
Whole-genome comparisons of Penicillium spp. reveals secondary metabolic gene clusters and candidate genes associated with fungal aggressiveness during apple fruit decay
- Published
- Accepted
- Received
- Academic Editor
- Thiago Venancio
- Subject Areas
- Agricultural Science, Genomics, Mycology, Plant Science
- Keywords
- Penicillium spp, Blue mold, Pome fruit, Comparative genomics, Gene profiling
- Copyright
- © 2019 Wu et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2019. Whole-genome comparisons of Penicillium spp. reveals secondary metabolic gene clusters and candidate genes associated with fungal aggressiveness during apple fruit decay. PeerJ 7:e6170 https://doi.org/10.7717/peerj.6170
Abstract
Blue mold is a postharvest rot of pomaceous fruits caused by Penicillium expansum and a number of other Penicillium species. The genome of the highly aggressive P. expansum strain R19 was re-sequenced and analyzed together with the genome of the less aggressive P. solitum strain RS1. Whole genome scale similarities and differences were examined. A phylogenetic analysis of P. expansum, P. solitum, and several closely related Penicillium species revealed that the two pathogens isolated from decayed apple with blue mold symptoms are not each other’s closest relatives. Among a total of 10,560 and 10,672 protein coding sequences respectively, a comparative genomics analysis revealed 41 genes in P. expansum R19 and 43 genes in P. solitum RS1 that are unique to these two species. These genes may be associated with pome fruit–fungal interactions, subsequent decay processes, and mycotoxin accumulation. An intact patulin gene cluster consisting of 15 biosynthetic genes was identified in the patulin producing P. expansum strain R19, while only a remnant, seven-gene cluster was identified in the patulin-deficient P. solitum strain. However, P. solitum contained a large number of additional secondary metabolite gene clusters, indicating that this species has the potential capacity to produce an array of known as well as not-yet-identified products of possible toxicological or biotechnological interest.
Introduction
Postharvest “blue mold” is the most common and economically deleterious pome fruit rot worldwide (Rosenberger, 1990; Xiao & Boal, 2009). Many Penicillium species (P. auarantiogriseum, P. carneum, P. commune, P. brevicompactum, P. crustosum, P. solitum, P. verrucosum and P. expansum) cause blue mold during long-term storage of apples, pears, quince and sometimes other fruits (Raper & Thom, 1949; Sanderson & Spotts, 1995; Sholberg & Haag, 1996; Pianzzola, Moscatelli & Vero, 2004). The fungus gains access to stored fruits primarily through stem punctures/wounds and bruises. On apples, P. expansum is the most cosmopolitan and aggressive species (Pianzzola, Moscatelli & Vero, 2004) while P. solitum is less aggressive and causes significantly less decay in storage (Pitt et al., 1991).
Isolates of P. expansum regularly produce patulin, a regulated mycotoxin, while all tested P. solitum isolates are non-toxigenic (Frisvad, 2004; Jurick et al., 2010). Comparisons of the patulin biosynthetic pathway have been conducted between P. expansum, P. italicum and P. digitatum but not other blue mold causing fungi (Li et al., 2015). By comparing the genomes of the aggressive and toxigenic P. expansum, the less aggressive and nontoxigenic P. solitum, and other closely related Penicillium species, we sought to identify differences that provide insights into blue mold decay of stored pome fruits. With the aim of devising novel strategies for management of these economically important fungi, it is important to understand the genetic basis of the secondary metabolite production which is facilitated by a well sequenced and annotated genome.
Penicillium expansum R19 was the first P. expansum strain to be sequenced, published and released to the public domain (Yu et al., 2014). Although several other strains of P. expansum have been sequenced since that time (Li et al., 2015; Ballester et al., 2015), to the best of our knowledge, there has been no comparative genomic analysis amongst Penicillium species causing blue mold on pome fruit. Therefore, we conducted a detailed study comparing different species of these fungi at the genome level. Recently, we sequenced the genome of P. solitum strain RS1 and achieved a high-quality assembly (Yu et al., 2016). Here, we re-sequenced the highly aggressive and patulin-producing P. expansum strain R19 to achieve a better assembly, and then compared the two blue mold genomes to each other, as well as to the genomes of five other Penicillium species that inhabit different ecological niches and hosts.
Our analysis included the genomes of two citrus pathogens, Penicillium digitatum (“pdi”) (Marcet-Houben et al., 2012) and Penicillium italicum (“pit”) (Ballester et al., 2015); two cheese making species, Penicillium camemberti (“pca”) and Penicillium roqueforti (“pro”) (Cheeseman et al., 2014); and the penicillin-producing industrial strain Penicillium chrysogenum (“pch”) (Van den Berg et al., 2008). We report differences and similarities in the gene repertoires of P. expansum and P. solitum, and discuss the potential functions of some of their unique genes. A robust phylogenetic analysis was performed using nearly four hundred core eukaryotic genes (CEGs) to compare the two blue mold species to the five other Penicillium genomes. Our data provides a solid platform to further explore the genetic basis of the differences in the ability of P. expansum and P. solitum to cause postharvest decay of pome fruits, uncover new insights into secondary metabolic gene clusters, and strengthen our understanding of genome-wide phylogenetic relationships amongst economically important Penicillium species.
