
Genomic diversity of prevalent Staphylococcus epidermidis multidrug-resistant strains isolated from a Children’s Hospital in México City in an eight-years survey
- Published
- Accepted
- Received
- Academic Editor
- Mario Alberto Flores-Valdez
- Subject Areas
- Genomics, Microbiology, Infectious Diseases
- Keywords
- Genomes, Pangenome, Prophages, CRISPR, Recombination, Antibiotic resistance, Insertion sequences, Clonal structure, Staphylococcus epidermidis, prevalence
- Copyright
- © 2019 Cabrera-Contreras et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2019. Genomic diversity of prevalent Staphylococcus epidermidis multidrug-resistant strains isolated from a Children’s Hospital in México City in an eight-years survey. PeerJ 7:e8068 https://doi.org/10.7717/peerj.8068
Abstract
Staphylococcus epidermidis is a human commensal and pathogen worldwide distributed. In this work, we surveyed for multi-resistant S. epidermidis strains in eight years at a children’s health-care unit in México City. Multidrug-resistant S. epidermidis were present in all years of the study, including resistance to methicillin, beta-lactams, fluoroquinolones, and macrolides. To understand the genetic basis of antibiotic resistance and its association with virulence and gene exchange, we sequenced the genomes of 17 S. epidermidis isolates. Whole-genome nucleotide identities between all the pairs of S. epidermidis strains were about 97% to 99%. We inferred a clonal structure and eight Multilocus Sequence Types (MLSTs) in the S. epidermidis sequenced collection. The profile of virulence includes genes involved in biofilm formation and phenol-soluble modulins (PSMs). Half of the S. epidermidis analyzed lacked the ica operon for biofilm formation. Likely, they are commensal S. epidermidis strains but multi-antibiotic resistant. Uneven distribution of insertion sequences, phages, and CRISPR-Cas immunity phage systems suggest frequent horizontal gene transfer. Rates of recombination between S. epidermidis strains were more prevalent than the mutation rate and affected the whole genome. Therefore, the multidrug resistance, independently of the pathogenic traits, might explain the persistence of specific highly adapted S. epidermidis clonal lineages in nosocomial settings.
Introduction
Staphylococcus epidermidis (SE) is a typical commensal bacterium of the human skin microbiome (Byrd, Belkaid & Segre, 2018). However, some SE strains behave as pathogens colonizing surgery wounds, medical devices, and in some circumstances, they reach the human bloodstream causing severe bacteremia and potential mortality (Chessa, Ganau & Mazzarello, 2015; Otto, 2009). Children are especially prone to acquire methicillin-resistant SE strains in perinatal hospitals (Marchant et al., 2013; Villari, Sarnataro & Iacuzio, 2000). The genetic basis for pathogenicity of SE strains includes genes related to biofilm formation (adhesion), phenol soluble modulins (PSMs), and diverse Mobile Genetic Elements (MGEs) like phages, insertion sequences (ISs), and pathogenicity islands, that may be associated with the transfer of antibiotic and virulence traits (Bouchami, De Lencastre & Miragaia, 2016; Miragaia et al., 2009; Rolo et al., 2017) (Conlan et al., 2012; Kozitskaya et al., 2005; Meric et al., 2015). There is no clear genetic distinction between pathogenic and commensal non-pathogenic SE strains, even though nosocomial strains are enriched in virulence and antibiotic resistance genes (Conlan et al., 2012; Kozitskaya et al., 2005; Meric et al., 2018). It has been proposed that these genes are within the pool of accessory genome mobilized within and between species (Meric et al., 2015; Rolo et al., 2017). In this sense, the emergence of methicillin-resistant staphylococci has been linked to MGEs such as the Staphylococcal Chromosomal Cassette (SCCmec), and the Arginine Catabolic Mobile Element (ACME) that play a role in adaptation to the skin and other human surfaces (Miragaia et al., 2009; Planet et al., 2013; Rolo, Lencastre & Miragaia, 2012). Recent phylogenetic studies indicate that ACME having originated from SE and transferred horizontally to S. aureus (Barbier et al., 2011; Onishi et al., 2013; Planet et al., 2013), whereas SCCmec may have originated and diversified in different staphylococci species independently (Miragaia, 2018). Other factors, including, genomic rearrangements mediated by IS256 in pathogenic SE strains (Gu et al., 2005; Ziebuhr et al., 1999a), conjugative transfer of antibiotic resistance (Forbes & Schaberg, 1983) and the metabolic state of the staphylococci cell, may also be linked to pathogenicity (Bosi et al., 2016). Thus, pathogenicity in staphylococci may be viewed as a set of evolving genetic traits for adaptation to specific niches (Ben Zakour, Guinane & Fitzgerald, 2008; Ziebuhr et al., 1999b).
The population structure of SE is essentially clonal as determined by Multilocus Sequence Typing (MLST) (Miragaia et al., 2002; Miragaia et al., 2007; Thomas et al., 2007). Although, nosocomial SE strains show high genetic diversity among isolates from distant geographic locations (Miragaia et al., 2007) several clonal complexes are disseminated globally including the ST2, ST5, and ST23 which indeed are the most frequently found in clinical environments (Lee et al., 2018; Miragaia et al., 2007). Besides the clonal structure, recombination has been estimated to occur two times more frequently than mutation (Miragaia et al., 2007). Genomic analysis had calculated that about 40% of the core genes of SE had undergone recombination (Meric et al., 2015). It suggests that recombination might be a general property of SE for rapid evolution in clinical settings but conserving high linkage disequilibrium between alleles. In this case, recombination might act as a cohesive force, maintaining the clonal population structure but allowing clones to diverge.
Multidrug resistance is a significant concern in most health care settings worldwide because of the difficulties associated with clinical treatment and the potential of dissemination. It has been reported that the prevalent SE clonal lineages ST5, ST12, and ST23 often display high resistance toward most of the antibiotic classes (Martínez-Meléndez et al., 2016; Widerstrom et al., 2012). Furthermore, there is evidence of increasing of rifampicin resistance over time in clinical SE isolates that belong to ST2 and ST23 from the USA, Australia, and some European countries (Lee et al., 2018). Mutations in the rpoB gene conferring rifampicin resistance have emerged independently, suggesting the view that a limited number, well adapted multi-resistant clonal SE lineages, prevail in clinical settings (Lee et al., 2018; Martínez-Meléndez et al., 2016). Reports on the profiles of antibiotic multi-resistance changes over time are scarce (Lee et al., 2018). Surveillance of rifampicin resistance in a study involving 24 countries and 96 institutions suggests annual local variations for this antibiotic (Lee et al., 2018). Therefore, the correct identification of pathogenic SE strains and its drug resistance profile will contribute to prevent and treat these bacterial infections in the clinic.
In this work, we evaluated the prevalence of SE in comparison with other staphylococci species isolated in a single hospital in México City during eight years period. Then, we aimed to assess the antibiotic resistance profiles of SE isolates, and through genomics, to know the genome structure of a selected set of antibiotic multi-resistant SE strains. In this context, we provide a genome-wide measure of recombination, to shed light on the mechanisms of SE genetic diversification and the evolution of multidrug-resistance in local hospital settings.
Materials & Methods
Ethical considerations
This study was carried out following the recommendations of the ethics review committee of the Facultad de Medicina-UNAM. The SE strains used in this work were obtained by a donation from the microbiology collection of the Instituto Nacional de Perinatología “Isidro Espinosa de los Reyes” (INPer). A consent form was not required. Original identification keys and clinical data concerning the isolates are maintained under the control of INPer. In the present work, new strain identifiers were assigned to the INPer strains. Authors do not have access in any form to the specific clinical information of strains and patients.
Clinical isolation of staphylococci and characterization
Staphylococci strains were recovered from primus isolates taken from patients at INPer. They were conserved frozen (−80 °C in an ILSHIN ultra-freezer). Characterization of staphylococci was carried out by streaking the cultures on tryptic soy agar plates (BD Diagnostic Systems, Germany). Plates were incubated for 18–24 h at 37 ° C to obtain isolated colonies. Individual colonies were used to inoculate tryptic soy broth and grown overnight after which genomic DNA was extracted (see below). Identification of Staphylococcus species was carried out using the VITEK®2 equipment (bioMérieux SA, 376 Chemin de l‘ Ome. France). Briefly, staphylococci colonies were resuspended in 0.45% saline solution and adjusted to a turbidity of 0.5 in a McFarland nephelometer. The bacterial suspension was transferred into the VITEK®2 GP ID card testing 64 biochemical properties for gram-positive bacteria, and eight specific tests for SE species (phosphatase and urease production, growth in 6% NaCl, resistance to novobiocin and polymyxin B, arginine hydrolysis, catalase, D-mannitol fermentation). Additional growth characteristics, biochemical (D-mannitol fermentation, coagulase, and catalase), and molecular tests (PCR detection of coa and mecA; see below) were performed at the arrival of the staphylococci collection to the Pathogenicity Laboratory at Faculty of Medicine, UNAM.
Molecular tests
To confirm the presence/absence of the coagulase gene (coa), and the methicillin-coding gene (mecA) in SE and S. aureus strains, we did a duplex PCR according to Hookey (Hookey et al., 1999). In brief, a PCR amplification of a 177 bp fragment of the coa gene, with primers coa-forward 5′-AACTTGAAAT-AAAACCACAAGG-3′, and coa-reverse 5′TACCTGTACCAGCATCTCTA-3′. In the same reaction, a 458 bp fragment of the mecA gene was amplified using primers mecA-forward 5′-ATGGCAAAGATATTCAACTAAC-3′and mecA-reverse 5′-GAGTGCTACTCTAGCAAAGA-3′. PCR reactions were performed on a BIO-RAD C1000 Thermal Cycler (BIO-RAD, USA). The amplification conditions were as follows: 94 °C for 10 min and 30 cycles at 94 °C for 30 s, 56.2 °C for 30 s and 72 °C for 30 s and a final amplification at 72 °C for 10 min; PCR products were separated by electrophoresis in a 1% agarose gel stained with ethidium bromide and visualized with UV light.
