
Identification of diabetes related phenotype and diagnostic biomarkers in coronary artery disease
- Published
- Accepted
- Received
- Academic Editor
- Fanglin Guan
- Subject Areas
- Bioinformatics, Biotechnology, Cardiology, Diabetes and Endocrinology
- Keywords
- Diabetes mellitus, Coronary artery disease, Mouse CAD model, Biomarkers
- Copyright
- © 2025 Aihemaiti et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2025. Identification of diabetes related phenotype and diagnostic biomarkers in coronary artery disease. PeerJ 13:e20117 https://doi.org/10.7717/peerj.20117
Abstract
Background
Diabetes mellitus (DM) and coronary artery disease (CAD) are closely interrelated clinical conditions. However, the combination analysis based on DM related CAD diagnostic model remains a gap. The primary objective of this study was to identify diagnostic models and diagnostic markers for CAD based on the association of diabetic phenotypes and attempt to explore them further in a mouse model.
Methods
We used data integration as well as multiple datasets for both coronary artery disease and diabetes to exclude bias as well as to improve reliability. We employed the least absolute shrinkage and selection operator (LASSO) regression algorithms to construct the CAD diagnostic model. Furthermore, we established mouse CAD model (low-density lipoprotein receptor deficient mice with high fat diet) to explore the crosstalk between the screened biomarkers and severe CAD progress.
Results
The intersecting genes from differential analysis and weighted correlation network analysis (WGCNA) results yielded 32 diabetes-related biomarkers. We then identified two diabetes-related phenotypes through the consensus clustering in CAD patients. Microenvironmental analysis revealed that phenotype 1 exhibited higher expression of most cytokines, inflammatory factors, interleukins, and related receptors. Immune cell composition in phenotype 1 showed increased infiltration compared to phenotype 2. The LASSO regression identified 16 diabetes-related genes and we further constructed a diagnostic model based on these genes, which the area under the curve (AUC) reached 0.8. Additionally, single cell immune analysis exhibited the location of these genes. KCNQ1, ATP6V1B1, MTDH, and ITPK1 were predominantly located in macrophages, indicating their potential in regulating macrophage during myocardial injury. Furthermore, We elucidated that KCNQ1 and ITPK1 exhibited high expression level in mouse CAD model in tissue level. exhibited similar expression trends with macrophage biomarkers (CD31 and CD68). The result of qPCR also indicated the elevated level of KCNQ1 and ITPK1, which exhibited crosstalk with CD31 and CD68 in mouse CAD model.
Conclusion
This study delves into the microenvironmental characteristics of diabetes-related phenotypes in CAD, constructing an optimal diagnostic model and validated the significance of diagnostic markers in mouse CAD model, which may offer insights that could be beneficial for clinical management in the near future.
Introduction
Coronary artery disease (CAD) is predominantly characterized by atherosclerosis, a complex disorder characterized by damage to the artery intima (Ben Braiek et al., 2021). It manifests in forms such as stable angina, unstable angina, myocardial infarction (MI), or sudden cardiac death (Iida, Harada & Takebayashi, 2019). Key risk factors like hypertension, hyperlipemia, diabetes mellitus (DM), and smoking are identified as significant contributors to the disease at the population level (Malakar et al., 2019). In 2017, CAD was accountable for roughly 8.93 million deaths, making up about 16.0% of global death, which is expected to remain the leading cause of death worldwide until 2030 (Xu et al., 2023). Coronary angiography is acknowledged as the definitive method for diagnosing the presence and extent of CAD (Li & Kronenberg, 2021). Despite considerable progress in medical treatments, CAD remains a critical public health issue globally (Tseng et al., 2020). Additionally, individuals diagnosed with coronary atherosclerotic heart disease (CHD) are at an increased risk of subsequent cardiovascular events, including MI, stroke, and chronic heart failure (CHF) (Xia et al., 2021), which may reduce the possibility of long-term survival of patients. Hence, Identification of effective diagnostic markers for CAD patients remains an urgent problem.
DM is identified by a variety of heterogeneous disorders resulting from either insulin deficiency or insulin resistance, leading to high blood glucose levels (Bao, 2022). Both type 1 and type 2 diabetes cause organ-specific tissue damage due to multiple metabolic factors (Fan et al., 2022). Interestingly, DM is also a significant risk factor for CHD. Chronic hyperglycemia in DM (Eid et al., 2019) not only harms various organs and tissues but also leads to microvascular complications such as retinopathy (Pignalosa et al., 2021), nephropathy, and neuropathy, as well as macrovascular complications like cardiovascular disorders (Darenskaya, Kolesnikova & Kolesnikov, 2021). The mechanisms behind hyperglycemia-induced vascular damage are complex and not fully understood. However, it is thought that increased intracellular glucose levels enhance the production of reactive oxygen species, subsequently disrupting vital downstream pathways (Cole & Florez, 2020; Vergès, 2020). Additionally, the hyperglycemic state resulting from diabetes may activate protein kinase C signaling and the late glycation pathway, leading to vascular inflammation as well as CAD (Henning, 2018). Enhancement of the hexosamine biosynthetic pathway and activation of the protein O-GlcNAcylation have also been shown to play an important role in diabetes-associated vasculopathy (Wang et al., 2023). Macrophages may be the crosstalk between DM and CAD. Peng & Gao (2022) found that NLPR3+ macrophages can exacerbate in vivo inflammation in diabetic patients. Macrophages also play an important role in cardiac remodeling in patients with CAD (Yap et al., 2023). Interestingly, plaque in coronary arteries is usually larger in diabetic patients than in nondiabetic patients, mainly due to inflammation caused by macrophages and T lymphocytes (Yahagi et al., 2017). These fundamental studies involved cellular and clinical models reveals the deep mechanisms of the DM and CAD. However, it lacked the comprehensiveness of integrated large-sample data and combinational analyses to some extent. Ideal clinical markers may be identified form the perspective of DM related CAD analysis.
In our study, we first explored diagnostic markers as well as diagnostic models for CAD patients from crosstalk with diabetes. Based on multiple fused datasets, we try to validate the identified diagnostic markers in a mouse model to further explore their diagnostic value in patients with CAD.
Materials and Methods
Data sources and preparation
The datasets concerning diabetes (2025) and CAD (Chen et al., 2023) predominantly derive from the GEO database. The diabetes datasets include GSE20966 (20 samples with diabetes), GSE25724 (six of 13 total samples have diabetes), and GSE38642 (nine of 63 total samples have diabetes), and the CAD datasets include GSE12288 (222 samples in total, with 110 samples in CAD), GSE20680 (195 samples with CAD), GSE20681 (198 samples in CAD), and GSE42148 (24 samples, with 13 samples in CAD). The single cell dataset was GSE32678. Additionally, to increase the sample size and confidence level, various datasets of patients with diabetes mellitus and coronary heart disease were merged, with batch effects excluded by “combat” packages for optimal analysis. For missing values, we fill them with the mean value. For data cleansing, we correct formatting errors, remove duplicate values, and remove unneeded data to ensure data cleanliness. For data with large variance, we usually use the “normalize” function to normalize the data, making the data comparable. In addition, for outliers, we usually remove them or fill them with mean values. Specific dataset inclusions and workflow are detailed in the supplementary material.
WGCNA analysis and differential analysis to identify diabetes related markers
Weighted correlation network analysis (WGCNA) is a systems biology approach for characterizing gene association patterns between samples (Langfelder & Horvath, 2008). This analysis focuses on identifying co-expressed genes based on gene-set introgression and gene-set-phenotype associations. We utilized WGCNA to pinpoint modules co-expressed with diabetes and the core genes in the network.
Identification of diabetes phenotypes in CAD patients
The intersecting genes from WGCNA and differential expression analysis were employed to identify immune-related diabetes phenotypes. We identified these phenotypes using the unsupervised clustering algorithm (consensus clustering). The main basis for the classification is the CDF value and delta region in the consensus clustering algorithm, when the CDF curve is the most downward and the delta region has the largest change, it means that this algorithm has the best classification effect.
Identification diabetes related coronary artery disease diagnostic system
For more efficient results, we applied the LASSO regression algorithms to reduce dimensionality and identified 16 diabetes-related genes. These genes were used to construct the diagnostic monitoring model for CAD and to measure the area under the curve (AUC) to evaluate the diagnostic capacity. The model was constructed by “glmnet” package (seed(123), alpha = 1, nfold=100).
Single-cell landscape analysis
The “Seurat” package was applied for single-cell sequencing analysis. The criteria for cell screening were defined as nCount_RNA >100, nFeature_RNA <5,000, percent.mt <25, and nFeature_RNA >100. Only the top 3,000 variable genes were then chosen for additional examination. Using principal component analysis (PCA), significant dimensions with p < 0.05 were found. Dimensionality reduction was made possible via the t-distributed stochastic neighbor embedding (tSNE) approach, which was combined with an 11-cluster classification analysis conducted on all cells. The “singleR” package was used to annotate cell clusters based on trends in the composition of marker genes.
Vertebrate animal study methods
This study was approved by Animal Experimental Medicine Ethics Committee of the First Affiliated Hospital of Xinjiang Medical University, Approval number: 20210301-215. Twenty-eight-week-old male C57/B6J wild-type and LDLR−/− mice were obtained from the Xinjiang Medical University experimental animal center’s specialized pathogen-free (SPF) barrier system. A high-fat diet consisting of 78.85% chow diet, 21% lard, and 0. 15% cholesterol was provided to the model group whereas the control group was fed chow diet. An injection of pentobarbital was used to induce anesthesia. We use carbon dioxide for the euthanasia of mice.
Mouse euthanasia operation procedures
We placed the mice to be executed in empty boxes and used carbon dioxide asphyxiation to execute them. Criteria established for euthanizing animals prior to the planned end of the experiment was the State Standard of the People’s Republic of China, GB/T 39760-2021.
Gene silence of KCNQ1 and ITPK1
We knocked down of KCNQ1 and ITPK1 through siRNA. The sequence was as follow: KCNQ1(SiRNA): CCGCCUGAACCGAGUAGAATT; ITPK1(SiRNA): AUCUUGUCCAGCUCCGUCATT.