Materials and Methods
Fungal growth, genomic DNA extraction, genome sequencing, assembly, and annotation
Growth, both in culture and in vivo including inoculation of apple, and genomic DNA extraction of P. expansum R19 were conducted using the same approaches as previously published (Conway, 1982; Yu et al., 2014). The sequencing, assembly, and annotation pipeline previously described in our study on P. solitum RS1 (Yu et al., 2016) was used for the genome of P. expansum R19. Briefly, the assembly was conducted using HGAP3 under default settings, which applies long read correction algorithms and the Celera assembler to confidently produce high quality contigs (unitigs) using reads from PacBio Single Molecule Real Time (SMRT) sequencing technology (Ricker et al., 2016). The annotation software MAKER (Holt & Yandell, 2011) was implemented for four iterative runs starting with gene predictions from CEGMA (Parra, Bradnam & Korf, 2007). This newly sequenced P. expansum genome was 32,356,049 bp which is 97.3% of the currently published P. expansum MD-8 genome. The BUSCO (v. 3.0.2) analysis resulted in 289 complete BUSCOs out of 290 (dikarya odb9) and one fragmented BUSCO for a 99.7% complete assembly (Waterhouse et al., 2018). In the annotation step, P. solitum proteins were used as a part of the protein evidence set instead of P. expansum proteins.
Penicillium species phylogenomics
Whole genome phylogeny of seven Penicillium species and one Aspergillus flavus strain was built using peptide sequences of shared core eukaryotic genes (CEGs) as predicted by CEGMA (Parra, Bradnam & Korf, 2007). The source data for the other included genomes came from NCBI and EMBL, A. flavus NRRL 3357 (https://www.ncbi.nlm.nih.gov/nuccore/EQ963472), P. digitatum Pd1(http://fungi.ensembl.org/Penicillium_digitatum_pd1_gca_000315645/Info/Index),P. camemberti FM 013 (https://fungi.ensembl.org/Penicillium_camemberti_fm_013_gca_000513335/Info/Index), P. chrysogenum Wisconsin 54-1255 (http://fungi.ensembl.org/Penicillium_rubens_wisconsin_54_1255_gca_000226395/Info/Index), P. italicum PHI-1 (https://genome.jgi.doe.gov/Penita1/Penita1.home.html), and P. roqueforti FM 164 (https://fungi.ensembl.org/Penicillium_roqueforti_fm164_gca_000513255/Info/Index). We used a set of 399 CEGs that satisfy the following criteria: (1) They are present in all eight above-mentioned genomes; (2) all sequences are at least 90% the length of their Saccharomyces cerevisiae orthologs (Engel et al., 2014). Sequences were first aligned with MAFFT with parameters—localpair—maxiterate 1,000 (Katoh et al., 2002), and then concatenated. The phylogeny was inferred using RAxML (Stamatakis, 2006) with the parameters “–f a -# 400 –m PROTGAMMAJTT”.
Markov cluster algorithm (MCL) clustering
The protein sequences of the seven selected Penicillium species were subjected to BLAST comparison against each other (Altschul et al., 1990). The bitscores were then used to generate clusters of genes via the TRIBE-MCL algorithm (Enright, Van Dongen & Ouzounis, 2002) using default settings.
Gene Ontology (GO) annotation and Fisher’s exact tests
P. expansum R19 protein sequences were used in a BLAST comparison against the NCBI NR protein database and the results were used to predict GO annotation using Blast2go with default settings (http://www.blast2go.com). Further, GO enrichment in selected groups of genes was tested using Fisher’s exact tests. The same analysis was also performed on P. solitum RS1 protein sequences.
Secondary metabolite gene clusters
Secondary metabolite gene clusters were identified using the antiSMASH program for fungi at default settings (Weber et al., 2015).
Results
The growth of Penicillium expansum and P. solitum was compared, both on agar plates as well as the aggressiveness on apples incubated at different temperatures. When cultured on potato dextrose agar, both fungi grew on all temperatures ranging from 0 to 20 °C although P. solitum grew slightly slower than P. expansum (Figs. 1A, 1B). However, when inoculated onto apple fruit, the differences in aggressiveness were much more pronounced (Figs. 1C, 1D). Both pathogens are necrotrophic and require a wound for infection, they are unable to puncture the skin of the fruit due to the lack of an appressorium and rely on pectin degrading enzyme production for growth (Yao, Conway & Sams, 1996; Jurick et al., 2009, p. 20019). Decay was evident 7 days post inoculation (DPI) on apples inoculated with P. expansum at 10 and 20 °C, while the apples inoculated with P. solitum displayed only a small lesion around the inoculation point at 20 °C (Figs. 1C, 1D).
To achieve optimal genome sequence and ensure reliable downstream bioinformatic comparisons, the PacBio Single Molecule Real Time (SMRT) sequencing platform was utilized to resequence P. expansum R19, with sequencing coverage reaching 65-fold. An assembly of 16 unitigs with an N50 value of 8.17 Mbp was achieved and the genome assembly was deposited under BioSample SUB4649056. Gene annotation revealed 10,560 putative protein coding genes. Using CEMGA 99.2% complete core eukaryotic genes (CEGs) and 99.6% partial (including complete) CEGs were predicted in the genome. A genome-wide multi-gene phylogenetic approach was then implemented, involving 399 CEGS, using P. expansum, P. solitum, and several other related Penicillium species, with Aspergillus flavus as a designated outgroup (Fig. 2). The closest relative of P. solitum amongst the Penicillium spp. was P. camemberti, while for P. expansum, the most phylogenetically similar were the two citrus pathogens P. digitatum and P. italicum (Fig. 2). P. roqueforti, a cheese making species associated with blue cheeses and P. chrysogenum, the penicillin-producing species, formed another distant, yet distinct clade (Fig. 2).

Figure 1: Effect of temperature on fungal growth in culture and during apple fruit decay.