Antibiotic resistance
The antibiotypes for 17 antibiotics were determined using automatic VITEK®2 equipment with panel card AST-GP577 (VITEK®2 Biomerieux, Francia). Resistance and susceptibility patterns to 17 antimicrobials were determined according to the M100S Performance Standards for Antimicrobial Susceptibility Testing (Clinical and Laboratory Standards Institute C, 2019). The following antibiotics were tested using the respective reference MIC: Amoxicillin/Clavulanic Acid: ≤4/2 µg/mL, Ampicillin: >8 µg/mL, Cefazolin: ≤2 µg/mL, Ciprofloxacin: ≥4 µg/mL, Clindamycin: >4 µg/mL, Erythromycin: >8 µg/mL, Gentamicin: >16 µg/mL, Imipenem: 8 µg/mL, Levofloxacin: ≥4 µg/mL, Linezolid: ≥8 µg/mL, Oxacillin: ≥0.5 µg/mL, Penicillin: ≥0.25 µg/mL, Rifampicin: ≥4 µg/mL, Synercid: > µg/mL, Tetracycline: ≥16 µg/mL, Trimethoprim/Sulfamethoxazole: >4/76 µg/mL, and Vancomycin: ≥32 µg/mL.
Methicillin resistance was evaluated by the disc diffusion method for cefoxitin on Mueller-Hinton agar plates (OXOID Ltd, Basingstoke, Hampshire, England), and with the plate method in Mueller-Hinton agar plates supplemented with 6 µg/ml of oxacillin, according to the standards from the Center for Disease Control and Prevention, USA (Centers for Disease Control and Prevention C, 2019)
Genome sequencing
Genomic DNA was isolated by using the Lysostaphin-Lysozyme method of Hookey (Hookey et al., 1999), only modifying the lysis step. Briefly, bacterial cells were resuspended in 250 µl of lysis solution (lysozyme 50 mg/ml; lysostaphyn 1 mg/ml in 25 nM Tris-HCl- 10 mM EDTA; all reagents from SIGMA-ALDRICH, St. Louis MO USA), and incubated at 37 °C by 90 min. The sample was then warmed up at 95 °C for 10 min and centrifuged at 8,000× g, after which the sample was processed as indicated in the method.
DNA integrity and purity were assessed by agarose gel electrophoresis and spectrophotometry (NanoDrop 2000; ThermoFisher, Waltham, MA, USA); final concentrations were assessed using the Qubit high sensitivity dsDNA assay (ThermoFisher). Libraries were generated with Nextera XT DNA library preparation kit (Illumina, San Diego, CA, USA), according to the manufacturer’s protocol, using 1 ng of DNA. Libraries quality were evaluated on an Agilent 4200 TapeStation system (Santa Clara, CA, USA) and sequenced using a NextSeq 500/550 mid output kit (2 × 150 bp) (Illumina), at an estimated depth of coverage of 100x.
Genome assembly and annotation
Assemblies were made with Spades 3.6.0 in genomes sequenced in high coverage (50x to 100x). Genome annotations were obtained from the PATRIC server (Wattam et al., 2018) (https://www.patricbrc.org/), through the automated bioinformatic method RAST (Rapid Annotation Subsystem Technologies) (Antonopoulos et al., 2019). Gene annotation of antibiotic resistance and virulence-related genes were obtained from the section of Special Genes of PATRIC. Annotations for virulome were supported with other sources of information such as PATRIC VF, VFDB, and Victors. For the prediction of antibiotic resistance genes, the PATRIC server was used to search specialty genes using the databases CARD (Comprehensive Antibiotic Resistance Database) and NDARO (National Database of Antibiotic Resistant Organism) (Davis et al., 2016).
Genome comparisons and pangenome modeling
Average Nucleotide Identity by MUMmer (ANIm) and Genomic Coverage (G cov) were calculated with the JSpecies program, with MUMmer used as a pairwise comparison tool for pairs of SE genomes (Richter et al., 2016). The pangenome was modeled using the GET_HOMOLOGUES package and its default values (Contreras-Moreira & Vinuesa, 2013). Briefly, groups of orthologues proteins were computed using the Ortho-MCL integrated into GET_HOMOLOGUES. Accessory genes (unique genes and genes present in at least in two genomes but not in all genomes), were obtained from the cloud, shell, and soft-core pangenome components according to GET_HOMOLOGUES (Contreras-Moreira & Vinuesa, 2013). The distribution of accessory genes in SE genomes was illustrated with the heatmap.2 function of the R’s ggplot2 package.
Phylogeny
A consensus core genome calculated from the pangenome model of 29 SE genomes was obtained with GET_HOMOLOGUES (Contreras-Moreira & Vinuesa, 2013). The resulting 1575 core protein clusters were subject to multiple alignments with MUSCLE (Edgar, 2004), gaps removed with TrimAl v2.1, and were concatenated using homemade Perl scripts (Capella-Gutiérrez & Gavaldón, 2013). Phylogenetic trees were constructed by the maximum likelihood (ML) method based on the substitution matrix of WAG (Whelan and Goldman model), with 1000 bootstrap replicates, using RaxML program (Stamatakis, 2015). To draw and edit the phylogenetic trees, we used the iTool program (Letunic & Bork, 2019).
Recombination
The inference of recombination was performed with ClonalFrameML (Didelot & Wilson, 2015). First, we ran GET-HOMOLOGUES to obtain the common protein clusters encoded by the genomes of each set of SE strains (Contreras-Moreira & Vinuesa, 2013). Second, they were converted to nucleotide sequences and concatenated using homemade Perl scripts. Multiple alignments were made as described above for phylogeny construction. Third, a RAxML (Stamatakis, 2015) tree was done to obtain the Newick format and the transition/transversion parameter κ for running ClonalFrameML under default parameters (Didelot & Wilson, 2015). Z-score statistics were obtained for all the sets of SE genomes and p-values using the web application Z Score Calculator (https://www.socscistatistics.com/tests/ztest/zscorecalculator.aspx). BoxPlots were performed with the R ggplot2 system.
Mobile elements identification
Prophages were identified with PHAST (Arndt et al., 2019). Only predictions ranked as “intact prophages” were considered for analysis. IS, CRISPR-Cas elements and spacer sequences were obtained from PATRIC annotations (Antonopoulos et al., 2019). Then, IS were classified into families by BLASTx comparison with the ISfinder (Siguier et al., 2006). The phage identity of the spacers within the CRISPR-Cas elements was determined by BLASTn comparisons with the NCBI virus database. Only identical matches with a phage sequence in the database were recorded (Páez-Espino et al., 2016).
Identification and classification of SCC mec and ACME proteins
All the predicted proteins that compose the 12 types of SCCmec and the six ACME types were downloaded from GenBank (Table S1) and compiled in a local protein database (SE-17db). Then, all encoded proteins of the 17 SE genomes were used as a query in BlastP similarity searches with the SE-17db. Best-blast hits with a minimum similarity of 80% and 60% of coverture over the length of the smaller protein were taken as evidence of homology with the respective SCCmec or ACME type. To classify the SCCmec types, we used the phylogenies of the recombinases ccr A, B, and C, performed according to the method described above. ACME types were classified by the presence/absence of the genes of the operons arg, opp, ars, and kpd according to a reference table (Table S2).
GenBank accession numbers
S. epidermidis of the INPer collection used in this work were uploaded in GenBank with the following Biosample identifiers: SAMN11086744, SAMN11086745, SAMN11086746, SAMN11086747, SAMN11086748, SAMN11086749, SAMN11086750, SAMN11086751, SAMN11086752, SAMN11086753, SAMN11086754, SAMN11086755, SAMN11086756, SAMN11086757, SAMN11086758, SAMN11086759, SAMN11086760. The accession numbers for the genomes of reference S. epidermidis strains are listed in Table S3.
Results
Survey of antibiotic multidrug-resistant intrahospital staphylococci in eight years period
We studied the incidence of staphylococci species and strains multi-resistant to antibiotics in the INPer children hospital from 2006 to 2013. A total of 822 staphylococci strains were recovered from distinct infection sites of newborns and adults. Staphylococcus species were identified using the VITEK®2 system and standard clinical methods, including tests for biofilm formation, coagulase reaction, presence of coa and mecA gene, and resistance to 17 antibiotics (see methods). The analysis showed that SE strains were the most abundant (573 strains), followed by S. aureus (146 strains), and other coagulase-negative Staphylococcus (CoNS) species in low proportion (81 strains) (Fig. S1). The SE group was composed mainly of strains recovered from blood samples and catheters (Fig. S1).
The number of antibiotic multi-resistant SE strains per year showed similar profiles through the studied period (Figs. 1A–1H). Methicillin resistance was found in about 80% of the SE isolates. SE strains multi-resistant to nine antibiotics were the most frequently found throughout the eight years. In contrast, S. aureus strains showed resistance to about four antibiotics as the most common profile (Fig. S2). Similarly, the resistance to each antibiotic remained without change, during the eight-year study (Figs. 1I–1P). These results suggest the persistence of a multidrug-resistant clone or few clones of SE at the hospital during the eight years, or frequent horizontal exchange of genes encoding antibiotic resistance between SE clones. To study these alternatives, we choose one or more SE strains per year to totalize 17 clinical SE coagulase-negative (CoNS) strains (Fig. 1). These particular strains came from nosocomial infections of 14 newborns and three adults, isolated from three different infection sites: blood (8 strains), catheters (7 strains), cerebrospinal fluid (1 strain), and soft tissue (1 strain) (Table 1).