CAD mouse model construction
Twenty-eight-week-old male C57/B6J wild-type and LDLR−/− mice were grown in the Xinjiang Medical University experimental animal center’s specialized pathogen-free (SPF) barrier system. Following randomization, a high-fat diet consisting of 78.85% chow diet, 21% lard, and 0. 15% cholesterol was provided to the CAD model group (LDLR−/− mice) continuously for 12 weeks, whereas the control group was fed chow diet. To create a model of a myocardial infarction, the anterior descending branch of the mice was clamped for seven days at the conclusion of the twelfth week. Changes in electrocardiogram before and after modeling were observed, and blood lipids and cardiac enzymes were measured 2 weeks after surgery to determine whether coronary artery disease occurred in mice. Sections of mouse heart tissue were obtained for immunohistochemistry (IHC) staining. The IHC staining of tissue slices from the cardiac tissues of the two groups (five mice in each group) was then examined (Fig. 1).
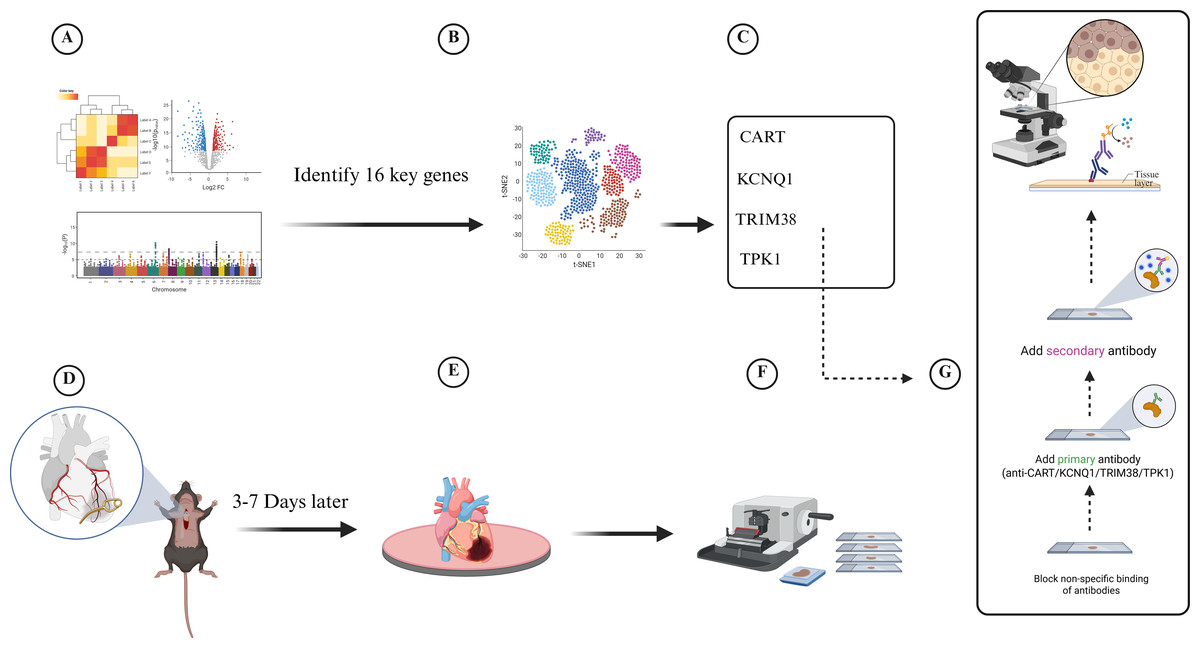
Figure 1: Experimental flowchart for transcriptomic and single-cell data to develop a diabetes-related phenotype and its microenvironment influence in coronary artery disease.
(A) Analyze the gene information of patients in GEO database, (B) Analyze single-cell data from patients with coronary myocardial injury, (C) Identify four key genes in diabetes-related CAD, (D) Establish myocardial infarction model, (E) Euthanasia of mice and removal of heart tissue, (F) Collect and section heart tissue, (G) Immunohistochemistry assay.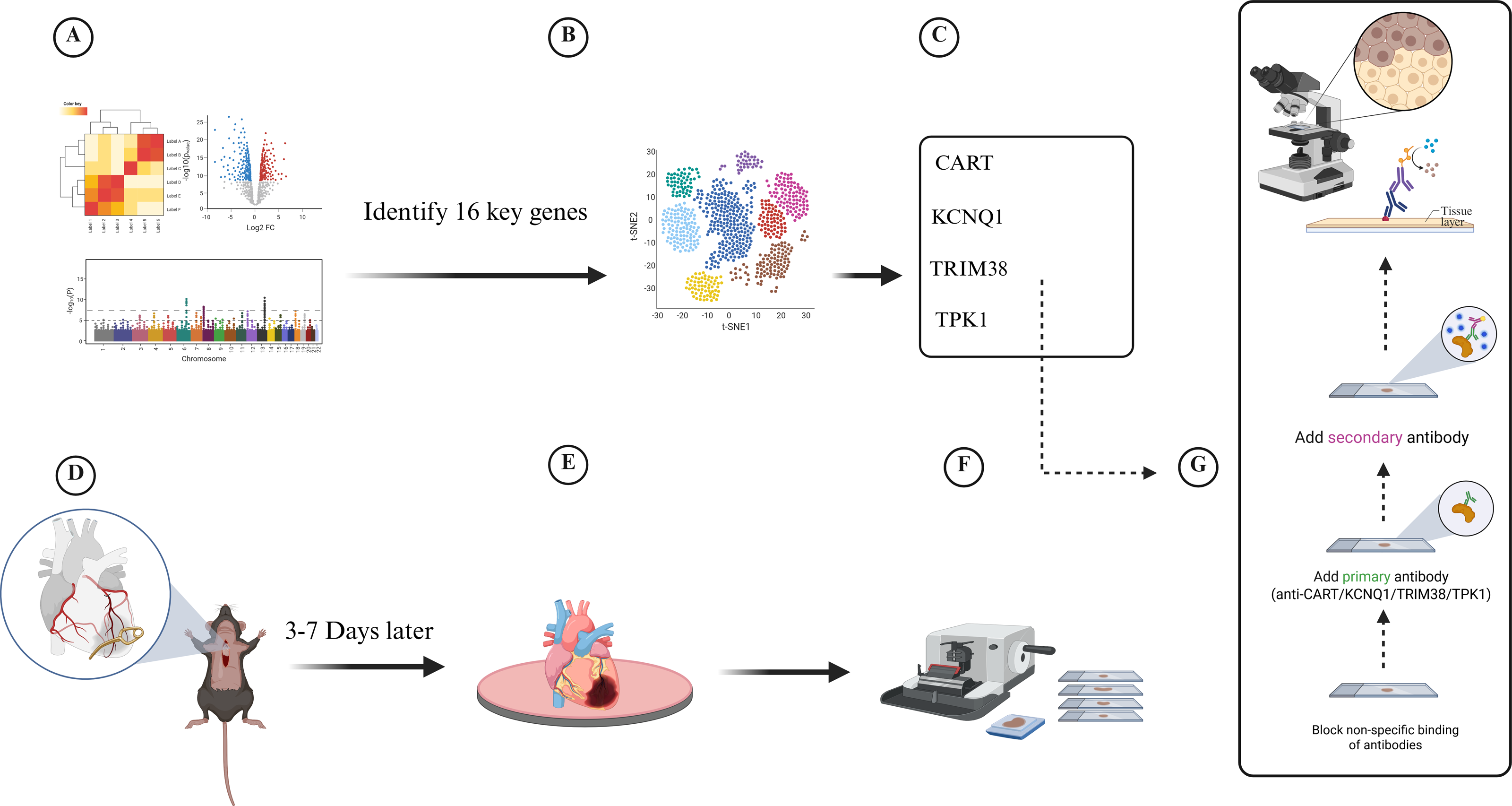{kind=link}
Flow cytometry analysis
Apoptosis rates were determined using the Annexin V/PE Apoptosis Detection Kit (BD Biosciences, Franklin Lakes, NJ, USA). Cells were collected with 1 × binding buffer. Subsequently, five µL of Annexin V-PE and five µL of 7-AAD were added, and the incubation was carried out for 5 min in the absence of light. The analysis of apoptosis was finally conducted using a FACSCalibur ffow cytometer.
Immunohistochemistry staining
Detailed procedures for immunohistochemistry can be found in the classic literature on Tavitian et al. (2025). Dimethylbenzene was used to dewax pathological sections, and gradient alcohols were used to hydrate them. Sections were then subjected to boiling sodium citrate (pH 6.0) for antigen retrieval and 3% H2O2 to deactivate endogenous peroxidase. Sections were then treated in turn with primary antibody, HRP-linked secondary antibody, and 5% goat serum. A DAB substrate was used to further visualize the indicated antibody staining. Sections were then examined under a bright-field microscope after being restained with hematoxylin and sealed with neutral glue. The staining intensity (negative, 0; weak, 1; moderate, 2; strong, 3) and the percentage of positive signal (negative, 0; 1–30%, 1; 31–60%, 2; above 60%, 3) were used to assess the protein expression. The IHC score was obtained by multiplying the two scores mentioned above. The main antibodies we used were KCNQ1 (A2174, 1:400; ABclonal, Waltham, MA, USA), ITPK1 (10568-AP-1, 1:500: Proteintech, Rosemont, IL, USA), CD31 (11265-1-AP, 1:600: Proteintech, Rosemont, IL, USA), CD68 (28058-1-AP, 1:300: Proteintech, Rosemont, IL, USA).
Statistical analysis
R version 4.1.0 was the primary tool for all bioinformatics analyses and related statistical calculations. For biological experiment statistical analysis, GraphPad Prism version 8.1 was used. Regarding the multiple testing methods for the statistical analysis, we have used FDR control methods (the BH method). Regarding cross-validation of diagnostic models. We divided the fused dataset into training and testing datasets according to 1:1, and performed cross-validation and correction in the two datasets,
Results
WGCNA and differential analysis to identify key diabetes-related gene markers
To reduce data analysis inaccuracies due to small sample sizes, data from three GEO platform datasets were merged, carefully excluding batch effects to create a combined dataset of diabetic patients and control samples. Initial analysis showed significant data dimension disparities between datasets (Fig. 2A). However, post-fusion, the data exhibited nearly uniform distribution patterns (Figs. 2B, 2C). Subsequent analysis of variance identified differential genes between diabetic patients and healthy controls (p < 0.05 and abs (log (FC)) >1), revealing 2,250 differential genes (Fig. 2D). Further, WGCNA analysis was conducted to isolate modular genes strongly linked to diabetes (Fig. 2E). In the WGCNA procedure, the soft threshold β was set at 9. For network visualization, 400 of these genes were randomly selected (Fig. S1). The WGCNA network was constructed using the partitioning method, merging and analyzing modules with high similarity, ultimately identifying 22 different gene modules (Fig. 2E). Correlation analysis between gene modules and clinical data of diabetic patients indicated that the yellow module had the strongest correlation with diabetes (r = 0.53, p = 5e−6) (Fig. 2F). To pinpoint hub genes within the yellow module, 368 genes with GS >0.2 and MM >0.6 were classified as central diabetes-related genes. The intersecting genes from differential analysis and WGCNA results yielded 32 diabetes-related biomarkers (Fig. 2G).