(A) Penicillium expansum growing on Potato Dextrose Agar. (B) Penicillium solitum growing on Potato Dextrose Agar. (C) Penicillium expansum causing blue mold decay on apple fruit. (D) Penicillium solitum causing blue mold decay on apple fruit.{kind=link}
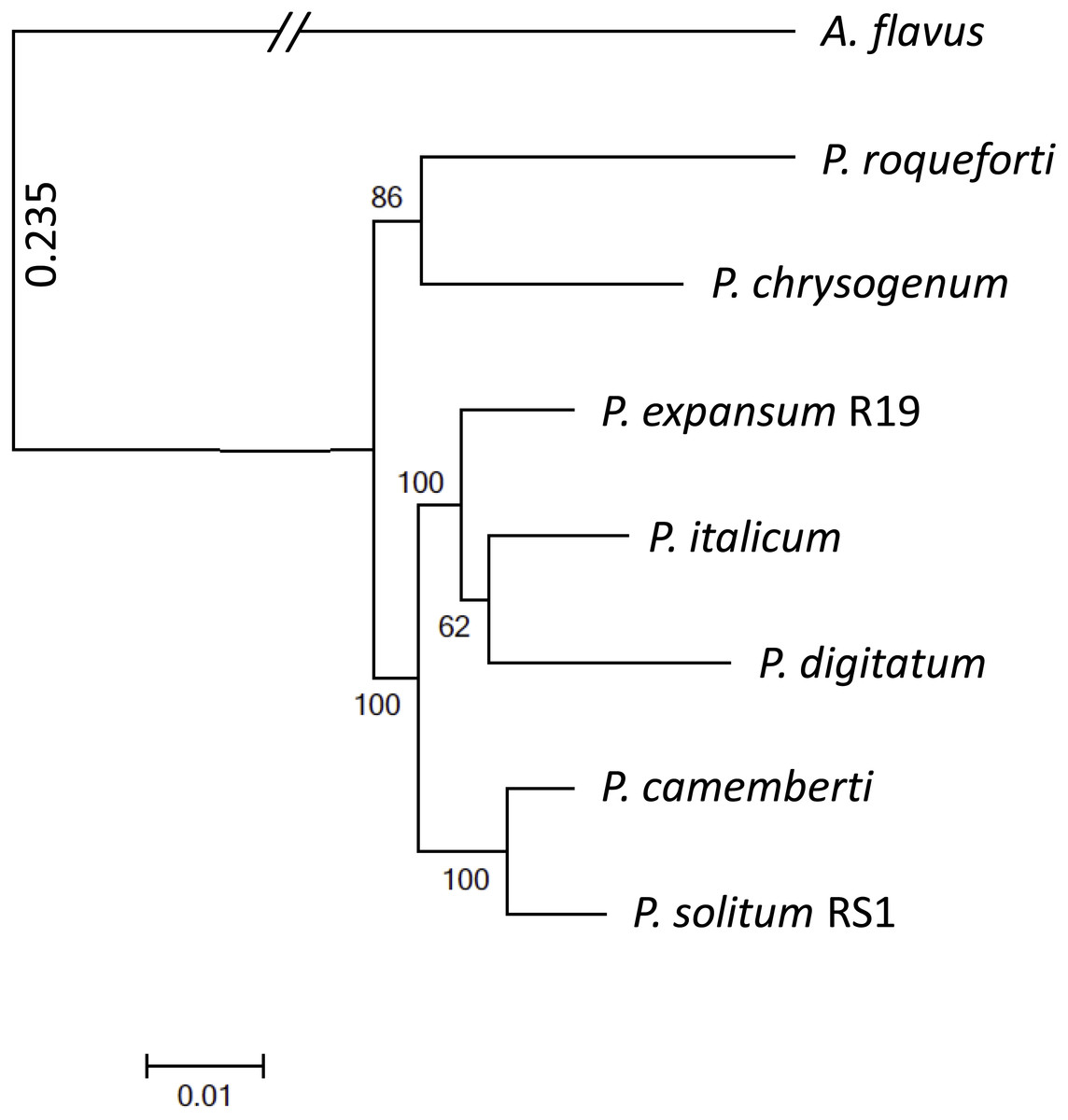
Figure 2: Phylogenomic analysis of seven Penicillium species.
A. flavus was used as outgroup and bootstrap values are indicated on the branches.{kind=link}
For P. expansum R19, 7,921 proteins were annotated (75% of all proteins) with a total of 37,962 GO entries (4.8 GO entries per protein), representing 6,000 unique GO categories. For P. solitum RS1, 7,920 proteins were annotated (74.2% of all proteins) with a total of 37,858 GO entries (4.8 GO entries per protein), representing 5,993 unique GO categories. We analyzed GO enrichment in a group of 877 proteins that were over-represented in P. expansum, defined as having ≥ 2X more copies than detected in P. solitum (Table 1). We also tested the GO enrichment in 550 P. expansum proteins that were over-represented in both blue mold fungi, defined as having on average ≥ 2X copies in P. expansum and P. solitum than in the five Penicillium species included in this study (Table 2). An analysis using P. solitum proteins over-represented in the two pome blue mold Penicillium species revealed similar results. Also, we examined the GO enrichment in a group of 1,096 proteins that are represented more frequently in P. solitum, defined as having ≥ 2X copies in P. solitum than in P. expansum (Table 3). The seven Penicillium species included in this study, with a total of 9,960 protein families, encompassing 78,479 proteins, were identified. All species shared 5,308 protein families, encompassing 64,598 proteins, or 82.3% of the total proteins also referred to as the proteome. For P. expansum 86.4% (9,119 out of 10,560), and for P. solitum 85.3% (9,099 out of 10,672) of the proteins were shared with the other species. However, a small set (36) of protein families were unique to the two blue mold species isolated from pome fruits, accounting for 41 proteins in P. expansum and 43 proteins in P. solitum (Table S1). Amino acid sequences from both fungi that cause blue mold of pome fruits were also compared. The more aggressive species, P. expansum R19, contained 222 gene families, encompassing 261 proteins that were not present in the less aggressive species P. solitum RS1. Similarly, there were 299 gene families, accounting for 375 proteins present in P. solitum but not in P. expansum, (Table S1). In addition, P. expansum R19 contained significant numbers of proteins with copy numbers that are one to several fold more than P. solitum RS1 (Table S1).