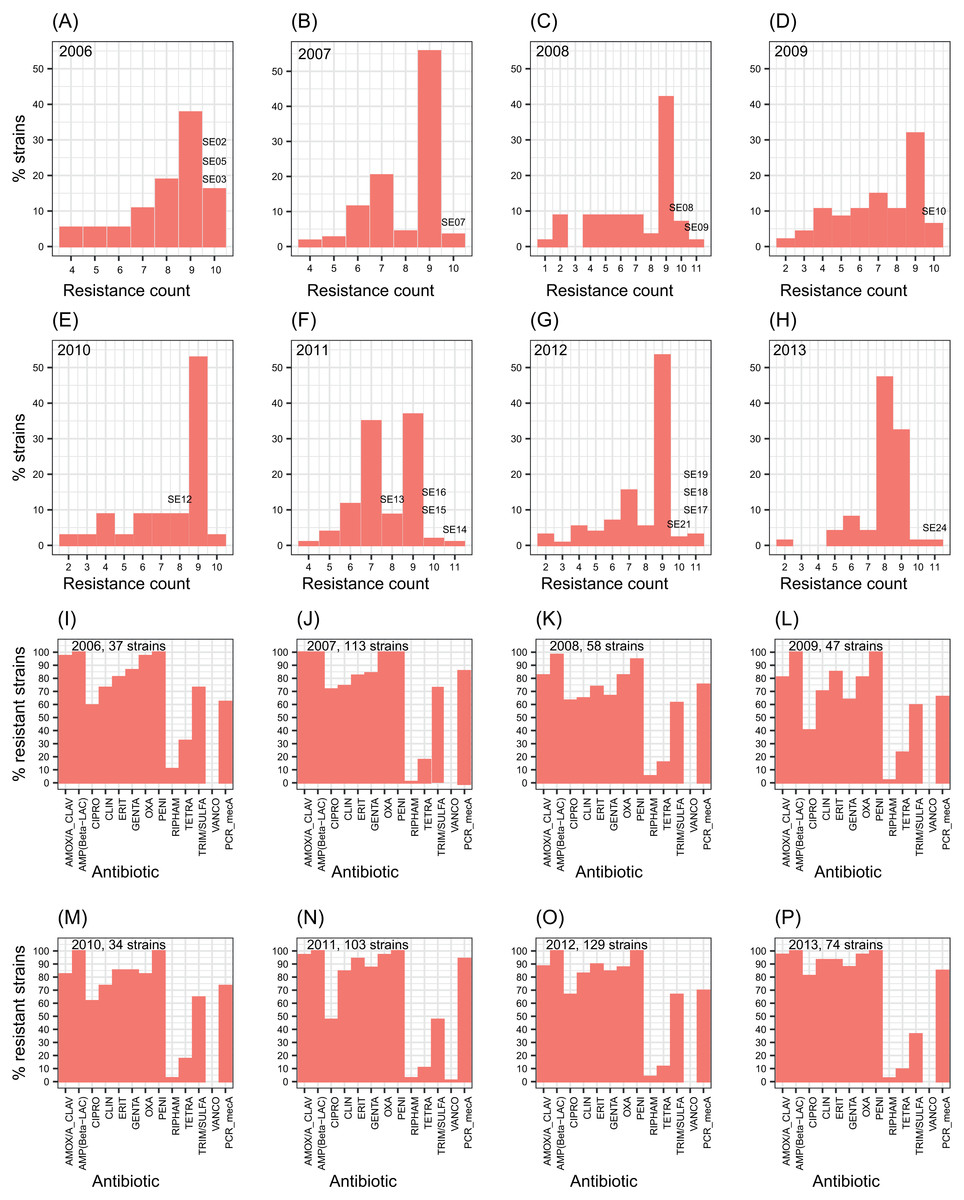
Figure 1: Profiles of S. epidermidis antibiotic multi-resistance in an eight years period.
(A–H) percentage of SE strains respect the count of antibiotic resistances by year. (I–O) number of strains resistant to each one of twelve antibiotics by year. Percentage of mecA strains are shown in the last columns of each plot. The SE strains selected for genome analysis are indicated over the bars.{kind=link}
| ccrrecombinases inSCCmectype | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Straina | Originb | STc | ST profiled | ica | CRISPR-Cas | Prophages | IS256e | mecA | ccrA | ccrB | ccrCf | ||
| S02 | N-Catheter | 23 | 7,1,2,1,3,3,1 | + | 1 | – | 1 (1) | + | IV | – | IV | – | IV |
| S03 | N-Catheter | 89 | 1,1,2,1,2,1,1 | – | 0 | + | 1 (1) | + | IV | – | IV | – | – |
| S05 | N-Catheter | 23 | 7,1,2,1,3,3,1 | + | 1 | – | 1 (1) | + | IV | – | IV | – | IV |
| S07 | A-STA | 59 | 2,1,1,1,2,1,1 | – | 0 | + | – | + | IV | VIII | IV | VIII | – |
| S08 | N-Blood | 23 | 7,1,2,1,3,3,1 | + | 1 | – | 1 (1) | + | IV | – | IV | – | – |
| S09 | A-Blood | 59 | 2,1,1,1,2,1,1 | – | 1 | – | – | + | IV | VIII | IV | VIII | – |
| S10 | A-Blood | U-ST | 65,48,5,5,8,5,11 | – | 0 | – | – | – | – | – | – | – | – |
| S12 | N-Blood | 5 | 1,1,1,2,2,1,1 | – | 3 | + | 1 (1) | + | IV | – | IV | – | IV |
| S13 | N-Blood | 81 | 2,17,1,1,2,1,1 | – | 1 | – | 1(0) | + | IV | VIII | IV | VIII | NA |
| S14 | N-Blood | 2 | 7,1,2,2,4,1,1 | + | 0 | + | 2 (1) | + | IV | – | IV | – | – |
| S15 | N-Catheter | 5 | 1,1,1,2,2,1,1 | – | 0 | + | 1 (1) | + | IV | – | IV | – | NA |
| S16 | N-Blood | 2 | 7,1,2,2,4,1,1 | + | 0 | + | 1 (1) | – | – | – | – | – | – |
| S17 | N-Catheter | 2 | 7,1,2,2,4,1,1 | + | 0 | – | 2 (1) | + | IV | – | IV | – | – |
| S18 | N-Blood | 2 | 7,1,2,2,4,1,1 | + | 0 | + | 2 (1) | + | IV | – | IV | – | – |
| S19 | N-Catheter | 2 | 7,1,2,2,4,1,1 | + | 0 | – | 2 (1) | + | IV | – | IV | – | – |
| S21 | N-Catheter | 35 | 2,1,2,2,4,1,1 | + | 1 | – | – | + | – | – | – | – | – |
| S24 | N-CSF | 5 | 1,1,1,2,2,1,1 | – | 3 | + | – | + | IV | – | IV | – | – |
Notes:
Broad gene catalog from draft genomes
We obtained the whole genome sequence of 17 nosocomial SE strains (see ‘Material & Methods’). After testing different parameters with the assembler programs Velvet and Spades (Bankevich et al., 2012; Zerbino, 2010), we got draft genomes assemblies consisting of about 92 up to 432 contigs with a 60–70×average sequence coverture per genome (Table S4). To assert that the 17 assemblies represent a substantial part of the SE genomes, we compared the total genome length, and the number and length of the predicted ORFs, with 11 complete genomes of SE downloaded from GenBank (Table S5). There were no differences between the genome length of the SE INPer genomes and the complete genomes from the GenBank (unpaired t-test = 2.33; p-value = 0.022), in ORFs number (unpaired t-test = −1.68; p-value = 0.117), or ORFs length (unpaired t-test = 2.60; p-value = 0.014) (Table S5). Therefore, we can conclude that the SE INPer genome sequences obtained here provide a broad catalog of genes per genome useful for comparative genomics.
Genomic, pangenomic, and phylogenetic relationships among SE isolates
To define the genomic similarity between the 17 clinical SE strains, we performed whole-genome pairwise nucleotide identity estimates (Average Nucleotide Identity by Mummer, ANIm) (Richter et al., 2016). The 17 SE showed high genomic ANIm values of about 99%, covering more than 90% of the genome length (Fig. S3). One exception is strain S10, which showed ANIm values of about 97% concerning the rest of the strains.
Besides the genomic identity between SE isolates, we wanted to investigate the extent of their genetic variability by performing a pangenome model. To this end, the SE collection was complemented by the inclusion of 12 complete genomes of SE strains downloaded from GenBank (Table S3, NCBI reference complete genomes). The model obtained using the GET_HOMOLOGUES software package (Contreras-Moreira & Vinuesa, 2013), indicated an open pangenome (Fig. S4). The core genome component for the 29 SE strains was predicted to consist of about 1,575 gene clusters, whereas the sum of genes unevenly distributed in the 29 SE genomes (accessory component) contains 4,360 gene clusters.
To know the phylogenetic relationship of the SE from INPer in the context of reference SE strains, we did an un-rooted ML phylogenetic tree using the predicted 1,575 concatenated core proteins (Fig. 2). There were three clades separated by the largest branches in the tree that comprise most of the SE local strains, and one or more SE strains isolated worldwide (Fig. 2, clades B, C, D). The clade marked as D in the tree consisted of two groups, one of which includes only reference SE strains, whereas the other had most of the SE strains of the collections studied. Strain S10, the most different strain by ANIm in the SE collection, was grouped in the clade A with the commensal strains SE ATCC12228 (2) and SE 14.1.R1 isolated from USA and Denmark.