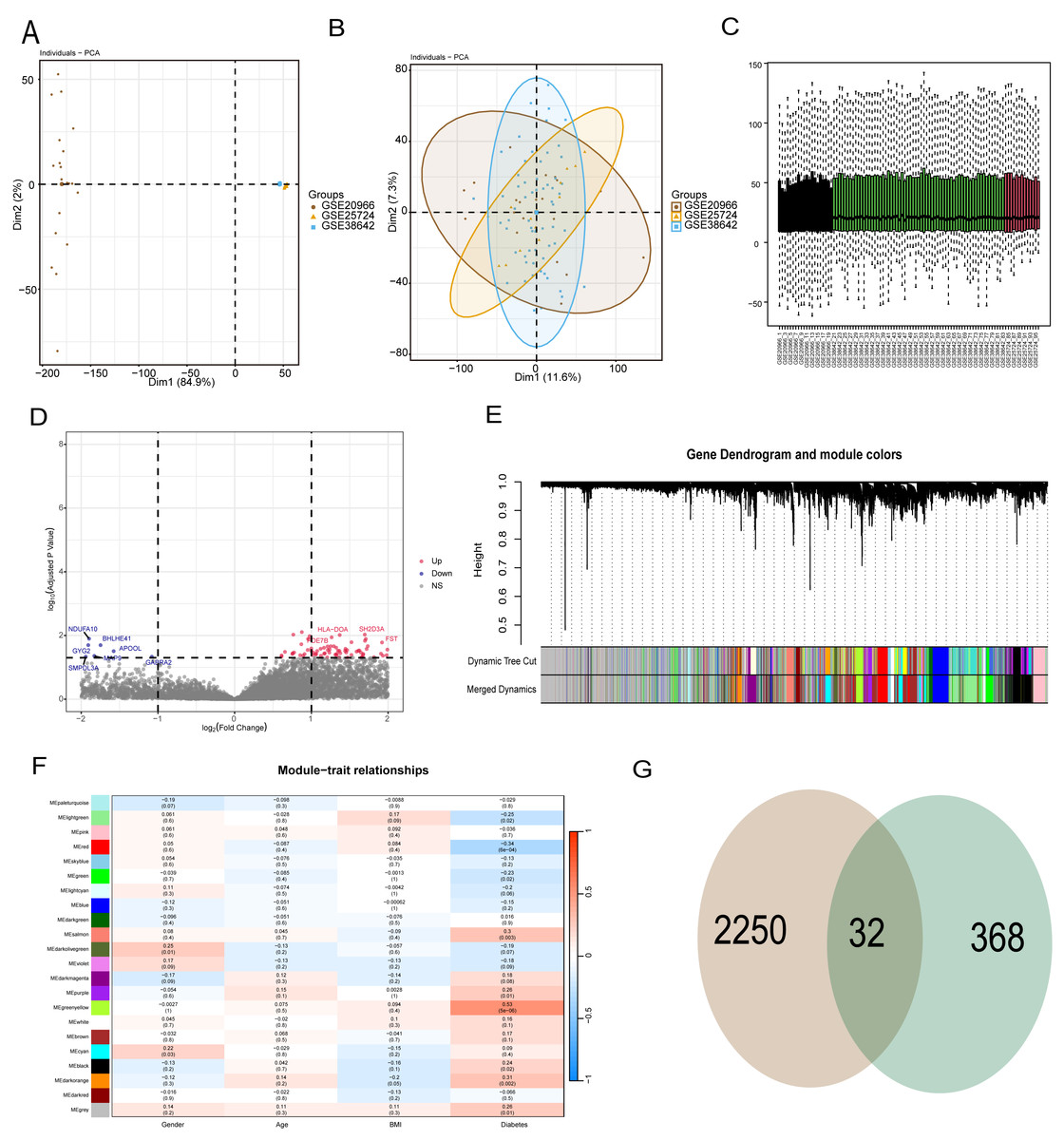
Figure 2: WGCNA and differential analysis to identify key diabetes-related gene markers.
(A) The data difference among datasets. (B, C) The data distribution after fusion. (D) The volcano plot between diabetic patients and healthy controls. (E) WGCNA analysis to identify different gene modules. (F) Correlation analysis between gene modules and clinical data of diabetic patients. (G) The intersecting genes from differential analysis and WGCNA results.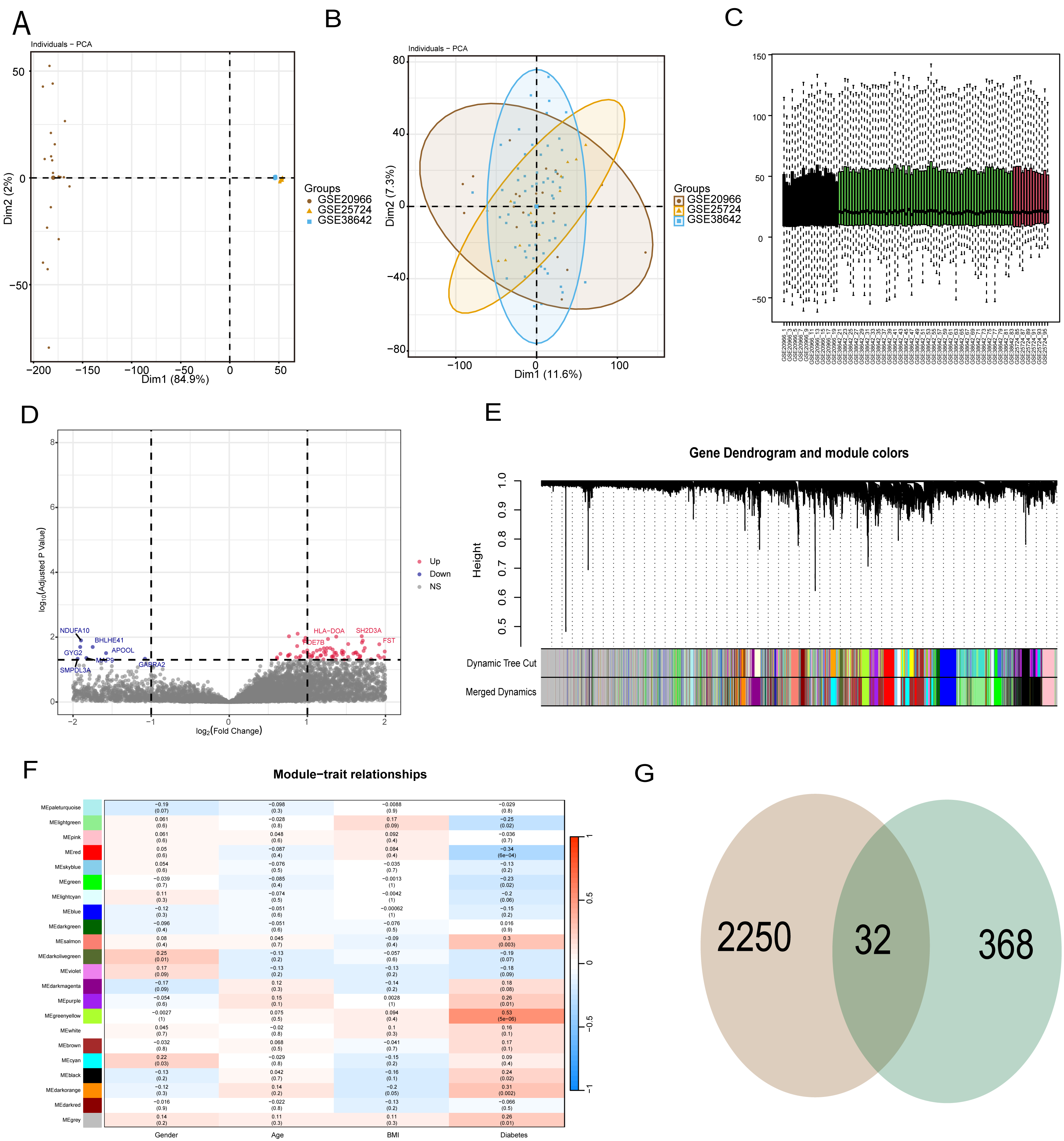{kind=link}
Diabetes related phenotype and characteristics in coronary artery disease
We employed the same methodology from the analysis of diabetes to boost sample size and lower error in the bioinformatics analysis of CAD patients. We reduced the batch effect by combining four datasets from the GEO platform. When the batch effect was excluded, the initial examination of the four datasets showed significant differences (Fig. 3A) that eventually became negligible (Fig. 3B). We also looked into the capacity of diabetes-related markers to classify patients with CAD, given the possible influence of diabetes on the development of CAD. We classified the gene expressions of 32 diabetes biomarkers in CAD patients using the consensus clustering method, which allowed us to identify diabetes-related phenotypes in CAD patients. Binary typing proved to be the most efficient method when comparing the delta areas and cumulative distribution function (PDF) curves of various categorization outcomes (Fig. S2). Consequently, CAD patients were divided into phenotype 1 and phenotype 2. Furthermore, we discovered that phenotype 1 had marginally higher expression of genes linked to diabetes than phenotype 2 (Fig. 3C). Additional examination of the microenvironmental scores revealed that phenotype 1 outperformed phenotype 2 in terms of stromal, immune, and estimation scores (Figs. 3D–3F).