| GO-ID | Term | Type | FDR | P-Value | SG | NSG | SNG | NSNG |
|---|---|---|---|---|---|---|---|---|
| GO:0003824 | catalytic activity | F | 2.37E–05 | 2.80E–09 | 319 | 3,992 | 155 | 3,455 |
| GO:0016829 | lyase activity | F | 1.59E–03 | 3.75E–07 | 37 | 218 | 437 | 7,229 |
| GO:0008152 | metabolic process | P | 1.81E–03 | 6.39E–07 | 369 | 5,015 | 105 | 2,432 |
| GO:0046271 | phenylpropanoid catabolic process | P | 1.34E–02 | 1.27E–05 | 4 | 0 | 470 | 7,447 |
| GO:0046281 | cinnamic acid catabolic process | P | 1.34E–02 | 1.27E–05 | 4 | 0 | 470 | 7,447 |
| GO:0034396 | negative regulation of transcription from RNA polymerase II promoter in response to iron | P | 1.34E–02 | 1.27E–05 | 4 | 0 | 470 | 7,447 |
| GO:0034395 | regulation of transcription from RNA polymerase II promoter in response to iron | P | 1.34E–02 | 1.27E–05 | 4 | 0 | 470 | 7,447 |
| GO:0009803 | cinnamic acid metabolic process | P | 1.34E–02 | 1.27E–05 | 4 | 0 | 470 | 7,447 |
| GO:0019748 | secondary metabolic process | P | 3.10E–02 | 3.29E–05 | 21 | 111 | 453 | 7,336 |
| GO:0016813 | hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds, in linear amidines | F | 3.93E–02 | 5.32E–05 | 6 | 7 | 468 | 7,440 |
| GO:0071281 | cellular response to iron ion | P | 3.93E–02 | 6.03E–05 | 4 | 1 | 470 | 7,446 |
| GO:0010039 | response to iron ion | P | 3.93E–02 | 6.03E–05 | 4 | 1 | 470 | 7,446 |
| GO:0009698 | phenylpropanoid metabolic process | P | 3.93E–02 | 6.03E–05 | 4 | 1 | 470 | 7,446 |
| GO:0071241 | cellular response to inorganic substance | P | 4.27E–02 | 7.05E–05 | 7 | 12 | 467 | 7,435 |
Notes:
Type indicates the category of GO terms (P: biological process; F: molecular function).
- FDR
-
False Discovery Rate
- SG
-
the number of proteins that are in the tested group and have the GO term
- NSG
-
the number of proteins that are not in the tested group, and have the GO term
- SNG
-
the number of proteins that are in the tested group, and do not have the GO term
- NSNG
-
the number of proteins that are not in the tested group, and do not have the GO term
| GO-ID | Term | Type | FDR | P-Value | SG | NSG | SNG | NSNG |
|---|---|---|---|---|---|---|---|---|
| GO:0009812 | flavonoid metabolic process | P | 3.48E–03 | 4.11E–07 | 4 | 0 | 198 | 7,719 |
| GO:0047661 | amino-acid racemase activity | F | 8.53E–03 | 2.01E–06 | 4 | 1 | 198 | 7,718 |
| GO:0036361 | racemase activity, acting on amino acids and derivatives | F | 1.25E–02 | 5.92E–06 | 4 | 2 | 198 | 7,717 |
| GO:0016855 | racemase and epimerase activity, acting on amino acids and derivatives | F | 1.25E–02 | 5.92E–06 | 4 | 2 | 198 | 7,717 |
| GO:0010333 | terpene synthase activity | F | 4.49E–02 | 2.65E–05 | 4 | 4 | 198 | 7,715 |
Notes:
Type indicates the category of GO terms (P: biological process; F: molecular function).
- FDR
-
False Discovery Rate
- SG
-
the number of proteins that are in the tested group and have the GO term
- NSG
-
the number of proteins that are not in the tested group, and have the GO term
- SNG
-
the number of proteins that are in the tested group, and do not have the GO term
- NSNG
-
the number of proteins that are not in the tested group, and do not have the GO term

Figure 3: Patulin gene cluster in Penicillium expansum (A) and P. solitum (B).