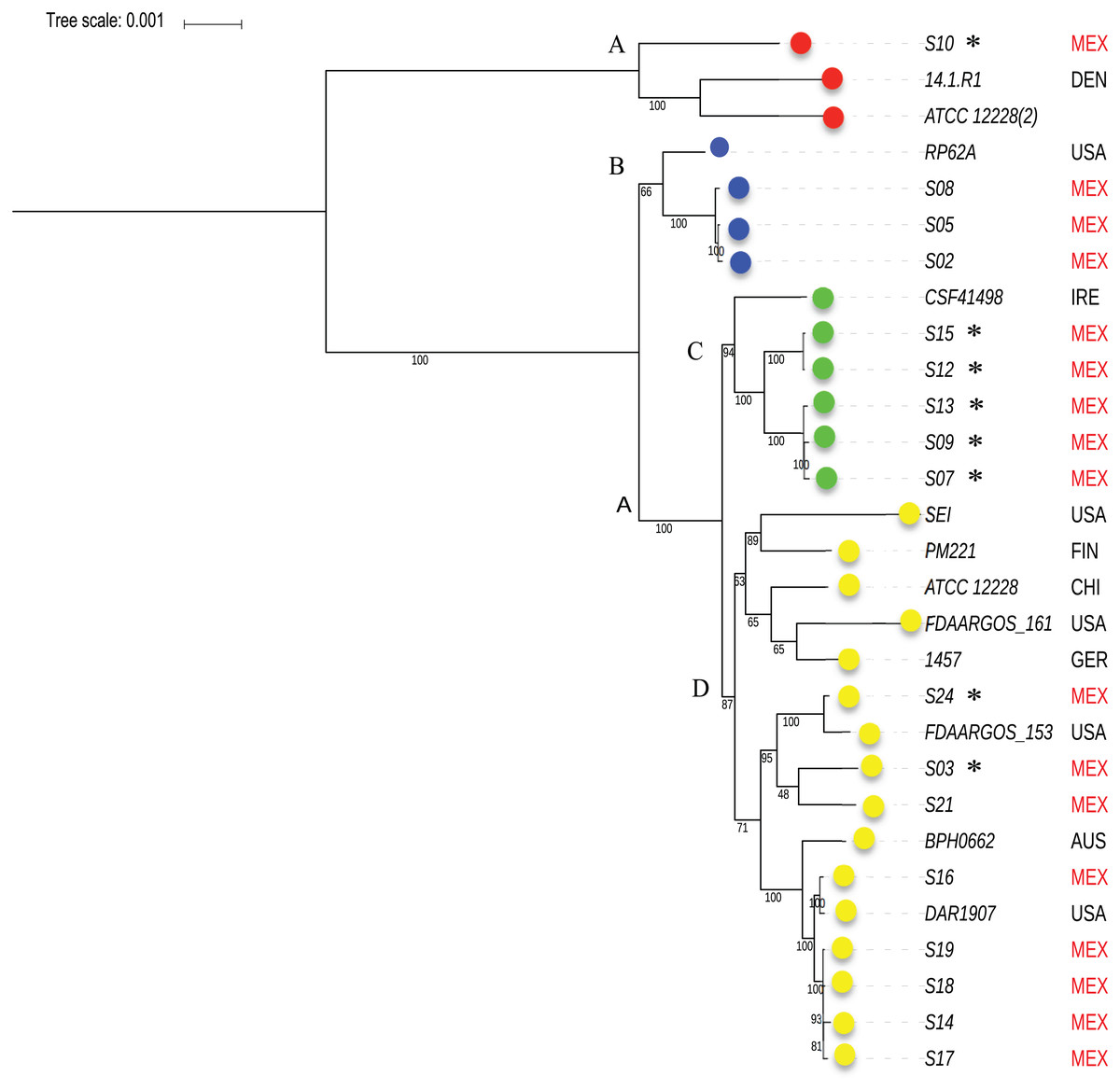
Figure 2: Phylogenetic relationships of S. epidermidis from the Children’s Hospital (INPer) and S. epidermidis selected from GenBank.
Phylogenetic relationships of S. epidermidis from the Children‘s Hospital (INPer) and S. epidermidis selected from GenBank. The ML tree consists of four main clades defined by the longest branches (A to D). They are indicated with color dots: red, clade A; blue, clade B; green, clade C; yellow, clade D. The tree was constructed with 1,575 common core proteins using RaxML program as described in methods. The results of bootstrap performed with 1,000 replicates are indicated in the branches. Acronyms specify the isolation site of the SE strains: MEX, México; USA, United States of America; FIN, Finland; GER, Germany; DEN, Denmark; IRE, Ireland; CHI, China; AUS, Australia. SE INPer strains are denoted in red. Asterisks indicate ica− strains.{kind=link}
The distribution of accessory genes in individual SE strains coincided with the phylogenetic clades. The SE strains grouped in three phylogenetic clades are closely related by their similitude in the gene presence/absence profile (Fig. 3). Despite the high identity of the SE strains, there is still considerable individual variation that may account for adaptations to local milieu.
Clonal structure
To investigate to which clonal ST complex the SE strains belong, we looked for the seven proteins of the S. epidermidis MLST scheme, and compared them with their respective alleles in the Staphylococcus epidermidis MLST database (https://pubmlst.org/sepidermidis/; Table 1; see methods) (Feil et al., 2004; Thomas et al., 2007). The analysis showed a total of 8 different STs; seven of them already recorded in the database. The S10 strain had an unassigned ST in the database and only differed by a single amino acid substitution in the YqiL protein (L370C). The ST2, ST5, and ST23 are worldwide distributed and are the most represented in our sample (Lee et al., 2018; Miragaia et al., 2007). In agreement with global data, ST35, ST59, ST81, ST89 are less frequently represented. The clonal relationships among STs determined by eBURST, indicate that founder clones are ST2 and ST5, whereas the other four STs (ST 59, 81, 89 and 23), are peripheral clones mostly related to each than to the primary founder clones (Fig. S5).
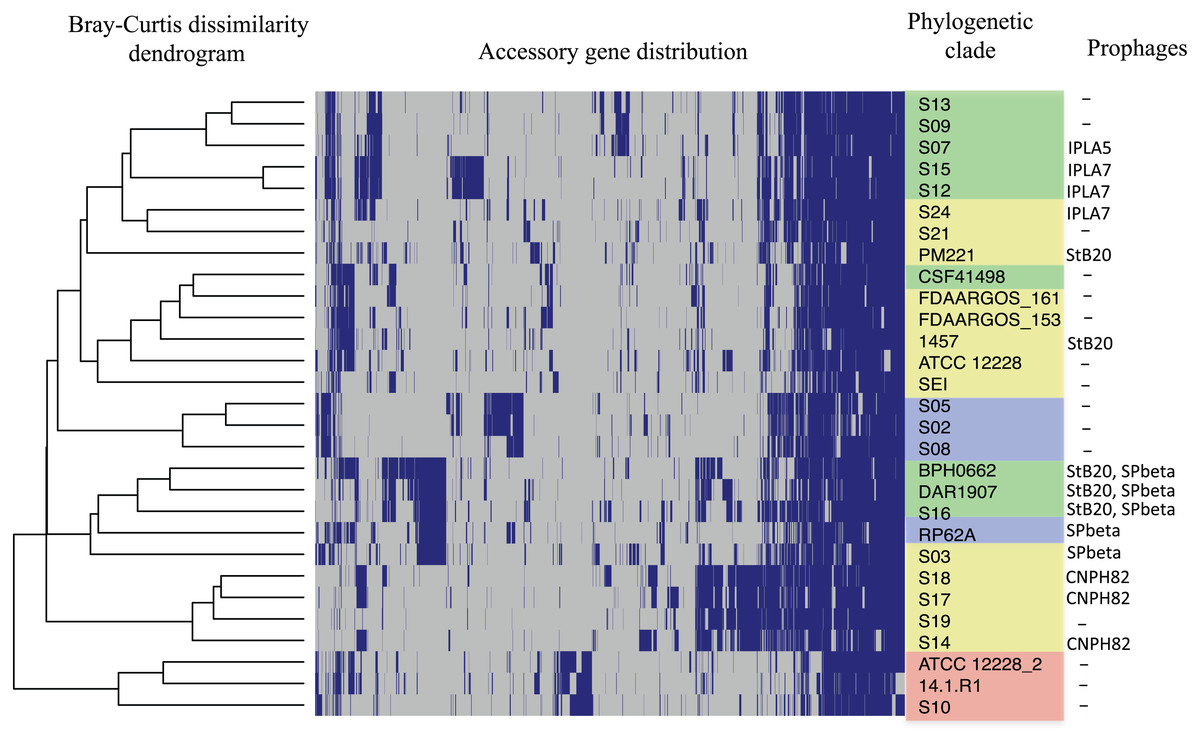
Figure 3: Distribution of accessory genes in the S. epidermidis genomes.