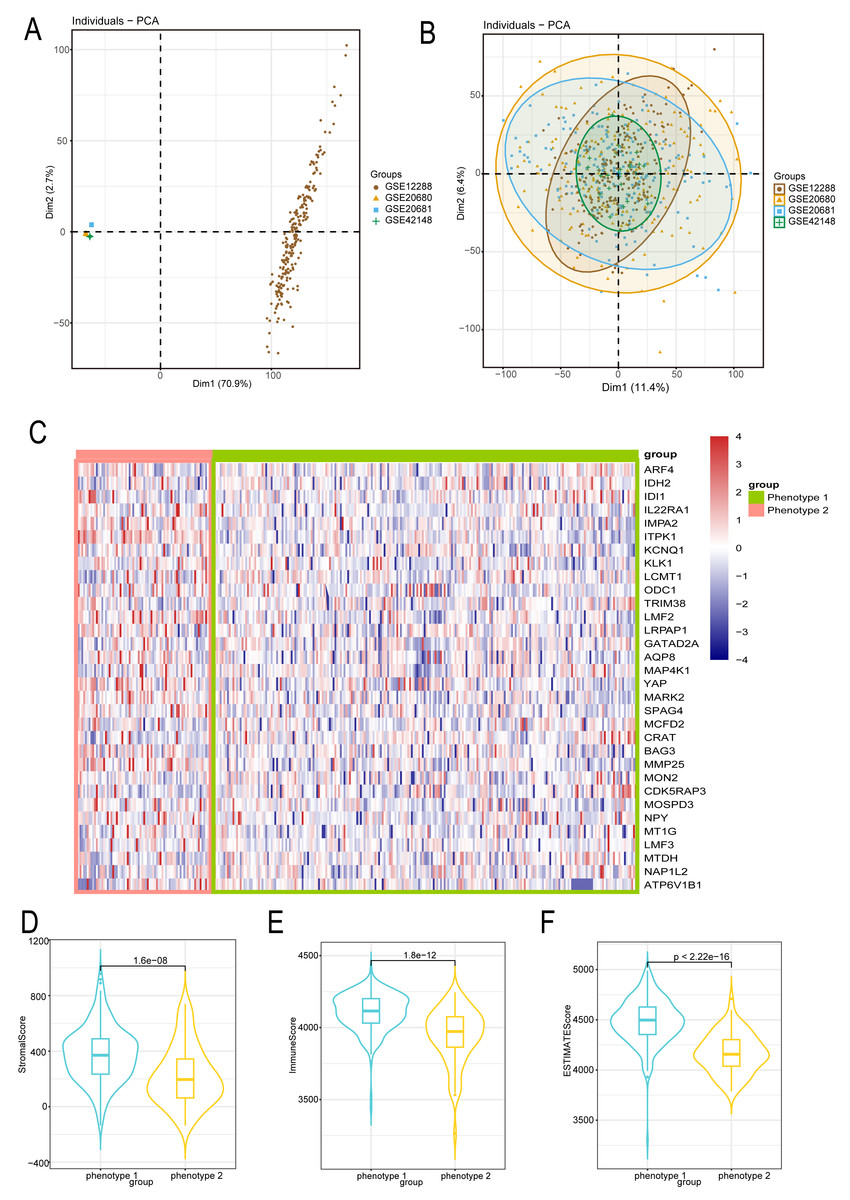
Figure 3: Diabetes related phenotype and characteristics in coronary artery disease (CAD).
(A) The data difference among datasets. (B) The data distribution after fusion. (C) The CAD phenotype based on consensus clustering. (D–F) The microenvironmental score analysis between different phenotypes.{kind=link}
Identification of microenvironment characteristics of diabetes related phenotype in CAD
We delved into the microenvironmental differences between diabetes-related phenotypes in CAD. We compared the phenotypes with regard to cytokines and inflammatory factors, which are important determinants of the immunological and inflammatory milieu. Typically, phenotype 1 had higher amounts of cytokines and inflammatory factors such CXCL4, CCL5, CXCR8, CXCR9, and other regulatory molecules, while phenotype 2 had lower levels of these factors (Fig. 4A). Furthermore, phenotype 1 also exhibited significant expression levels of numerous interleukins and associated receptors, such as IL2, IL3, PDGFD, etc., which are known to regulate inflammation (Fig. 4A). We also examined each phenotype’s immune cell composition. The estimation of immune cell infiltration was made using several algorithms. Phenotype 2 showed less immune cell infiltration than phenotype 1, which showed greater infiltration of immune cells such as CD8+ T cells, NK cells, etc. (Fig. 4B). Additionally, we examined the strength of endogenous and exogenous immunomodulation between the phenotypes. While endogenous immunity strength was similar between phenotypes, exogenous immunity was significantly stronger in phenotype 1 compared to phenotype 2 (Fig. 4C). Comparative analysis of classical immune features showed that promotive immune features were significantly more robust in phenotype 1 than in phenotype 2 (Fig. 4D).
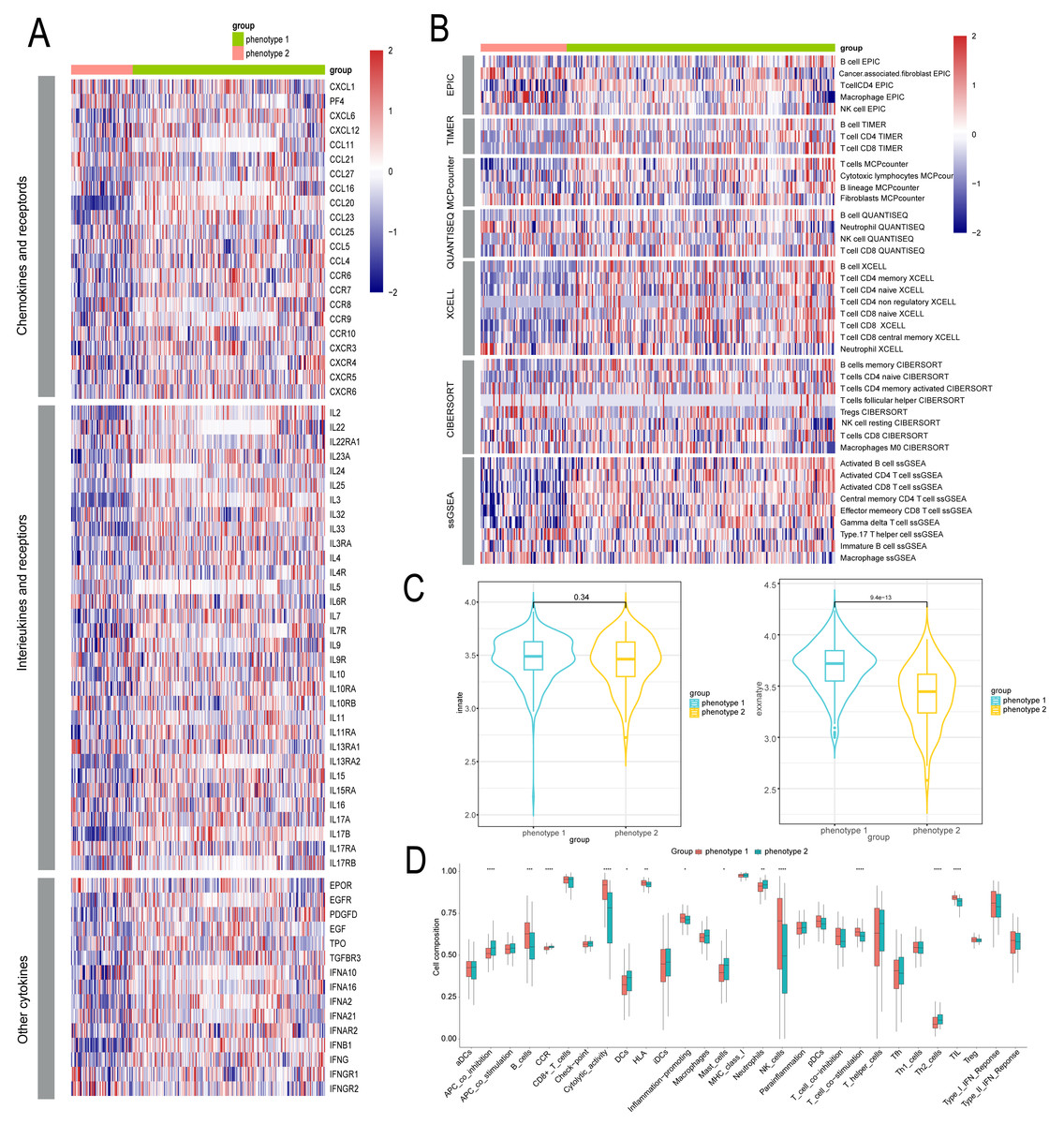
Figure 4: Identification of microenvironment characteristics of diabetes related phenotype in CAD.
(A) The inflammation feature between different phenotypes. (B) The immune microenvironment feature between different phenotypes. (C) The endogenous and exogenous immunity feature between different phenotypes. (D) The classical immune features between different phenotypes.{kind=link}
Diabetes related phenotypes differ in myocardial lesion signaling and metabolic characteristics
Recognizing the significant distinctions between the two diabetes-related phenotypes, we looked at the differential pathways in the 2,180 differential genes that were found. Figure 5A shows a volcano diagram of these genes. We conducted Gene Set Variation Analysis (GSVA) for 10 changed pathways common to CAD and compared these between the two phenotypes, indicating considerable route variations (Fig. 5B). The subgroups’ different metabolic and myocardial injury-related pathways were revealed using enrichment analysis. Gene Ontology (GO) analysis identified biological process differences primarily in “fatty acid metabolic process,” “response to glucocorticoid,” “heart morphogenesis,” and other metabolism-related processes. Additionally, immune-related processes like “leukocyte cell–cell adhesion” and “regulation of innate immune response” were significantly enriched (Fig. 5C). KEGG analysis further enriched metabolic signals such as “Lipid and atherosclerosis” and “Carbon metabolism,” along with myocardial lesion signals like “Diabetic cardiomyopathy” and “Cardiac muscle contraction” (Fig. 5D). We investigated metabolic differences using the MSigDB database, taking into account the variation in metabolic signals among different phenotypes. The most important route variations are represented by a heatmap (Fig. 5E), which mainly shows the metabolic transport and absorption of proteins, fatty acids, glucose, and heart disease-related pathways. The significant distinctions between these symptoms, as revealed by the GSEA analysis, include alterations in metabolism, chemokines, and diabetic cardiomyopathy (Fig. 5F).