Gene IDs and gene names are indicated below the genes. Reciprocal best matches are linked with a line. Red colored genes are known members of the patulin gene cluster.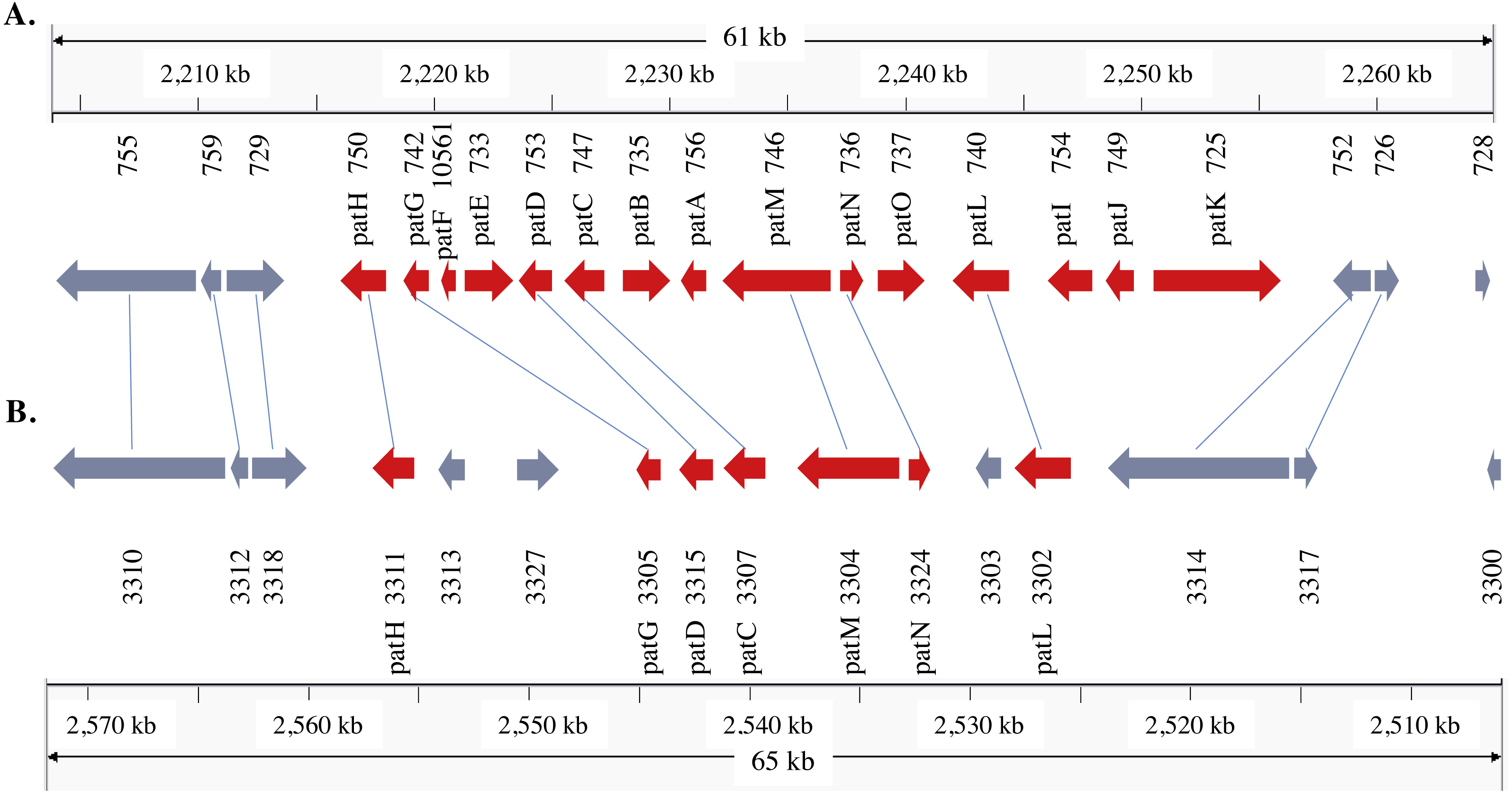{kind=link}
| GO-ID | Term | Type | FDR | P-Value | SG | NSG | SNG | NSNG |
|---|---|---|---|---|---|---|---|---|
| GO:0046983 | protein dimerization activity | F | 1.49E–09 | 1.77E–13 | 29 | 63 | 478 | 7,350 |
| GO:0042559 | pteridine-containing compound biosynthetic process | P | 1.23E–04 | 2.91E–08 | 10 | 8 | 497 | 7,405 |
| GO:0006729 | tetrahydrobiopterin biosynthetic process | P | 7.47E–04 | 4.43E–07 | 6 | 1 | 501 | 7,412 |
| GO:0046146 | tetrahydrobiopterin metabolic process | P | 7.47E–04 | 4.43E–07 | 6 | 1 | 501 | 7,412 |
| GO:0008124 | 4-alpha-hydroxytetrahydrobiopterin dehydratase activity | F | 7.47E–04 | 4.43E–07 | 6 | 1 | 501 | 7,412 |
| GO:0042558 | pteridine-containing compound metabolic process | P | 1.29E–03 | 9.17E–07 | 10 | 14 | 497 | 7,399 |
| GO:0015074 | DNA integration | P | 5.73E–03 | 4.76E–06 | 6 | 3 | 501 | 7,410 |
| GO:0046654 | tetrahydrofolate biosynthetic process | P | 1.56E–02 | 1.66E–05 | 4 | 0 | 503 | 7,413 |
| GO:0003934 | GTP cyclohydrolase I activity | F | 1.56E–02 | 1.66E–05 | 4 | 0 | 503 | 7,413 |
| GO:0016829 | lyase activity | F | 1.99E–02 | 2.36E–05 | 35 | 227 | 472 | 7,186 |
| GO:0000981 | RNA polymerase II transcription factor activity, sequence-specific DNA binding | F | 2.12E–02 | 2.76E–05 | 41 | 289 | 466 | 7,124 |
Notes:
Type indicates the category of GO terms (P: biological process; F: molecular function).
- FDR
-
False Discovery Rate
- SG
-
the number of proteins that are in the tested group and have the GO term
- NSG
-
the number of proteins that are not in the tested group, and have the GO term
- SNG
-
the number of proteins that are in the tested group, and do not have the GO term
- NSNG
-
the number of proteins that are not in the tested group, and do not have the GO term
In the P. expansum R19 genome, we identified the fifteen-gene patulin cluster (Fig. 3). In the P. solitum genome, a sequence similarity search (BLAST) identified a partial patulin gene cluster, spanning 31 kb (Fig. 3). Within the P. solitum partial gene cluster, only seven of the identified patulin biosynthetic genes were present (Fig. 3), with amino acid sequence identity >91% and e-value = 0. The seven genes identified from the partial cluster were patC, patD, patG, patH, patL, patM, and patN (Fig. 3). Five other genes (patA, patB, patE, patK, and patO) were found dispersed elsewhere in the genome with much lower amino acid sequence identity (between 29% and 49%). Three genes, patF, patI and patJ, were not found anywhere in the genome (e-value cutoff of 1e−5). Using the antiSMASH platform, all seven of the Penicillium spp. genomes were analyzed to determine their secondary metabolism genes. Within the P. solitum genome, we identified 66 gene clusters that were putatively involved in secondary metabolism (SM), more than any other of the Penicillium species analyzed in this study (Fig. 4). In contrast, 58 gene clusters were found in P. expansum, 21 gene clusters in P. italicum, and 35 gene clusters in P. digitatum.