The heat-map profile was performed with the ggplot2 function in R using the Bray-Curtiss dissimilarity matrix. The Bray–Curtiss dendrogram is indicated at the left. The heat-map at the middle indicates gene presence (blue color); empty cells represent the absence of genes. S. epidermidis strains are shown at the left with the same colors of the clades in the core proteins phylogeny (Fig. 1): red, clade A; blue, clade B; green, clade C; yellow, clade D. In the last column. the presence/absence of genomic regions similar to known prophages (Table S5) is indicated.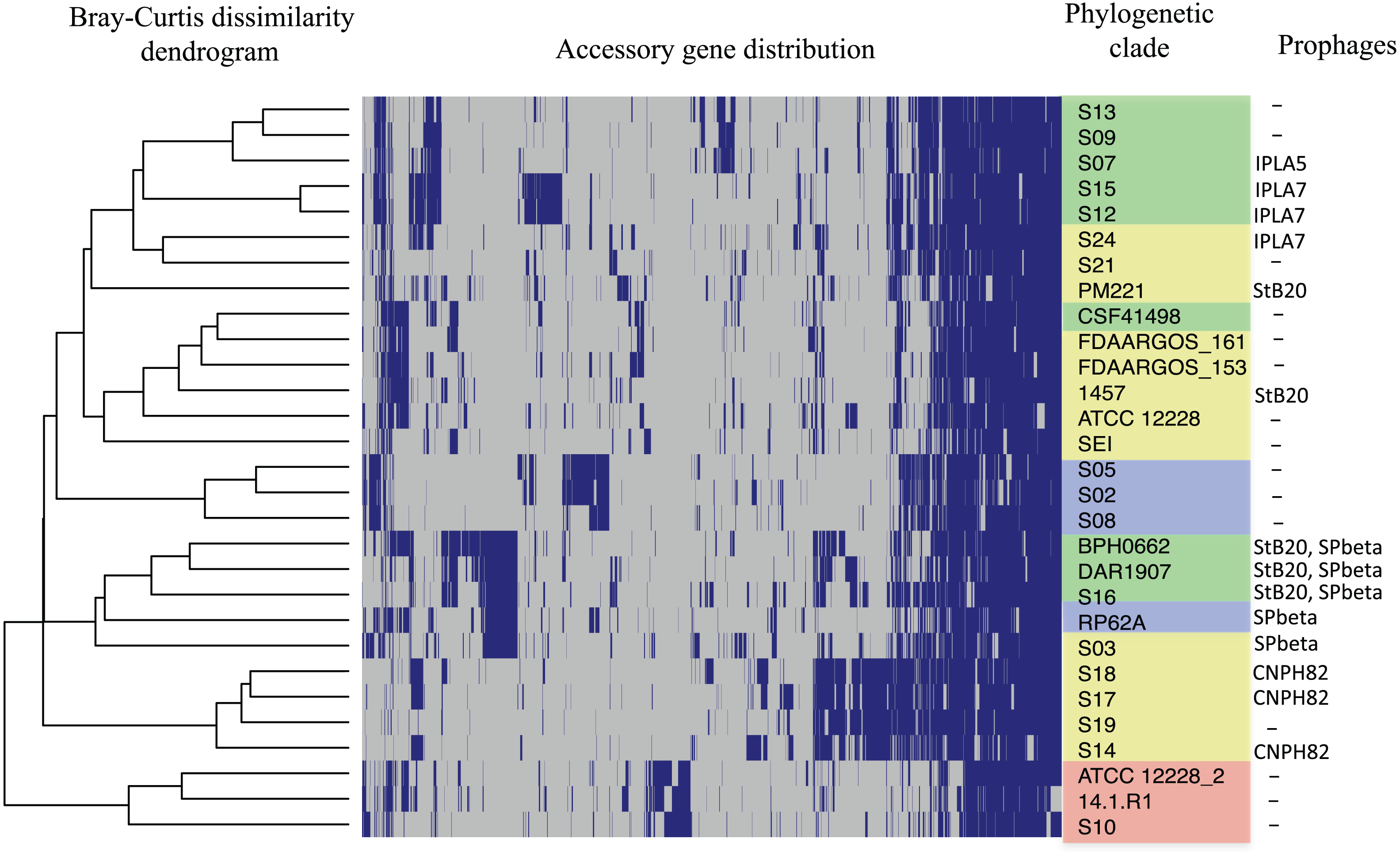{kind=link}
Virulence genes
Several known virulence genes of SE are shared by commensal and pathogenic strains (Otto, 2009). Among them, we found the cluster icaADBCR (biofilm formation) in nine out of the 17 SE genomes analyzed, except for icaC, absent in strain S05 (Table 1). The phenol-soluble modulins (PSMs), involved in inflammatory response and lysis of leukocytes were present in the genomes of all SE strains (Peschel & Otto, 2013). The novel ESAT 6 (esaAB, essABC, and esxAB genes) secretion system implicated in immune system evasion and neutrophil elimination was only present in S10 strain (Burts et al., 2005; Dai et al., 2017; Wang et al., 2016).
Frequently, SE isolates contain the genomic island ACME, composed by arginine deaminase catabolic (arg) and an oligopeptide permease operons (opp) (Miragaia et al., 2009). ACME has been associated with successful adaptation to the skin and mucosal surfaces outcompeting other related bacteria (Planet et al., 2013). The diversity of ACME has been organized in six distinct types according to their gene composition (Table S2 and references therein). We looked for the presence of ACME genes in the 17 SE genomes of INPer collection. By means of BlastP comparisons we identified genes belonging to the ACME Type I (arg+ opp+) in genomes of SE strains S03, S17, and S21, and the ACME Type V that contain the arg+ opp+, and ars+ (arsenate resistance operon), and kpd+, a potassium transporting operon in S24 (Centers for Disease Control and Prevention C, 2019; O’Connor, McManus & Coleman, 2018) (Table S2). Even though some other SE strains present genes of the arg and ars operon, they lacked the genes of the opp operon; therefore, they were not assigned to ACME known types.
Antibiotic resistance genotype and phenotype
The SE strains were tested for their susceptibility to methicillin and other β-lactams antibiotics as described in methods. All the 17 SE strains have the β-lactamase gene (blaZ) and their regulators (blaR and I), which are probably responsible for the broad resistance spectrum to penicillin, carbapenems, and cephalosporins determined by the VITEK®2 system (Table 2). Methicillin resistance was evaluated by the disc diffusion method for cefoxitin and corroborated by the presence of mecA in the genomes (Table 1 and Table 2). 12 out of 17 SE strains showed agreement between the cefoxitin resistance phenotype and the mecA genotype. Although strains S03, S05, S14, S15, and S21, have the mecA gene in the genome, they were scored as susceptible for cefoxitin (disc diffusion ≥ 25 millimeters). To support that the 15 mecA positive strains are Methicillin-Resistance SE (MRSE) strains, we evaluated their resistance to oxacillin. Commonly, the Methicillin-Resistance S. aureus (MRSA) strains are evaluated by the resistance to oxacillin (Centers for Disease Control and Prevention C, 2019). In the SE collection, only the S07 strain was susceptible to oxacillin (6 µg/ml) in plate assays, despite the detection of mecA in the genome. Altogether, these results indicate that most of the SE strains (15) studied here are MRSE, identified by the presence of the mecA gene; any phenotypic inconsistencies could be due to lack of expression of the mecA, heteroresistance, or the limitation of the current antibiotic susceptibility testing methods (Band et al., 2019; Harrison et al., 2019; Nicoloff et al., 2019).
| S. epidermidisstrains | Genotype | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Antibiotic | Class | S02 | S03 | S05 | S07 | S08 | S09 | S10* | S12 | S13 | S14 | S15 | S16* | S17 | S18 | S19 | S21 | S24 | Genes probably involved |
| Ampicillin | Penicillin | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | blaZ (17), mecA (15) |
| Penicillin | Penicillin | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | blaZ (17), mecA (15) |
| Amoxicillin | Penicillin | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | blaZ (17), mecA (15) |
| Oxacillin | Penicillin | + | + | + | – | + | + | – | + | + | + | + | – | + | + | + | + | + | blaZ (17), mecA (15) |
| Cefoxitin | Penicillin | – | – | + | – | + | + | – | + | + | – | – | – | + | + | + | – | + | blaZ (17), mecA (15) |
| Imipenem | Carbapenem | + | + | + | + | + | + | + | + | + | + | + | + | blaZ (17), mecA (15) | |||||
| Cefazolin | Cefalosphorin | + | + | + | + | + | + | + | + | + | + | + | + | blaZ (17), mecA (15) | |||||
| Ciprofloxacin | Quinolone | + | – | + | – | + | + | + | – | – | + | + | + | + | + | + | + | + | gyrA S84F (10) |
| Levofloxacin | Quinolone | + | – | + | + | + | + | – | – | + | + | + | + | + | + | + | + | gyrA S84F (10) | |
| Erythromycin | Macrolide | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | mphB (3), ermC (11) |
| Clindamycin | Lyncosamide | + | + | + | + | + | + | + | + | – | + | + | + | + | + | + | + | + | linA (1), ermC (11) |
| Tetracycline | Tetracicline | – | + | – | + | – | + | – | – | + | + | – | – | + | + | + | – | – | tetL (4) |
| Gentamycin | Aminoglycoside | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | aac/aph (15), aph3 (2), ant9 (3), aadD (11) |
| Trimethoprim/SMX | Sulfonamina | + | + | + | + | + | + | + | – | – | + | + | + | + | + | + | + | + | sul3 (17) |
| Chloramphenicol | Phenicol | + | – | + | + | + | + | + | – | + | + | + | catB (6) | ||||||
| Rifampicin | Miscellaneus | + | + | + | + | + | + | + | + | + | + | + | – | + | + | + | + | + | rpoB D471E, I527M (3); I527M (4) |
| Vancomycin | Glycopeptide | – | – | – | – | – | – | – | – | – | – | – | + | – | – | – | – | – | vanRI (17) |
The gene mecA encodes for a penicillin-binding protein (PBP) carried in a mobile element known as Staphylococcal Chromosomal Cassette or SCCmec (International Working Group on the Classification of Staphylococcal Cassette Chromosome E, 2009). By localizing the recombinases ccrA, B, and C, as well the mecA genes in the contigs of the respective genomes, and then constructing phylogenies including known SCCmec recombinase genes, we classified the SCCmec types of the SE INPer strains (Table 1; Fig. S6). The analysis demonstrated the presence of the community-acquired SCCmec type IV cassette in 13 out of 14 methicillin-resistant strains. The SE INPer strains S10 and S16 lack the SCCmec cassette, and no mecA gene was detected. Although the S21 strain has a mecA gene, we were unable to find other gene elements to show the presence of a mec cassette. Moreover, strains S07, S09, S13 strains contain an additional SCCmec type VIII cassette in tandem with the SCCmec IV cassette. The S07 strain carries a contig of 36 535 bps of the SCCmec type IV and VIII, suggesting the probable structure of the recombined cassette (Fig. S7).
The genomic analysis revealed that some SE strains studied here included genes for resistance to fluoroquinolones, macrolides, sulfonamides, aminoglycosides, tetracycline, and other antibiotics not used as the first choice in clinical therapy (Table 2). The results given in Table 2 corroborate that in most of the cases, the probable gene responsible for the resistance is present in the genomes. Besides, non-synonymous mutations in the antibiotic target proteins GyrA and RpoB were identified in some SE strains resistant to quinolones and rifampicin. Despite other SE strains lack these mutations, they still were resistant to these antibiotics. Then, other mutations in the antibiotic target protein or other genetic mechanisms not yet known would be responsible for these resistances.