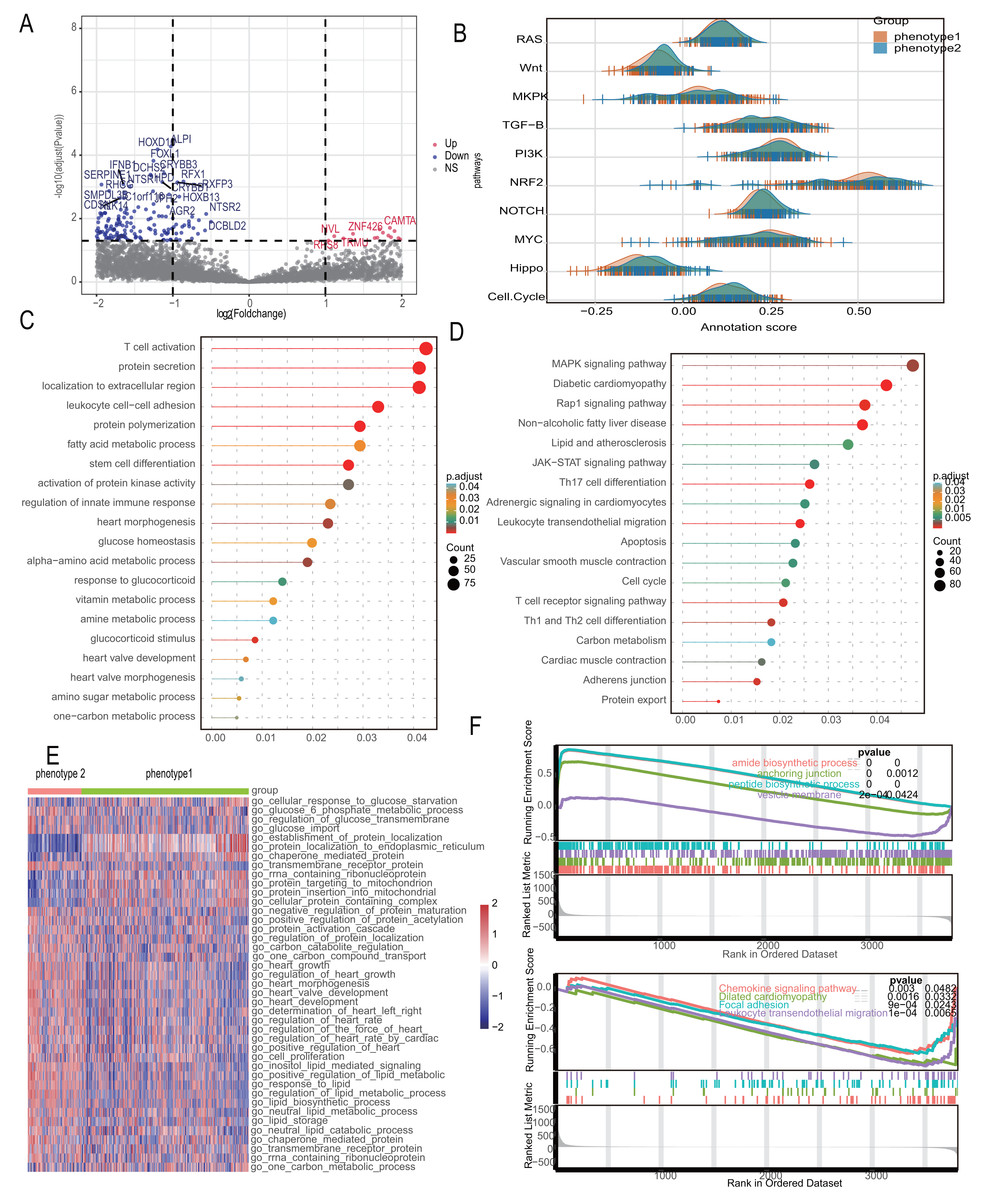
Figure 5: Diabetes related phenotypes differ in myocardial lesion signaling and metabolic characteristics.
(A) Volcano diagram to show the differential gene between diabetes-related phenotypes. (B) Pathway variances in different phenotypes. (C) GO analysis based on the differential genes. (D) KEGG analysis based on the differential genes. (E) A heatmap to visualize the metabolic difference. (F) GSEA analysis between the phenotypes.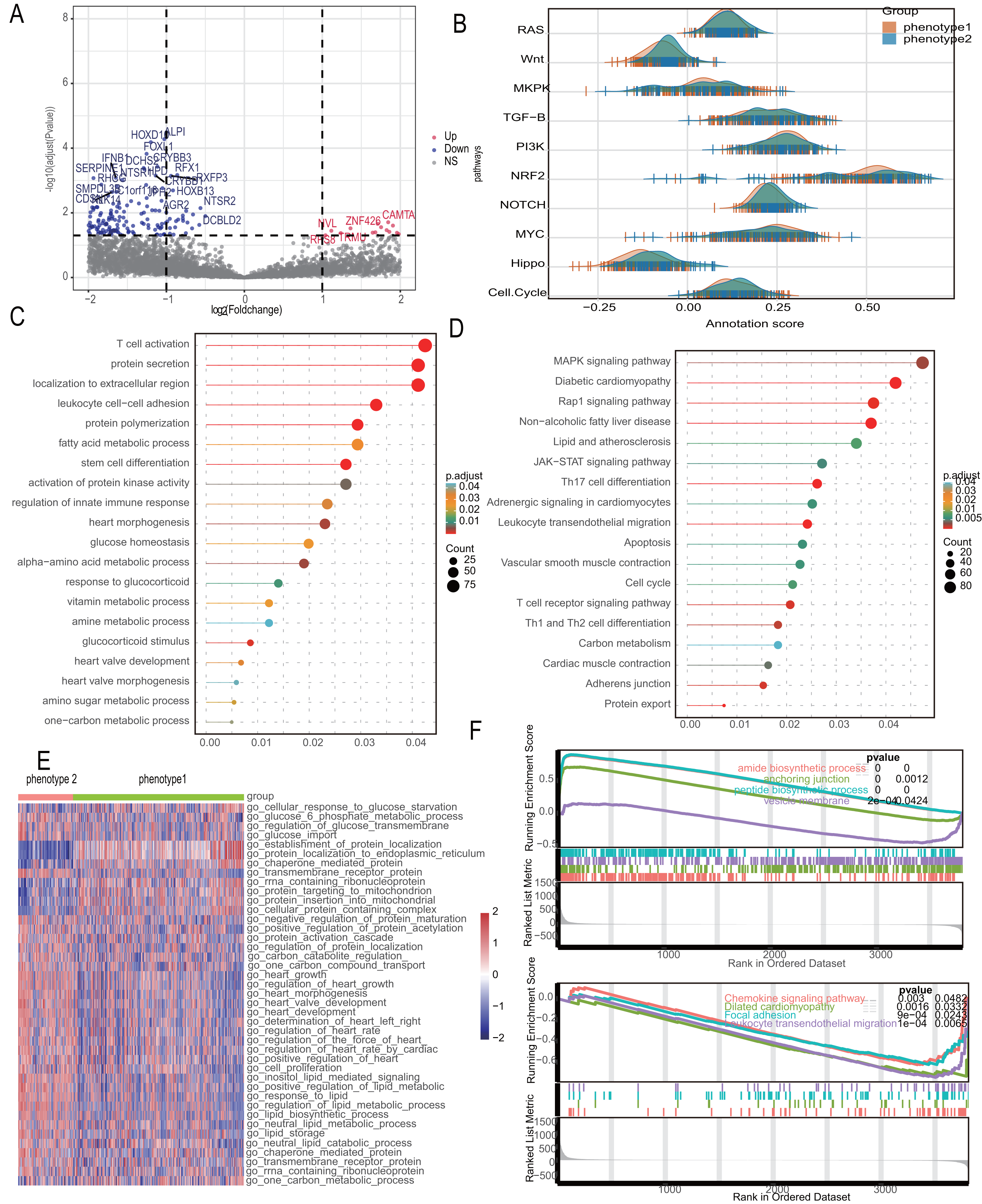{kind=link}
CAD diagnostic model constructed with diabetes-related genetic features shows excellent performance
To develop a more precise CAD diagnostic system, we further selected key genes for constructing a diabetes-related CAD typing using Lasso regression. A total of 32 core genes were included in the screening model (seed= 1, alpha=1, family=“gaussian”), and final model scores were derived using the predict algorithm. The final model incorporated 16 diabetes-related genes: “TRIM38,” “SAPG4,” “ODC1,” “NPY,” “MTDH,” “MT1G,” “LMF2,” “KCNQ1,” “ITPK1,” “GATAD2A,” “CRAT,” “CDK5RAP3,” “BAG3,” “ATP6V1B1,” “ARF4,” “AQP8.” (Figs. 6A, 6B). The modeling algorithm is as follows: model score= −0.0012369*exp (TRIM38) + 0.0017723*exp(SAPG4)+ 0.0014256*exp(ODC1)+ 0.0014256*exp(NPY)+ 0.0014256*exp(MTDH)+ 0.0016259*exp(MT1G) −0.0050854*exp(LMF2)+ 0.0015382*exp(KCNQ1)+ 0.0011388*exp(ITPK1)+ 0.0087858exp(GATAD2A)+ 0.0020088*exp(CRAT) −0.0065762*exp(CDK5RAP3) −0.00023941*exp(BAG3)+ 0.00817318exp(ATP6V1B1)+ 0.0065567*exp(ARF4)+ 0.0075323*exp(AQP8).