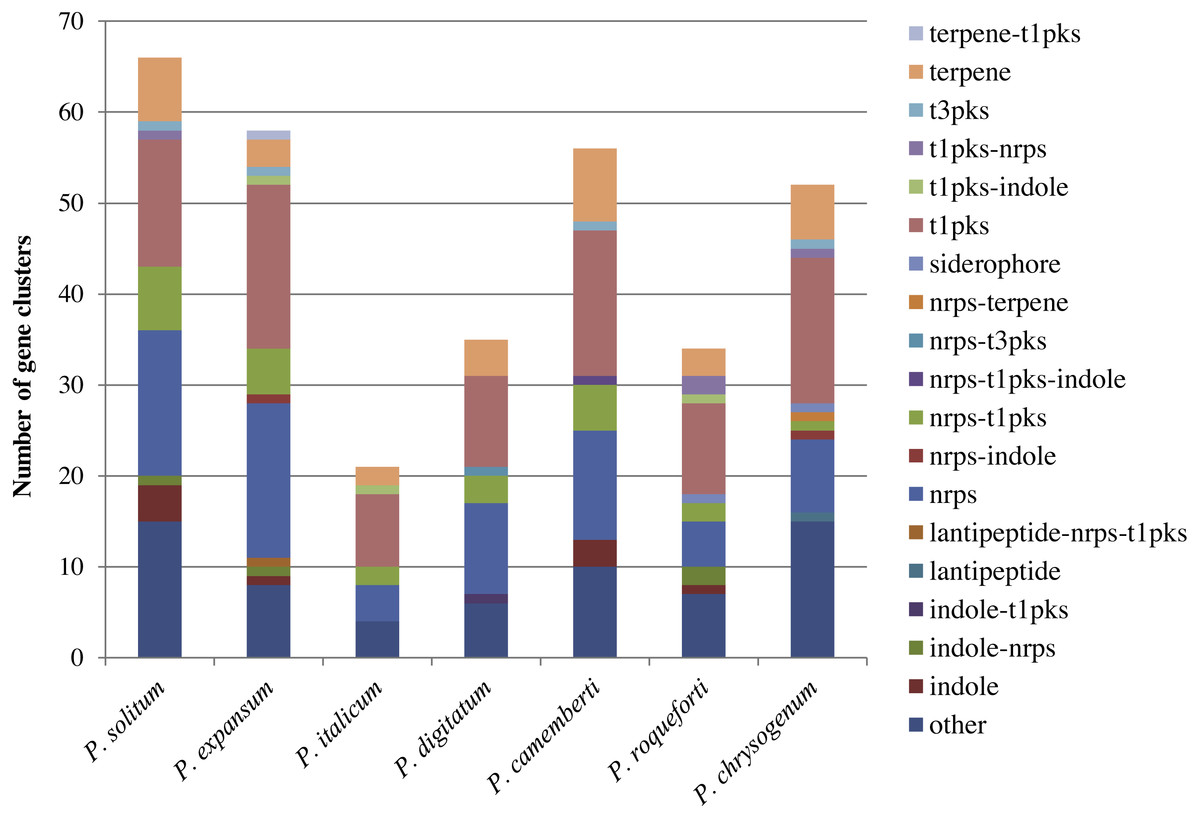
Figure 4: Composition of secondary metabolism gene clusters and their backbone genes in seven different Penicillium species.
SM gene cluster types in seven different Penicillium spp.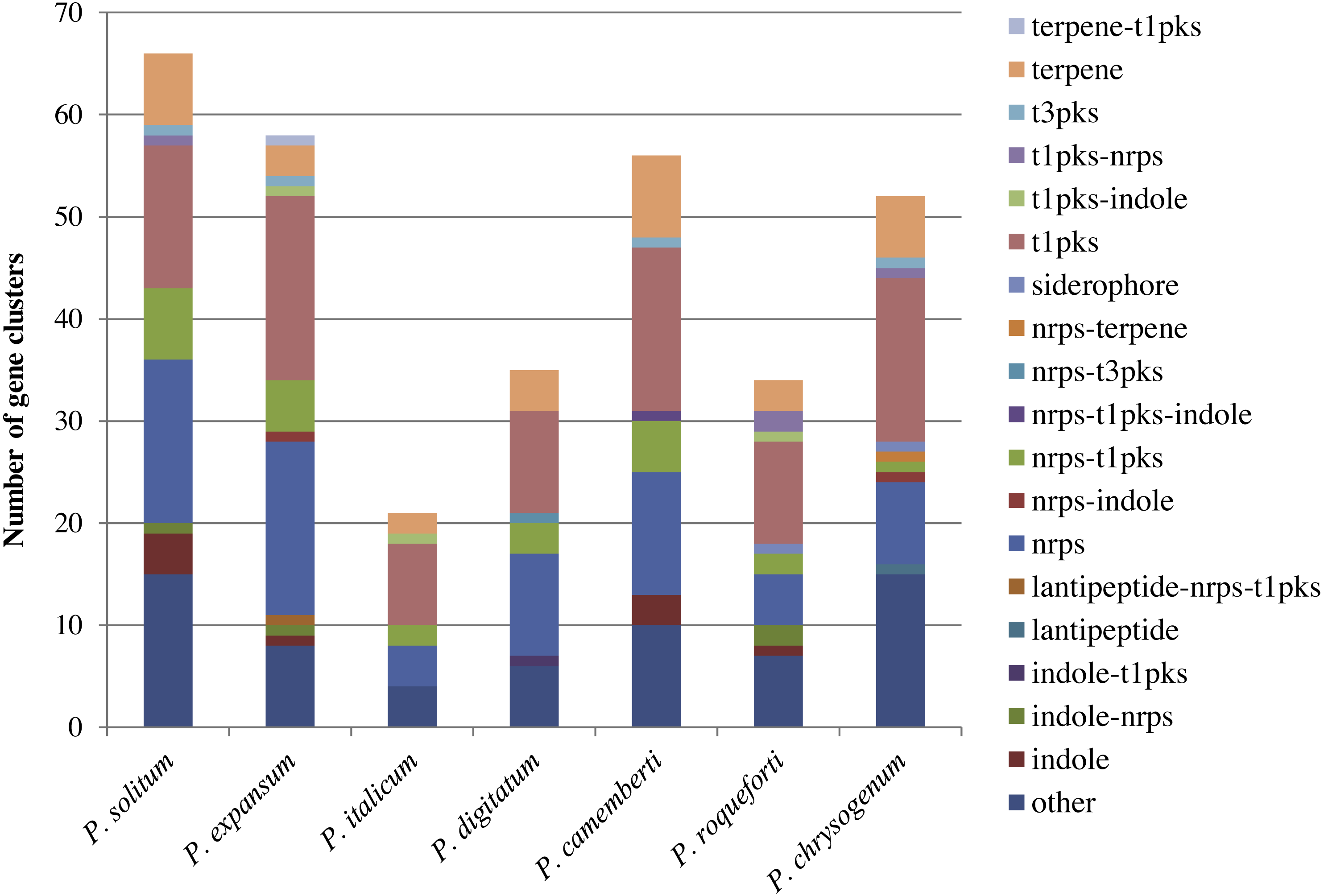{kind=link}
Discussion
The first aim of this study was to provide a high quality, annotated, and assembled genome sequence of P. expansum (R19), so that it could be used for further downstream bioinformatic analyses. The longer reads achieved by the PacBio platform enabled better assembly and resulted in fewer gaps than illumina sequencing has produced. PacBio SMRT sequencing technology allows for Hence, an assembly of 16 unitigs with an N50 value of 8.17 Mbp was achieved, a major improvement over the 48.5 kbp previously achieved using Illumina (Yu et al., 2014).
Although P. expansum and P. solitum cause blue mold decay of pome fruits (Frisvad, 2004; Jurick et al., 2010), the closest relative of P. solitum was determined to be P. camemberti, a species named after the distinctive soft cheese with a white rind (Cheeseman et al., 2014). In addition to causing blue mold on apple, P. solitum has been isolated from spoiled processed meats, cheeses, and margarine which are commonly stored at 4 °C like apples (Pitt et al., 1991; Hocking et al., 1998). Moreover, P. solitum has been isolated from several very cold, high salt environments and has been termed an “extremophile” (Stierle et al., 2012). In contrast, for P. expansum, the closest relatives in our analyses were P. digitatum (Marcet-Houben et al., 2012) and P. italicum (Ballester et al., 2015) indicating that ancestors of these three species may have occupied the same carbohydrate-rich niches associated with fruit decay.
| Species | Nr. of sequences | Group | Habitat |
|---|---|---|---|
| Amphimedon queenslandica | 1 | Sponge | Aquatic |
| Thelohanellus kitauei | 1 | Myxozoa | Aquatic/Parasitic |
| Thalassiosira pseudonana | 1 | Diatom | Aquatic |
| Chlamydomonas reinhardtii | 1 | Green algae | Aquatic |
| Volvox carteri | 2 | Green algae | Aquatic |
| Chlorella variabilis | 1 | Green algae | Aquatic |
| Ostreococcus tauri | 1 | Green algae | Aquatic |
| Auxenochlorella protothecoides | 2 | Green algae | Aquatic |
| Phytophthora sojae | 1 | Oomycetes | Plant pathogenic |
| Phytophthora ramorum | 1 | Oomycetes | Plant pathogenic |