Mobile genetic elements
The genomic variability observed in the SE strains suggests active processes of recombination and gene exchange. To study this concern, we first look for prophages and CRISPR-Cas related systems in the genomes. Prophage footprints were found in 10 out of 17 genomes of the SE strains. The most significant prophage hits detected by the PHAST program, were for genomic regions spanning about 28 to 95 kb that include an attachment site, a signature of lysogenic phages (Arndt et al., 2019) (Table S6). In this analysis, prophages were found integrated into the genomes of some SE strains, such as CNPH82 found in the SE strains S14, S17, and S18 (Daniel, Bonnen & Fischetti, 2007). Some other prophages such as StB20 and SpBeta were present in S03 and S16 strains respectively and, the prophages IPLA5 in strain S07 and IPLA7 in S12 and S15 strains (Gutiérrez et al., 2012). In the remaining SE strains, prophages sequences were not detected.
SE strains have also acquired defense mechanisms against phage infection. The search for CRISPR-Cas immune systems identified nine out of the 17 SE INPer strains carrying a CRISPR-Cas Type III system. The system III is composed by the Cas1 and Cas2, responsible for spacer processing and insertion; the ribonuclease Cas6, and the cascade proteins Csm1 to Csm6, involved in the processing of the target transcript (Table S7). The CRISPR-Cas Type III system has been already reported to confer immunity to phages as well as to conjugative plasmids in SE (Marraffini, 2015; Marraffini & Sontheimer, 2008). Although the enzymatic organization of the CRISPR-Cas systems is remarkably conserved in SE, there are variations in the array of repeats and spacers in the CRISPR loci. Three distinct types of identical repeated units of 30 or 36 nucleotides, associated with specific sequence spacers have been described (Marraffini, 2015). Three spacers that correspond to the CRISPR loci found in the strains S02, S05, and S24, match precisely with a sequence in the Staphylococcus phage PH15 genome for the first two strains and the Staphylococcus phage 6ec genome for the last (Aswani et al., 2014; Daniel, Bonnen & Fischetti, 2007).
SE strains harbor many ISs, belonging to different families (Table S8). The presence of IS256 has been found within pathogenic SE, associated with biofilm formation and virulence (Kozitskaya et al., 2004; Murugesan et al., 2018). Among the SE strains of our collection, the isolates having the IS256 also contain the ica operon, confirming previous observations. Exceptionally, only the strain S21 has the ica genes but lacks IS256.
Recombination
The above results suggest that high frequency of Horizontal Gene Transfer (HGT) and recombination might promote diversification of local SE populations. To evaluate this, we measured the ratio of recombination to mutation (r/m), using the ClonalFrameML program (Didelot & Wilson, 2015) in the 17 INPer SE genomes and 16 combined sets of SE genomes downloaded from GenBank (Fig. 2; Table S3). A median average r/m rate of about 6.9 was calculated when the 17 SE were tested, suggesting that nucleotide substitutions by recombination are more frequent than random point mutations (Vos & Didelot, 2009). Every COG class shows r/m values equal or higher than the estimate for the complete set of 17 SE genomes. Indeed, the r/m values on virulence or antibiotic resistance gene class result similar to the other COGs involved in housekeeping functions.
To determine whether or not the recombination estimates were affected by the sample composition of SE strains, we design several control tests, with distinct groups of genomes. First, we discarded the most divergent S10 strain of the SE collection and ran the ClonalFrameML test only with the 16 SE most related SE strains of the collection. As shown in Fig. 4, the r/m rate for this set reduced to a median of four, and this value is not significantly different respect to the r/m of SE of the complete genome of SE strains obtained from the GenBank (z-score = 1.4, P-value 0.135) (Fig. 4, boxplot 25). Second, we computed the r/m rate in eight different sets randomly selected among 260 complete and draft genomes of SE strains from the GenBank (Fig. 4, 26–33; Table S3). Some sets (Ctr2 and 8) display the lowest r/m values, whereas the rest control sets have r/m upper than two up to four. These results indicate that the strains sample composition influence the recombination estimates.
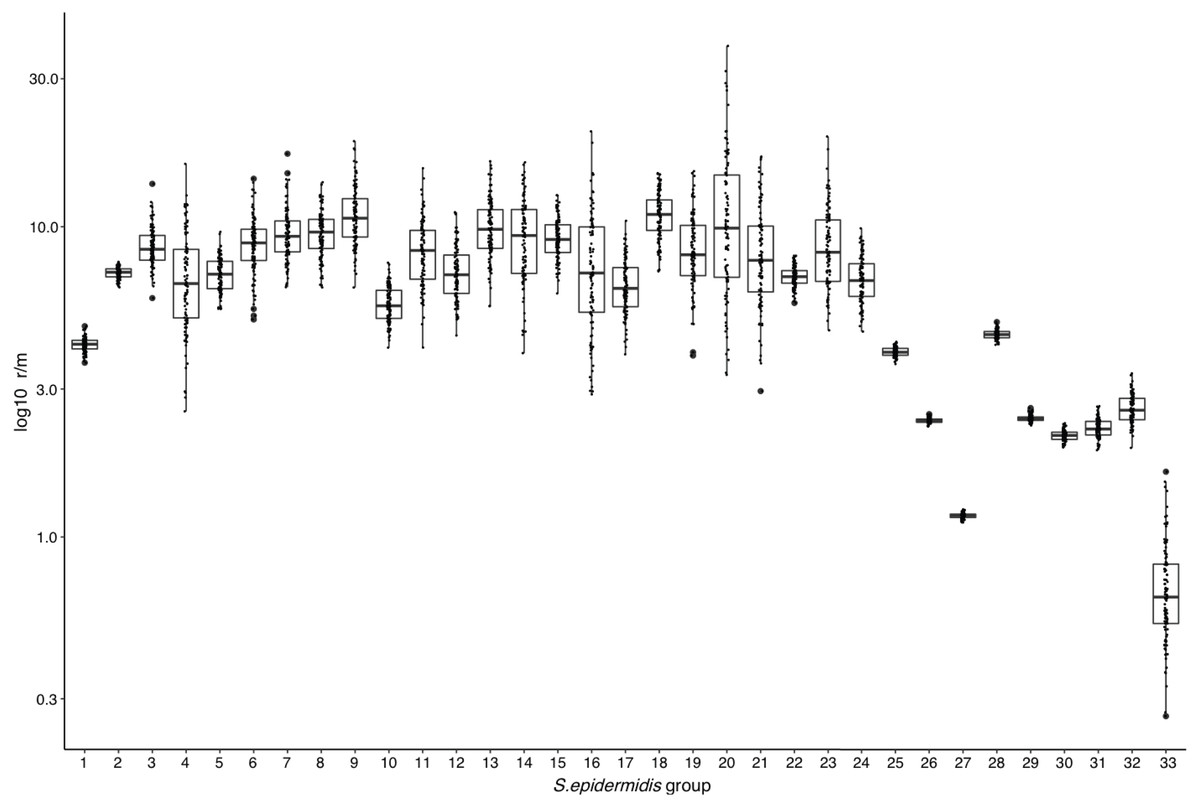
Figure 4: Rates of recombination/mutation for S. epidermidis INPer strains compared with sets of S. epidermidis from GenBank.