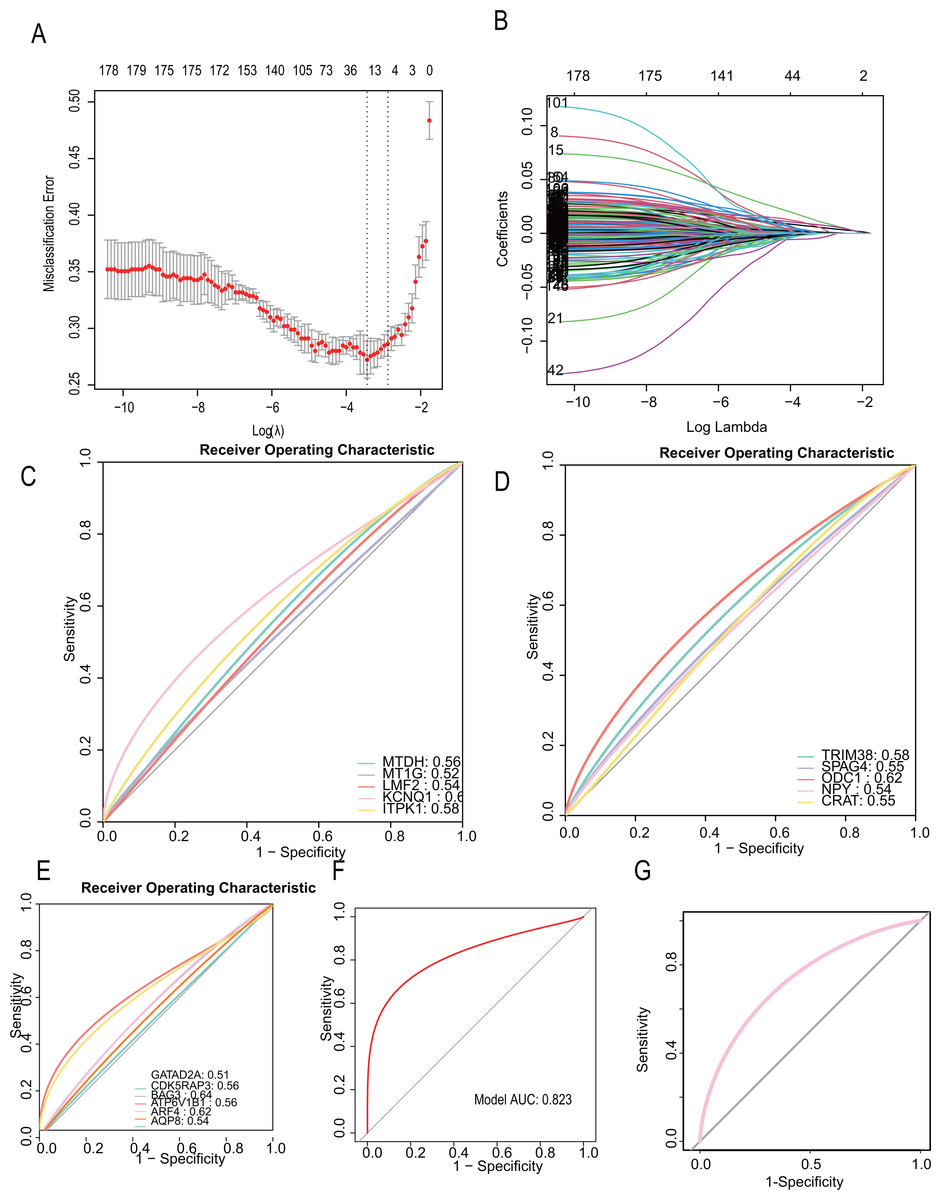
Figure 6: CAD diagnostic model constructed with diabetes-related genetic features shows excellent performance.
(A–B) LASSO regression to construct the diagnostic model (the model constructing gene are “TRIM38,” “SAPG4,” “ODC1,” “NPY,” “MTDH,” “MT1G,” “LMF2,” “KCNQ1,” “ITPK1,” “GATAD2A,” “CRAT,” “CDK5RAP3,” “BAG3,” “ATP6V1B1,” “ARF4,” “AQP8. ” Their Lasso coefficients are −0.0012369, 0.0017723, 0.0014256, 0.0016259, −0.0050854, 0.0015382, 0.0011388, 0.0087858, 0.0020088, −0.0065762, −0.00023941, 0.013747, 0.0081731, 0.0065567, 0.0075323). (C–E) The areas under the receiver operating characteristic (ROC) curves on the model constructing genes (MTDH (AUC = 0.56), MT1G (AUC = 0.56), LMF2 (AUC = 0.54), KCNQ1 (AUC = 0.62), ITPK1 (AUC = 0.58), TRIM38 (AUC = 0.58), SPAG4 (AUC = 0.55), ODC1 (AUC = 0.62), NPY (AUC = 0.54), CRAT (AUC = 0.55), GATAD2A (AUC = 0.51), CDK5RAP3 (AUC = 0.56), BAG3 (AUC = 0.64), ATP6V1B1 (AUC = 0.56), ARF4 (AUC = 0.62), AQP8 (AUC = 0.54)). (F, G) The ROC curve on the diagnostic model (Training dataset). (G) The ROC curve on the diagnostic model (Testing dataset).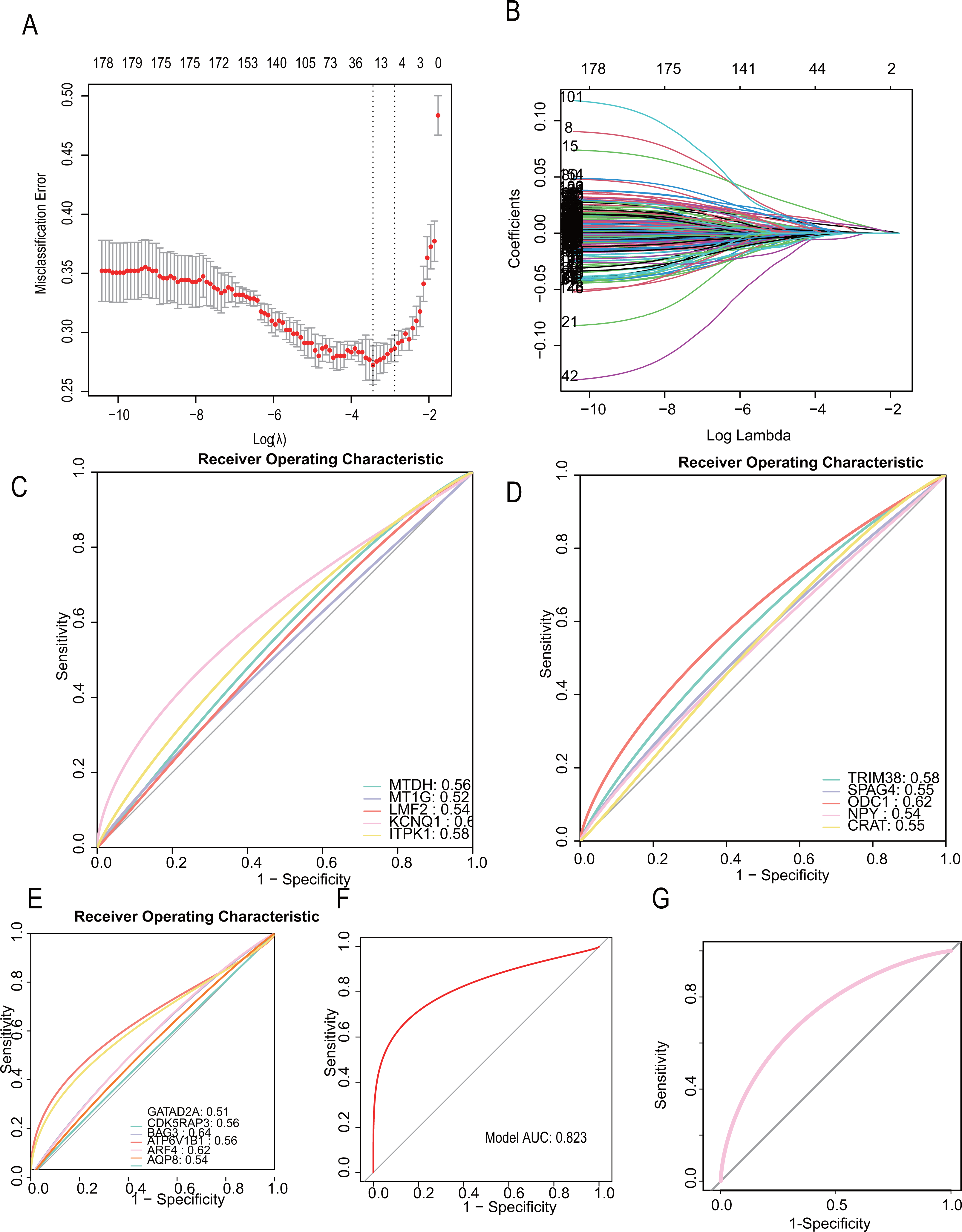{kind=link}
The predictive performance for each gene and the total LASSO diagnostic model was calculated. The areas under the receiver operating characteristic (ROC) curves were in Figs. 6C, 6D, 6E, respectively. Notably, when all 16 genes were included in a single diagnostic model, the AUC in the training dataset reached 0.823, and 0.758 in testing dataset (Fig. 6F, Fig. 6G).
Single-cell analysis explored CAD patient landscapes as well as expression patterns of modeled key genes
Further analysis was conducted on the expression landscape of diabetes-related genes using single-cell data from patients with coronary myocardial injury. After calculating and filtering the mitochondrial gene and ribosome ratios, as well as applying downscaling and tSNE clustering analyses (Fig. S3), we classified the patient cells into 11 subpopulations. These subpopulations were annotated using the “SingleR” package (Fig. 7A), identifying six cell types: macrophages, endothelial cells, monocytes, NK cells, muscle cells, and T cells (Fig. 7B). Macrophages and endothelial cells showed closer associations with other cells, indicating their potential roles in pathogenesis (Fig. 7C, Fig. S4). Dot plots were used to illustrate the top five genes differing among individual cells (Fig. 7D). The distribution of key markers in individual cells of the diabetes diagnostic model revealed that LMF2 and CDK5RAP3 were predominantly found in macrophages, while ARF4 was widely distributed among various immune and endothelial cells, as well as in muscle cells. KCNQ1, ATP6V1B1, MTDH, and ITPK1 were mainly distributed in macrophages (Fig. 7E, Fig. S5). Additionally, the heatmap exhibited the differential genes of various immune cells (Fig. S6).