| Fusarium graminearum | 4 | Ascomycetes | Plant pathogenic |
| Colletotrichum sublineola | 2 | Ascomycetes | Plant pathogenic |
| Penicillium expansum | 1 | Ascomycetes | Plant pathogenic |
| Penicillium solitum | 1 | Ascomycetes | Plant pathogenic |
| Setaria italic | 2 | Higher plants | Soil |
| Triticum aestivum | 2 | Higher plants | Soil |
By comparing the two blue mold fungi at the whole-genome scale, our aim was to identify differences in genetic factors that are linked to pathogen aggressiveness during pome fruit rot in addition to finding genes specific to the apple fruit pathogens. Thirty-six protein families were present only in the two pome fruit pathogens (P. expansum and P. solitum), but absent in the other five Penicillium species examined.. In P. expansum there were 222 gene families not found in P. solitum. of which 44 protein families were present only in P. expansum R19, but absent in the other species analyzed in this study. These protein families are of primary interest since they may represent specific genes involved in pome fruit maceration or account for P. expansum’s increased aggressiveness during apple fruit decay. Here, we discuss the potential role of several P. expansum and P. solitum-specific enzymes.
To examine similarities and differences in gene repertoires amongst the seven Penicillium species, we identified protein families using a Markov cluster algorithm called TRIBE-MCL that detects and categorizes eukaryotic protein families (Enright, Van Dongen & Ouzounis, 2002). Gene ontology (GO) enrichment analyses were performed using annotated protein sequences via Blast2go (Conesa et al., 2005). Several categories related to combating plant defense mechanisms were enriched, including phenylpropanoid catabolic process, cinnamic acid catabolism, and response to iron ion sequestration (siderophores). Flavonoid metabolism, a specific gene category involved in plant defense, was enriched, which may be beneficial for the fungus to overcome the phenolic-rich (quercetin) environment of the apple host tissue, known to be important in basal defense systems of apple fruits (Sun et al., 2017). These categories included lyase activity. It is well established that lyases degrade cell wall components like pectin and they have been shown to be involved in the maceration of pome fruit tissues by P. expansum and P. solitum (Yao, Conway & Sams, 1996; Jurick et al., 2009).