1. Sixteen strains out 17 S. epidermidis of the INPer collection; 2. Seventeen S. epidermidis strains of the INPer collection. 3–22, r/m rates for the genes encoding proteins classified in COGs in the 17 SE INPer: 3. COG C (energy production). 4. COG D (cell division). 5. COG E (amino acid transport and metabolism); 6. COG F (nucleotide transport and metabolism). 7. COG G (carbohydrate transport and metabolism). 8. COG H (coenzyme transport and metabolism). 9. COG I (lipid transport and metabolism). 10. COG J (translation, ribosomal structure, and biogenesis). 11. COG K (transcription). 12. COG L (replication, recombination, and repair). 13. COG M (cell wall, membrane, and envelope biogenesis). 14. COG O (post-translational modification, protein turnover, and chaperones). 15. COG P (inorganic ion transport and metabolism). 16. COG Q (secondary metabolites biosynthesis, transport, and catabolism). 17. COG R (general function predicted). 18. COG S (function unknown). 19. COG T (signal transduction mechanisms). 20. COG U (intracellular trafficking, secretion, and vesicular transport). 21. COG V (defense mechanism). 22. Unassigned COGs. 23. Antibiotic resistance genes predicted in PATRIC server for the SE INPer strains. 24. Virulence genes predicted in PATRIC server for the S. epidermidis INPer strains. 25. Reference set of 12 complete genomes of S. epidermidis strains used through this work. 26–33, subsets of draft SE strains from GenBank: 26. Ctr1 (n = 36). 27. Ctr2 (n = 26). 28. Ctr3 (n = 22). 29. Ctr4 (n = 35). 30. Ctr-5 (n = 36). 31. Ctr6 (n = 26). 32. Ctr7 (n = 36). 33. Ctr8 (n = 36). Descriptions of the SE strains included in the control sets and their GenBank accession numbers are shown in Table S3.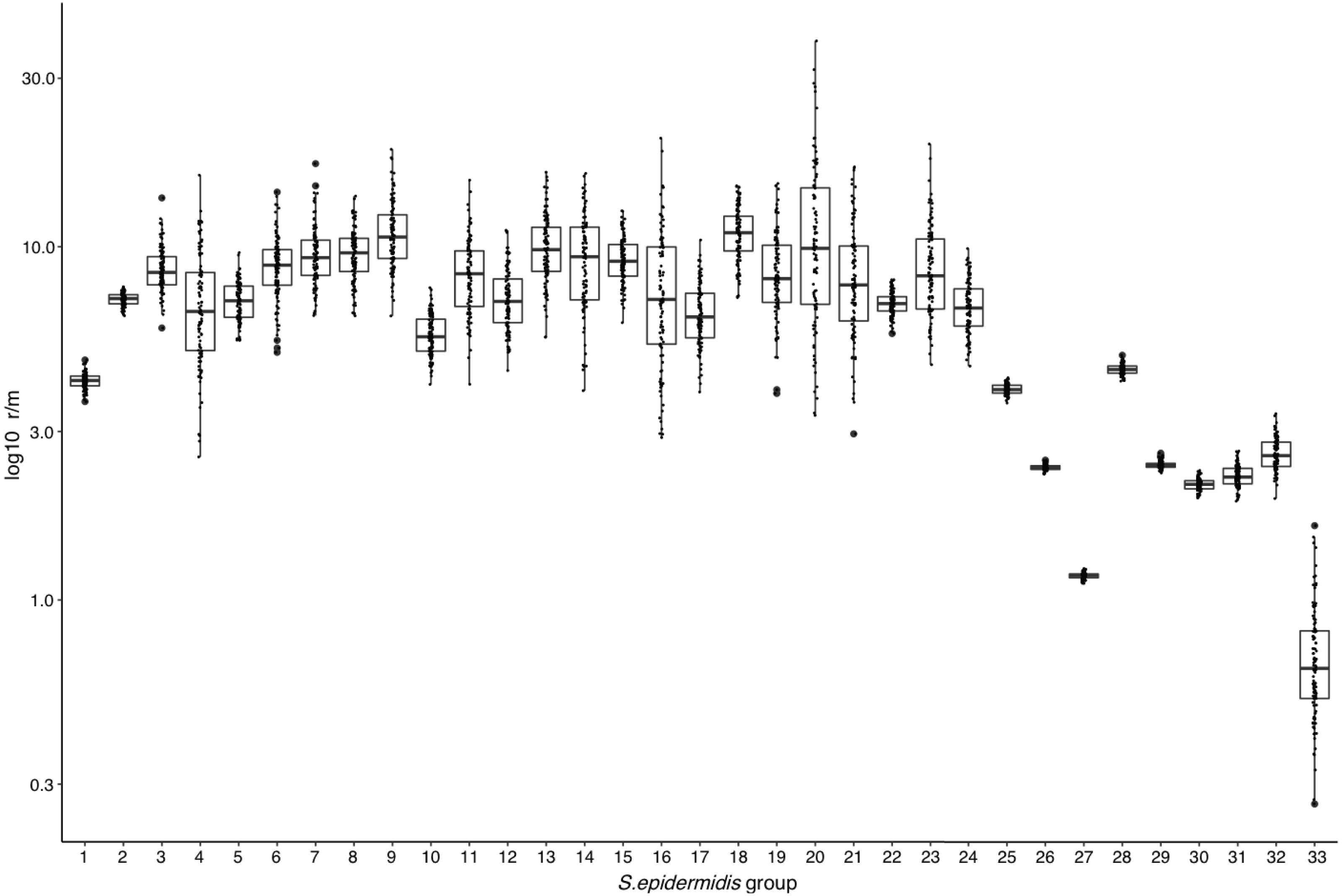{kind=link}
The RaxML nucleotide phylogenetic tree used as a reference to estimate recombination looks similar to the core protein phylogeny presented in Fig. 2; SE strains within clades maintain cohesive relationships (Fig. 5). However, multiple recombination events were detected in the ancestral nodes leading to the SE strains. The most prominent branch (red dot line in Fig. 5) divided the SE strains into two large clades, one including the clade D and the other constituted by clade B and C. Within different divergent lineages recombination introduces much more nucleotide variants than mutation as presented before (r/m). These results indicate that local hospital settings SE strains may contain enough genomic diversity despite their close relationship with the main clonal ST complexes of worldwide distribution.
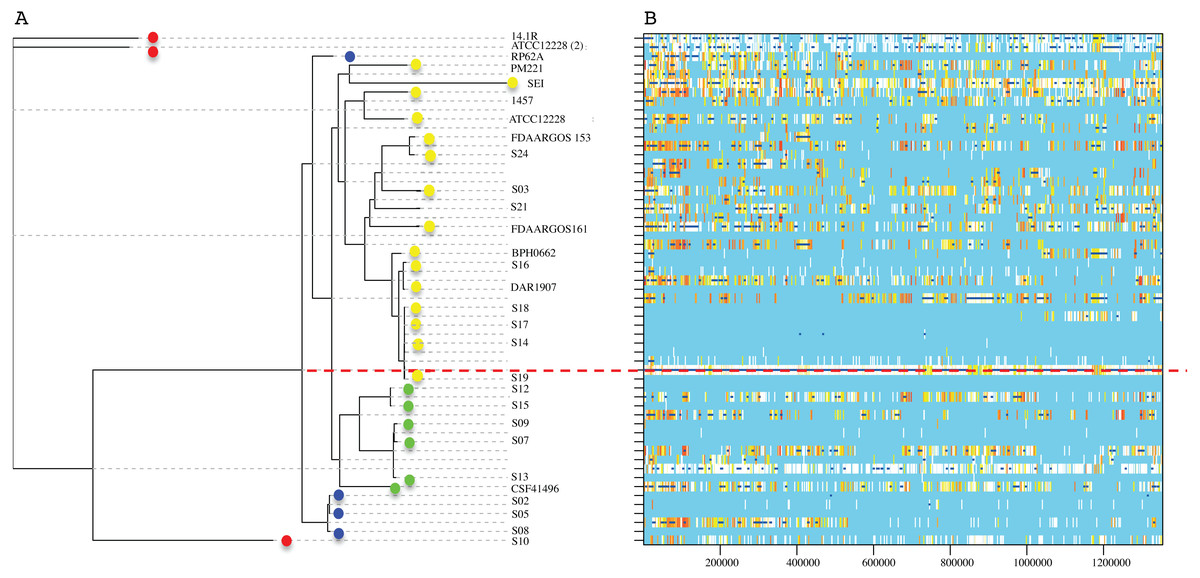
Figure 5: Genome-wide recombination between S. epidermidis strains.
ClonalFrameML program detected several events of recombination along 1,354,455 concatenated genomic regions of 29 S. epidermidis genomes (17 INPer genomes and 12 GenBank genomes). (A) RaxML nucleotide tree is shown at the left of the scheme. Color dots indicate the corresponding clades in the protein phylogeny (Fig. 1). (B) Blue bars indicate recombination events along the concatenated genome segments. White bars indicate non-homoplasic nucleotide substitutions; yellow to red bars are probable homoplasic nucleotide substitutions (Didelot & Wilson, 2015). Red dots line indicate an ancestral event of recombination.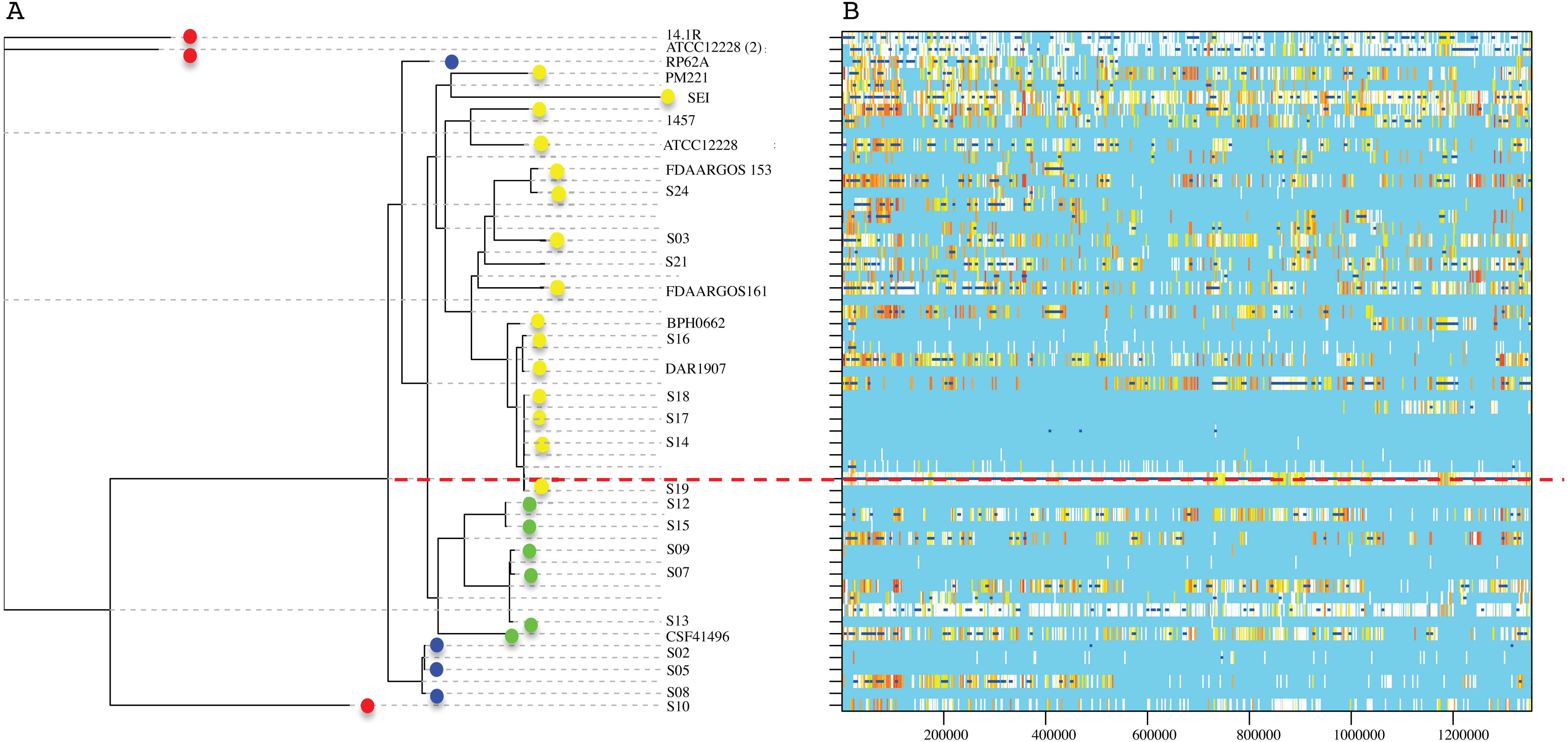{kind=link}
Discussion
SE is among the most common bacterial isolates found in the human skin microbiome (Byrd, Belkaid & Segre, 2018). It is also frequently recovered from bacteremia and sepsis samples in neonatal care clinic units, being its most probable etiological agent (Byrd, Belkaid & Segre, 2018; Otto, 2009). In clinical practice, it is difficult to assess if SE clones are the causal agents of the disease, are accidental, or opportunistic pathogens (Miragaia et al., 2008; Otto, 2017). In this work, we conducted a survey at the Instituto Nacional de Perinatología “INPer” in México City, over a period spanning eight years, to register changes in the antibiotic profile of staphylococci species and strains. We focus on SE because it was the bacteria most frequently found among the staphylococci isolates and showed a remarkable multi-resistance pattern. The number of antibiotic resistances remained very similar year by year with most SE strains being multi-resistant to nine antibiotics; a minor representation of low-resistant strains (<5 antibiotics) was found. Therefore, the SE population within the INPer hospital was highly stable and led us to question its genetic composition. Specifically, we address the hypothesis that a single or few clones are the basis of the normalization of all the SE multi-resistant strains in the children’s health care unit studied.
