Genes related to the anthocyanin synthesis pathway were chosen as target genes. Those genes were normalized to UBC23 and GAPDH2, respectively. Error bars represent standard deviation from theree independent biological replicates. “G”, “T” and “R” mean green stage, turning stage and red stage in fruit, respectively.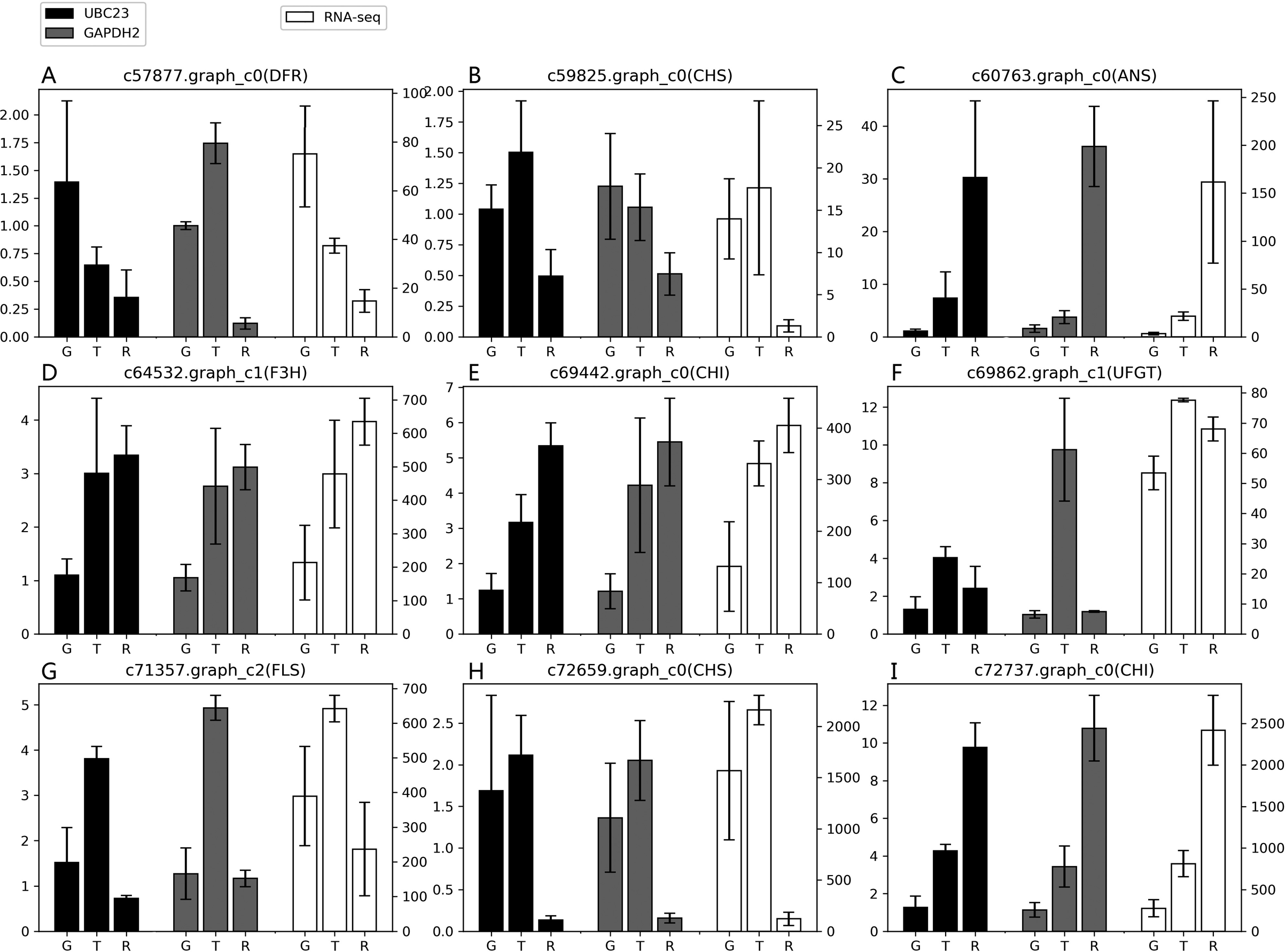{kind=link}
To ensure the reliabilty of result, We chosen EkSS3 gene related to glycometabolism pathway as target gene. Relative expression levels were normalized using two most stable reference genes (EkGAPDH2 and EkUBC23). As shown in Fig. S6, when normalized using EkGAPDH2 and EkUBC23 as reference gene respectively, the results had no significant difference. Compared with the green stage, EkSS3 expression was clearly downregulated at turning stage and red stage. The EkGAPDH2 and EkUBC23 performed well when normalizing target gene in different pathway.
Discussion
Many studies have shown that it was quicker and more efficient to screen appropriate reference genes based on transcriptome data. In addition, primers designed according to the transcriptome sequencing of the material itself are more stable than those using other materials. Selection of the reference gene based on transcriptome data has been done in many plant species, such as Corylus (Yang et al., 2017), Sedum alfredii (Sang et al., 2013) and Dendrocalamus sinicus (Guo, Chen & Yang, 2018), and so on. In the present study, we selected three stable reference genes (EkUBC23, EkCYP38 and EkGAPDH2) based on transcriptome data of E. konishii pericarp in three different developmental stages. This result validates and complements the results of reference gene screening by Liang. (Liang et al., 2018b).