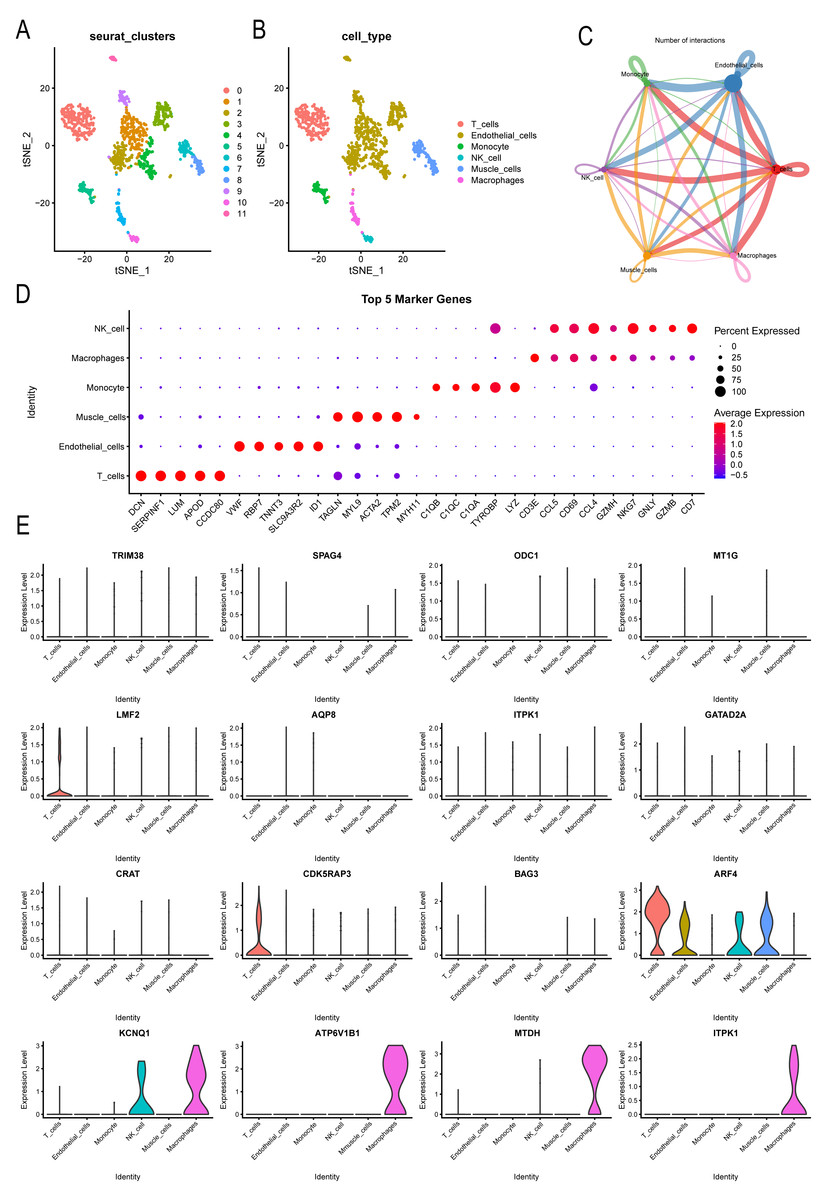
Figure 7: Single-cell analysis explored CAD patient landscapes as well as expression patterns of modeled key genes.
(A–B) The subgroup annotation based on single cell analysis. (C) The association between different immune cells. (D) Dot plots to illustrate the top five genes differing among individual cells. (E) The distribution of key markers in individual cells.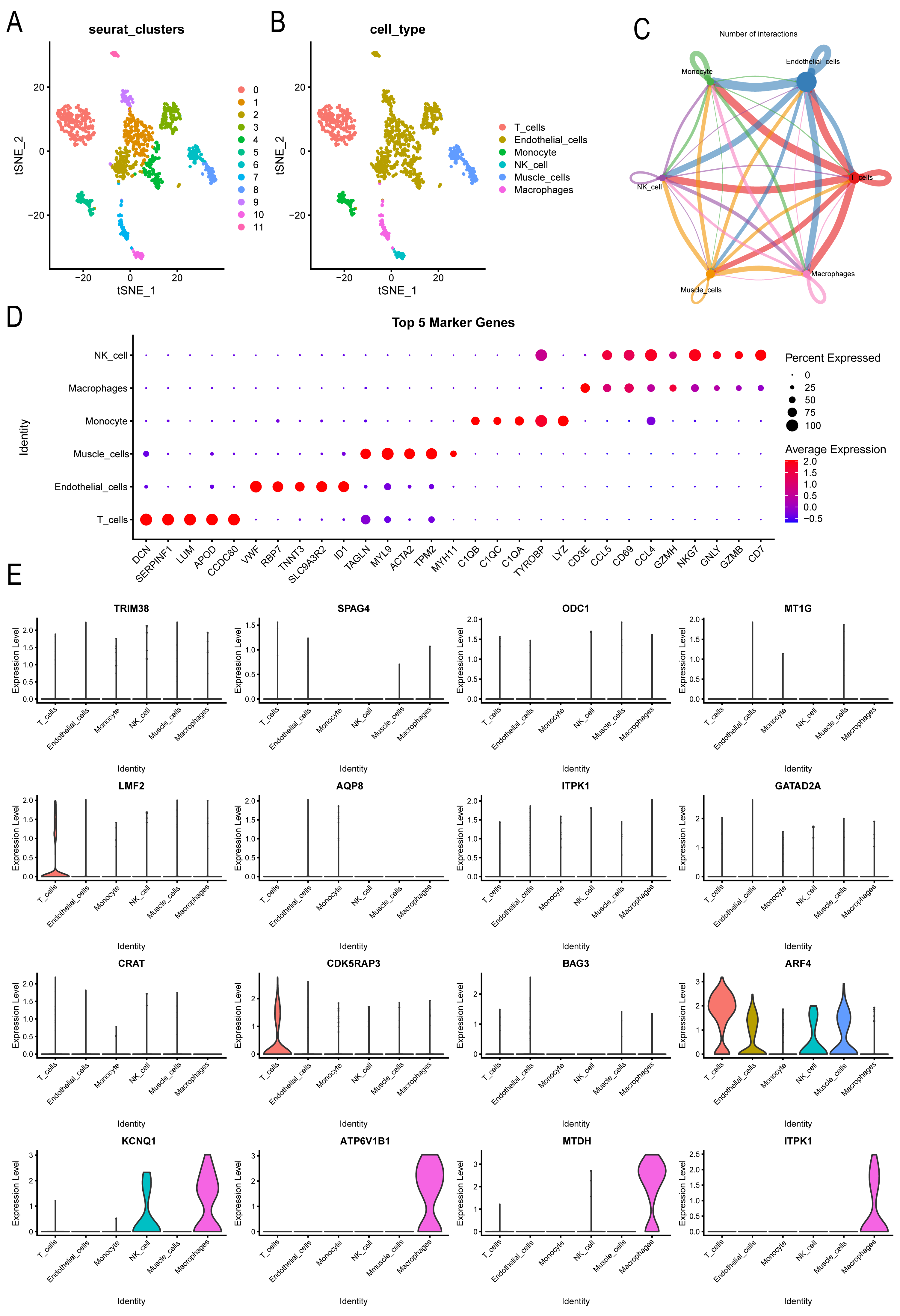{kind=link}
KCNQ1 and ITPK1 were found to influence immune microenvironment of coronary artery disease
Building on the diagnostic model analysis, we further investigated the clinical relevance of these diagnostic markers. A mouse coronary artery disease model and a normal mouse control were established, with myocardial tissues removed, fixed, embedded, and subjected to IHC staining (Fig. 8A). We assessed the differential expression of the 16 gene diagnostic markers and some common immunological indicators. The results indicated upregulated expression of KCNQ1 and ITPK1 in mice with coronary artery disease, along with increased expression of CD31 and CD68. This suggests that KCNQ1 and ITPK1 are significant in the pathogenesis of CAD, potentially influencing disease progression by affecting the immune microenvironment in patients with CAD (Fig. 8B). We further compared mRNA levels in different subgroups and found the results to be consistent with those of histochemical staining (Fig. 8C). We further treated cells with CHD inducing conditions such as high glucose and fat, and then knocked down ITPK1 or KCNQ1. We found that high-glucose and high-fat conditions promoted the apoptosis rate of HUVEC, which was reversed by knockdown of KCNQ1 or ITPK1, suggesting the important role of ITPK1 or KCNQ1 in the development of CAD (Figs. 8D–8F).
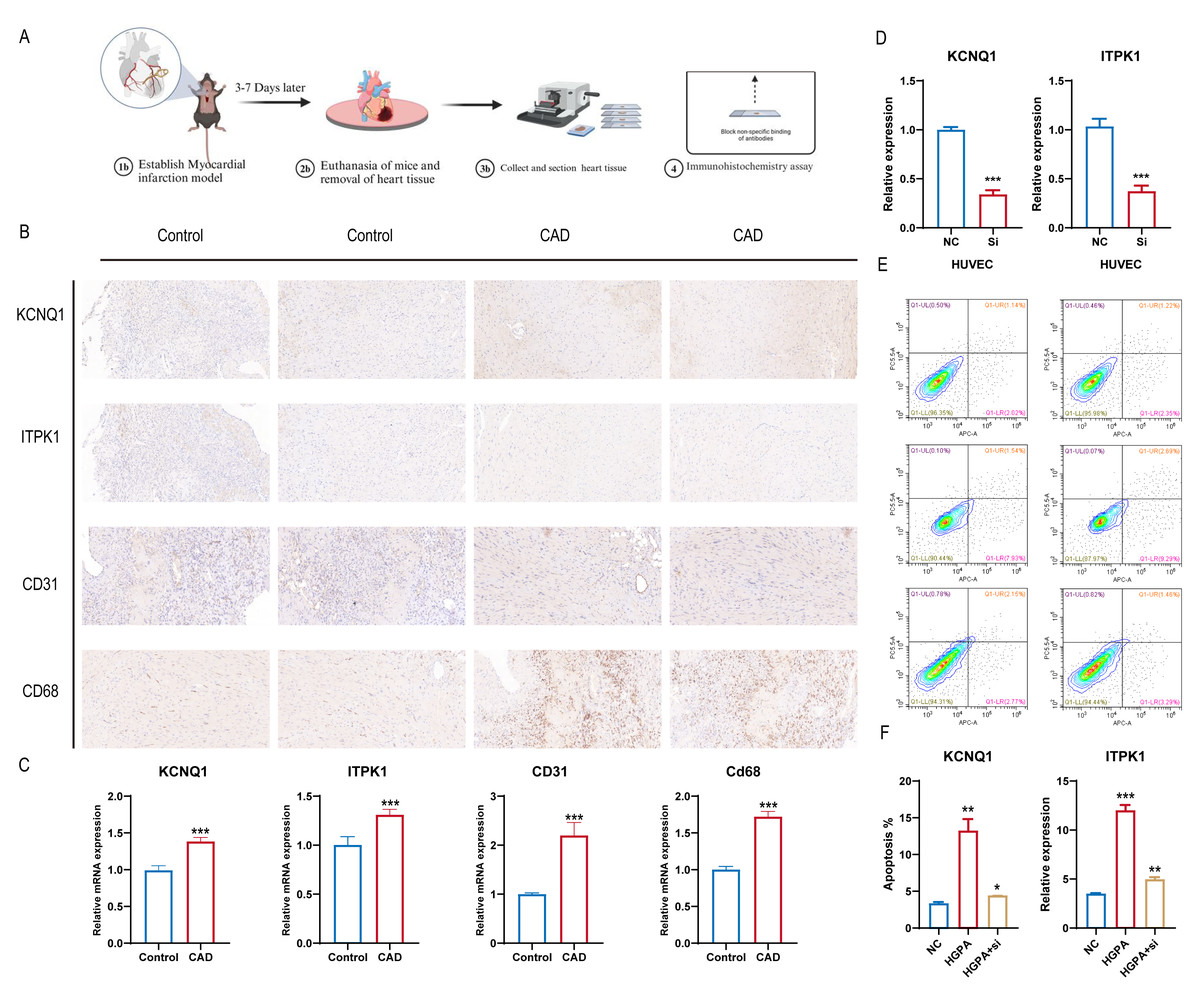
Figure 8: KCNQ1 and ITPK1 were found to influence immune microenvironment of coronary artery disease.