We analyzed the genomes of 17 selected SE strains isolated mostly from neonatal patients. Our findings regarding virulence genetic determinants are concordant with those found in SE isolates from hospitals and commensal strains isolated worldwide. The most prominent features of these isolates are their ability to form biofilms, the presence of PMSs, and the multidrug resistance profile displayed (Otto, 2014; Xu et al., 2018). Employing a phylogenetic strategy using the ccrA, B, C recombinases, we conclude that most of the INPer SE strains carry the SCCmec type IV. Surprisingly, eight strains of the SE collection analyzed here, harbor neither the ica operon nor the IS256, considered pathogenicity markers in SE (Kozitskaya et al., 2004; Murugesan et al., 2018). Likely, these are commensal SE strains that invaded the patients in the course of their hospital stay even though we cannot discard they use other pathogenicity mechanisms. These strains showed resistance to multiple antibiotics, and three of them contain a composite SCCmec cassette formed by type IV and VIII gene elements. We propose a probable structure of the combined SCCmec IV and VIII cassette (Fig. S7). It is an unusual combination of SCC elements, but there are reports in the literature of mosaic chromosomal staphylococcal mec cassettes (Heusser et al., 2007). However, this SCCmec genetic element should be subject to further corroboration. Classification of SCCmec is still hard to discern due to the variability and presence of repeated elements, which difficult the correct assembly of the region (International Working Group on the Classification of Staphylococcal Cassette Chromosome E, 2009). The chromosomal cassettes might be hot spots of recombination of heavy metal resistance genes, insertion sequences, and antibiotic resistance genes recruited by HGT (Xue et al., 2017).
The genomic antibiotic resistance spectrum of SE strains is very diverse, with some genes present in most strains and others only in few. Examples of diversification of the resistance mechanisms are the presence of membrane efflux pumps (NorAB), which may confer resistance to quinolones as well, and several modifying enzymes (AAC, APH) that inactivate aminoglycosides such as kanamycin. Indeed, the fosB gene, which encodes the resistance to fosfomycin was found in 9 out of 17 SE strains. In several of these instances, the resistance phenotype coincided with the presence of one or more genes, encoding modifying or degrading enzymes and mutant protein targets for some antibiotics (Table 2).
The genome analysis also indicates some mechanisms for antibiotic resistance, including non-synonymous substitutions in the housekeeping genes gyrA and rpoB. In the gyrase (gyrA) it was found the amino acid S84F change which confers quinolone resistance, whereas, in rpoB (β-subunit of the RNA polymerase), a double amino acid substitution D471E: I527M, and a single I527M were identified. The double mutant rpoB D471E: I527M has been recognized elsewhere as the most common cause of worldwide rifampicin resistance (Lee et al., 2018). Indeed, the presence of this rpoB variant reduces the susceptibility to vancomycin and teicoplanin. In this work, the rpoB double mutant was detected in S02, S05, and S08 all belonging to ST23 clonal type, while the single mutant I527M was observed in the strains S14, S17, S18, and S19, which are within the ST2 clonal type. Both are the clonal lineages worldwide distributed reported by Leeetal2019. These INPer strains can form biofilms and contain most of the virulence determinants (Table 1). Therefore, they are very adapted SE strains and continuously present in the INPer.
In the phylogenetic trees reported, pathogenic SE strains are intermingled with commensal SE strains with no pathogenic cluster found (Meric et al., 2018; Miragaia, Couto & Lencastre, 2005). Recently, Meric et al. suggested, that pathogenic SE subpopulations occur within the commensal SE strains, which contain genes and alleles necessary for colonization at distinct infection sites (Meric et al., 2018). These Genome-wide-association (GWAS) studies showed the enrichment of several genes involved with methicillin resistance, biofilm formation, cell toxicity, and inflammatory response in pathogenic SE isolates (Meric et al., 2018). Therefore, pathogenic and commensal SE strains likely coexist in the same infection site, but current clinical methods of isolation prevent us from distinguishing one from the other. Eight ica− out of 17 strains analyzed, could not form biofilms even thought they were recovered from ill patients (Table 1).
Furthermore, here are various footprints of MGEs, including prophages, ISs, and the phage immunity CRISPR-Cas systems in the SE genomes that likely contribute to the adaptability by the acquisition of virulence and antibiotic resistance factors. As expected, due to its mobile nature, these elements do not follow a uniform distribution in the phylogeny, indicating frequent genetic exchange in the SE population. Together with HGT, homologous recombination may be a factor for genetic diversification of SE in hospital settings. In our work, extensive genome analysis of the rates of recombination versus mutation suggests that recombination affects the whole genome and not only a particular class of genes. It has been estimated that recombination could involve 40% of the genome of SE, whereas in S. aureus recombination comprises the 24% portion (Meric et al., 2015). Although recombination rates depend strongly on the sample of strains used for the analysis, the estimated r/m values reported agrees with other recombination test performed with distinct samples of SE, few or whole-genome markers as well as reported r/m numbers in the literature (Meric et al., 2015; Miragaia, Couto & Lencastre, 2005). Therefore we can conclude that the SE population despite its whole low level of nucleotide variation (ANIm > 97%) shows cohesive clonal behavior but frequent gene exchange and recombination.
Conclusions
At local hospital settings, pathogenic, and commensal SE strains coexist, but it is hard to discern if they are contaminants, commensal colonizers, or virulent strains (Widerstrom et al., 2016). Indeed, single-colony testing for the identification of SE isolates limits to know the extent of multiclonal or multi-species infections (Harris et al., 2016; Van Eldere et al., 2000). In the present study, some analyzed SE strains came from nosocomial patients, but lack ica genes, a classical virulence marker. However, we cannot exclude that these putative commensal SE strains are pathogens in a non-determined manner, or in other conditions. Likely they are part of the intra-hospital non-pathogenic microbiome. These presumed SE commensal strains, as well as the biofilm formers considered pathogenic SE strains are multi-resistant to antibiotics. The results present here of an 8-year survey, suggest that the multi-resistance to antibiotics might drive adaptation and persistence of specific SE clones in hospital settings. HGT and recombination might play a crucial role in the origin of the pathogenic clones, by moving and recombining antibiotic resistance and virulence genes in distinct genomic clonal backgrounds, including non-pathogenic strains. Therefore, the clinical and genetic factors that influence the adaptability stability and change of SE community overtime should be addressed in detail in future studies.
Supplemental Information
Survey of staphylococci at Instituto Nacional de Perinatología, México City Alone eight years
A. Species classification and proportion. B. Origin of the isolates and proportion. C. Isolation sites proportion.
Frequency cumulation of antibiotic resistant strains in Staphylococcus
A. S. epidermidis. B. S. aureus. The absolute number of antibiotic resistances (x-axis) by the number of strains (y-axis) was counted from 2006 to 2013.
Genome identity (ANIm) between pairs of SE INPer strains
Pairwise whole genome alignments were done with Mummer within the JSspecies program (Richter et al., 2016). The percent of average nucleotide alignment (ANI) was illustrated by a heat-map constructed with ggPlot2 in R (see methods). ANI > 99% are in red color. S. epidermidis ATCC 12228 was included for comparison. S10 strain had ANI = 97% respect all the other SE strains.
Pangenome model of SE strains
The pangenome model of 29 SE strains was performe with GET_HOMOLOGUES (Contreras-Moreira & Vinuesa, 2013) as described in methods. (A) Pangenome size (number of gene family clusters, Y axis) as a function of the number of SE genomes (X axis). (B) Core genome according to the Tettelin equations.
Clonal relationships of the STs detected in INPer strains respect to the ST database
Alleles for the seven proteins used in the S. epidermidis MLST scheme (Thomas et al., 2007) were looked at the Staphylococcus epidermidis MLST database (https://pubmlst.org/sepidermidis/; Table 1; see methods) (Feil et al., 2004). The clonal relationships among STs were determined by eBURST (http://eburst.mlst.net). Six out of 8 ST complexes assigned to the SE INPer strains are denoted by numbers in violet color.
Phylogenetic tree of the SCCmec recombinases
A. ccrA. B. ccrB. C. ccrB
Proposed structure of the combined cassette SCCmec IV-VIII
The segment corresponding to SCCmec cassettes contained within the contig 12 of the S07 strain is annotated according to the best blastN matches against the nr database of the Genebank.