The expression of reference genes was related to organ type, developmental stages and external environmental conditions. ACT was commonly used as a reference gene. For example, it was the most stable gene in studies of Glycine max (Ma et al., 2013). In contrast, we confirm that ACT is the most unstable gene in our study. This consequence is consistent with studies in Setaria viridis (Martins et al., 2016) and E. konishii by Liang et al. (2018b). UBC has been widely accepted for normalization of gene expression as a reference gene (Yuan, Wan & Yang, 2012). UBC was selected as the reference gene in the study of Prunus pseudocerasus (2 (Zhu et al., 2015). However, it was unsuitable as a reference gene for Oryza sativa (Li et al., 2008). In this study, we identified UBC as a reference gene to quantify low expression target genes. CYP played important roles in Elaeis guineensis (Yeap et al., 2014) and Malus domestica (Kumar & Singh, 2015) as reference a gene. CYP with stability and low expression is also used for quantifying low expression target genes. GAPDH, an enzyme in glycolysis, has been widely used as a reference gene in different species (Kozera & Rapacz, 2013). However, GAPDH has different performances in different species and different experimental conditions. GAPDH shows stable expression in Citrus sinensis (Wu et al., 2014) and Lycopersicon esculentum (Mascia et al., 2010), but it did not perform well in S. viridis (Martins et al., 2016) and Panicum virgatum (Jiang et al., 2014). In previous study, our colleague Liang revealed EkGSTU1 performed better than EkGAPDH2 in root, leaf, branch and seed samples (Liang et al., 2019). However, in our study, EkGAPDH2 and EkUBC23 performed well in different stages of fruit ripening. Our study expanded the scope of reference genes screening for this species. In this study, we selected EkGAPDH2 as the reference gene to quantify the high expression target genes for further study. We also comfirmed EkGAPDH2 and EkUBC23 had stable expression in other tissues by semi-quantitative RT-PCR experiment. The RT-PCR experimental method was the same as described above and the details of result were show in Fig. S7. The result showed that EkGAPDH2 and EkUBC23 had stable expression in root, branch and leaf, it will facilitate to reference genes screening for whole plant.
The genomic information of E. konishii still unclear. The fruit of Euscaphis konishii has high medicinal value,which was used as medicinal source material in China. In order to analyze the expression patterns of medicinal compound related gene in fruit of Euscaphis konishii, we selected two reliable refenrece genes for qRT-PCR normalization. The results will contribute to future studies of the gene expression in E. konishii and genetic studies related to fruit developmental stages.
Conclusions
The development of high-throughput sequencing technology provides an accurate and efficient approach to molecular biology. It plays an important role in molecular breeding and specific gene research. In this study, we construct a screening system for reference genes of E. konishii based on an RNA-seq database from different developmental pericarp samples. We selected three stable reference genes (EkUBC2 3, EkCYP38, and EkGAPDH 2) from eight candidate reference genes. Among them, EkUBC23 and EkCYP38 with low expression are suitable for quantifying low expression target genes. However, EkGAPDH2 with a high expression is favourable for quantifying high expression genes. Our study will contribute to future studies of gene expression in E. konishii and genetic studies related to fruit. The results also provided reference for neighboring species.
Supplemental Information
The gene models of eight reference genes were designed by homology comparison of Arabidopsis.
The position of primers was denoted by arrow marker symbol.
The melting curves of candidate reference genes
The Derivative Reporter (X-axis) was plotted versus the reaction temperature of qRT-PCR (Y-axis). A single peak of the melting curve in qRT-PCR were used to ensure the specificity of the primers for the candidate reference genes.
Candidate reference genes for RT-PCR amplification
PCR products on 1.2% agarose gel stained with GoldView. Lanes 1, 2, 3, 4, 5, 6, 7 and 8 were the gene products of EkACT7, EkMDH, EkmMDH2, EkUBQ1, EkUBC23, EkCYP38, EkTUA3 and EkGAPDH2, respectively.
Simplified schematic of higher plant anthocyaninbiosynthesis
The study of our lab colleague Yuan was used as a reference that publish in International journal of molecular science: Sequencing of E. konishii endocarp transcriptome points to molecular mechanisms of endocarp coloration. The blue box indicated 9 genes related to the anthocyanin synthesis pathway.
Validation of the candidate reference genes
UBC23 and GAPDH2 were used to normalized EkSS3, respectively. Error bars represent standard deviation from theree independent biological replicates. “G”, “T” and “R” mean green stage, turing stage and red stage in furit, respectively.
PCR products of EkGAPDH2 and EkUBC23 checked on 1.2% agarose gel
(A) The gene expression pattern of EkGAPDH2 in root, leaf, branch and fruit. (B) The gene expression pattern of EkUBC23 in root, leaf, branch and fruit.