Single copies of SnoaL-like polyketide cyclases (PF07366) are found in both P. expansum and P. solitum (Table S1). A more detailed analysis of PF07366 on http://pfam.xfam.org/ reveals their presence in a number of prokaryotes, in two other fungal species, and a few other eukaryotes (Table 4). These eukaryotic members are either aquatic or plant related (Table 4), suggesting that this gene might have been laterally transferred between organisms living in close proximity or occupying the same ecological niche. These cyclases are backbone genes for polyketide secondary metabolite biosynthesis and thus involved in fungal secondary metabolism. A Glucose-6-phosphate isomerase (PF10432) is found in a number of prokaryotes and only two eukaryotes, P. expansum and P. solitum (http://pfam.xfam.org/, Table S1), suggesting that this gene might have been laterally transferred. The exact function it plays in pome fruit decay warrants further exploration via RNAi and /or gene deletion studies.
A thermolysin metallopeptidase (PF01447) was found only in P. expansum, but not in P. solitum or any of the other five Penicillium species included in this study. Peptidases are known virulence factors for fungal plant pathogens such as Fusarium graminearum. They break down plant host proteins to provide nutrients for fungal growth, reproduction, and colonization (Lowe et al., 2015). This P. expansum unique metallopeptidase may play a role in pome fruit decay and should be explored further. In addition to the above-mentioned examples, several putative transcription factors, hydrolases, and other enzymes that are unique to one or both blue mold Penicillium species were uncovered (Table S1). They provide a guide for future research to pinpoint the genes essential for postharvest apple fruit decay.
Patulin is a well-studied, polyketide-derived mycotoxin commonly found in apple products. Due to its carcinogenic effects, the European Union (EU) has set limits on the maximum allowable amount of patulin to 50 µg/L for juice and fruit derived products, 25 µg/L for solid apple products, and 10 µg/L for juices and food for babies and infants (European Union, 2003). The U.S. Food and Drug Administration (FDA), as well as regulatory agencies in other countries, have imposed limits on the permissible amounts of patulin in fruit juices and processed pome fruit products for human consumption at 50µg/L (Puel, Galtier & Oswald, 2010). There are 15 genes involved in patulin biosynthesis found within a well-defined gene cluster (Tannous et al., 2014; Li et al., 2015; Ballester et al., 2015). The structural organization of the cluster is shown in the same order in our P. expansum R19 strain as reported earlier (Tannous et al., 2014). The partial patulin gene cluster arrangement in P. solitum was very similar to that found in P. camemberti (Ballester et al., 2015), with the same seven patulin biosynthetic genes present and arranged in the same configuration. The incomplete patulin gene cluster found in both P. solitum and in P. camemberti may explain why these species are unable to produce patulin in either liquid or solid culture conditions (Frisvad, 2004). Curiously, some of the patulin genes were dispersed outside of the cluster, likely due to chromosomal rearrangements. Our genomic data suggests that the ability to produce patulin was lost in the ancestor of these two species.
While patulin is the mycotoxin of primary concern to apple producers, processors and packers in the U.S. and EU, it is only one of many secondary metabolites known to be produced by Penicillium species (Puel, Galtier & Oswald, 2010; Wright, 2015). Genes responsible for the synthesis of secondary metabolites are generally organized together in clusters in fungal genomes. Despite the large number of putative SM gene clusters in these Penicillium species, only a few gene products such as patulin, penicillic acid, and citrinin are well studied in agriculture and food science. In addition, because P. solitum is an extremophile, there are reports of several interesting secondary metabolites isolated from unusual environments that merit further study in order to determine if they are produced in apples with blue mold symptoms. For example, a strain of P. solitum isolated from The Berkeley Pit, a former copper mine located in Butte, Montana produces two drimane sesquiterpene lactones named Berkedrimanes A and B (Stierle et al., 2012). These secondary metabolites inhibit the signal transduction enzymes caspase-1 and caspase-3 and mitigate the production of interleukin 1- β in a leukemia cell line (Stierle et al., 2012). Another biologically active metabolite from P. solitum is solistatin, a phenolic compactin analogue related to Mevastatin, one of the classes of statins which inhibit HMG-CoA reductase, which are widely prescribed as cholesterol lowering agents (Sørensen et al., 1999).
Across the fungal kingdom, however, the products of bioinformatically discovered SM gene clusters are largely unknown. Some clusters are functional and expressed only under specific conditions (e.g., during competition for nutrients or stress conditions), but not expressed under routine conditions in the laboratory (Tannous et al., 2014; Li et al., 2015). In light of our recent findings, it remains unexplored whether the large number of bioinformatically detected SM gene clusters in the P. solitum genome translates into a greater capacity for producing various secondary metabolites on apple, and if any of these metabolites might pose hitherto undiscovered risks to human health. Further characterization of these identified gene clusters, via functional gene deletion studies, will help ascertain our understanding of SM gene cluster function and the mechanisms of secondary metabolism/biosynthesis.
Conclusions
Comparison of the genomes of P. expansum and P. solitum has provided a unique opportunity to explore the genetic basis of the differential ability of these two species to cause blue mold decay. Key genes identified here can now be analyzed functionally to confirm their involvement in apple fruit decay. Fungal gene networks, pathways, and regulators can be exploited to design specific control strategies to block decay. Additionally, our comparative genomic data has clarified phylogenetic relationships between Penicillium species that occupy different ecological niches. Additionally, the SM gene cluster repertoire has been elucidated in both P. expansum and P. solitum and provided evidence to explain the lack of patulin production in P. solitum (RS1), while also revealing the potential for discovery of hitherto unknown secondary metabolites of possible biotechnological use. In agriculture, these genomic findings will guide apple fruit producers to help maintain safe, high quality apples during long-term storage, as well as being of interest to the food processing industry, plant pathologists, and the broader scientific community.
Supplemental Information
Gene clusters in P. expansum (R19) and P. solitum (RS1)
Gene cluster identities in Penicillium expansum and P. solitum.