(A) Workflow of mouse coronary artery disease model. (B) The co expression of KCNQ1 and ITPK1 with macrophage biomarkers in immunohistochemical staining. (C) The mRNA level of ITPK1 and KCNQ1 in various models. (D) Knockdown of KCNQ1 and ITPK1 in HUEVC cells. (E–F) Apoptosis rate of various groups.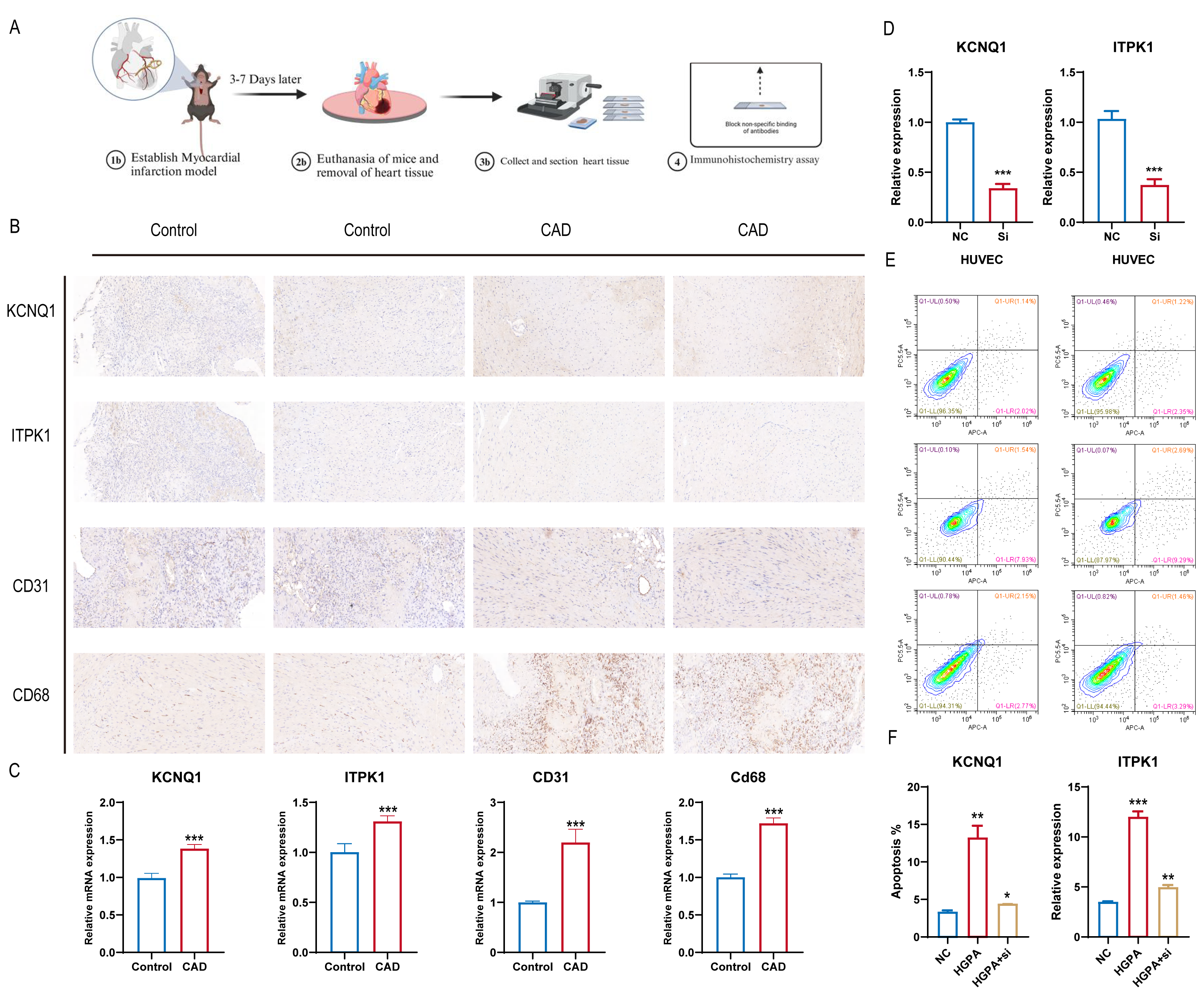{kind=link}
Discussion
In our current study, 32 diabetes-related biomarkers were identified based on differential expression analysis and WGCNA results. We further identified a diabetes related CAD phenotype based on the consensus clustering and exhibited its variation on cytokines, inflammatory factors, and immune cells infiltration. Utilizing a prediction algorithm, we identified 16 core diabetes related genes and construct a diabetes related CAD diagnostic model. Additionally, we found that KCNQ1 and ITPK1 may influence the immune microenvironment of CAD. CAD is a hazardous cardiovascular disease with complex pathology, the underage of which has made macro-control of the disease increasingly difficult (Vergès, 2020). Interestingly, patients with coronary artery disease often also have concomitant diabetes, which simultaneously exacerbates the progression of coronary artery disease. Previous literature indicated that CAD patients with type 2 diabetes mellitus (T2DM) often have increased epicardial adipose tissue (EAT) volume, correlating with higher cardiovascular mortality risk (Liu et al., 2023). Currently, combined diabetes and coronary heart disease analyses based on multiple datasets remain a vacuum; therefore, diagnostic models and biomarkers identified by combined analyses may lead to new directions and clinical practices for CAD treatment.
Several studies have underscored the importance of various genes in crosstalk between diabetes and CAD (Deda et al., 2022). A review highlighted CHI3L1, CD36, LEPR, RETN, IL-18, RBP-4, and RARRES2 as potential genetic markers for insulin resistance (IR) and atherosclerosis in T2DM patients, which ultimately promote the progression of CAD (Liu et al., 2023). Additionally, genes like LDLR, CETP, APOA5, Apo E, Apo B, and Apo A-I are linked to CAD onset in T2DM patients, suggesting the potential for personalized diet–gene intervention therapies (Raj et al., 2015). In our current research, differential analysis and WGCNA analysis were used to extract diabetes-related pathogenic genes, and the immune and inflammatory microenvironment differences of diabetic comorbidity phenotypes were verified in the CAD cohort, which further verified the potential regulation of diabetes-related genes on CAD. Additionally, we found similarities in endogenous immunity between the two subtypes as well as differences between exogenous immunity. The endogenous immune system consists of mast cells, eosinophils and basophils, and natural killer cells, while the exogenous immunity consists mainly of T cells and B cells (Sun et al., 2020). This suggests that T cells and B cells play a potential role in the differentiation of CAD subtypes. Interestingly, this is the first time to identify a coronary heart disease classification based on diabetes-related characteristics, filling the gap in the clinical practice. Our classification is based on the consensus algorithm and is based on gene expression levels. By evaluating the expression levels of different genes, it accurately locates the internal microenvironment of different patients, which is conducive to precisely identifying the immune infiltration status of the patients. Similarly, our subtypes also have some limitations, including confounding factors such as sample heterogeneity when identifying diabetes-related genes and coronary heart disease-related genes, which may interfere with our results to a certain extent. In future research, it might be possible to assess the expression levels of the corresponding genes through expression probe, thereby further promoting and applying our classification.
We also found that KCNQ1 and ITPK1 may influence the immune microenvironment of CAD through the crosstalk with macrophages. Previous studies have identified the involvement of macrophages in the pathogenesis of diabetes and related CAD (Henning, 2018; Peng & Gao, 2022; Yap et al., 2023). Dysregulation of macrophage polarization between M1 and M2 phenotypes alters the immune-inflammatory response in CAD, leading to plaque progression. Therefore, metabolic dysregulation of macrophages may be one of the key features of CAD progression (Wang et al., 2024). CD31 and CD68 are key regulators of macrophages (Andreata et al., 2018; Garaicoa-Pazmino et al., 2019), and our study identified a co-expression profile between KCNQ1 and ITPK1 and these two regulators, thus speculating that they may regulate the pathogenesis and progression of CAD by regulating macrophage metabolism. In previous study, Zhu, Deng & Zhou (2018) identified KCNQ1 as a potential immunomodulatory gene in gout, and identified the pharmacological value of KCNQ1 in type 2 diabetes. Abnormalities in KCNQ1 have also been found to be significantly associated with arrhythmia (Kekenes-Huskey et al., 2022; Khatami, Mohajeri-Tehrani & Tavangar, 2019). Interestingly, KCNQ1 also plays a significant role in heart-related diseases. KCNQ1 exerts a crucial function in regulating potassium ions within the heart. Herbert et al. (2002) reported that mutations in KCNQ1 are closely associated with the occurrence of long QT syndrome, and they may cause cardiac dysfunction through a dominant negative mechanism. Similarly, KCNQ1 has been found to be closely associated with the occurrence of gestational diabetes. This might be caused by the imbalance in the release of insulin related to KCNQ1 (Herbert et al., 2002; Majcher et al., 2022). In our research, we also discovered that KCNQ1 plays a certain role in the apoptosis of vascular smooth muscle cells, which opens up a new research direction. To conclude, KCNQ1 has a certain impact on the occurrence of diabetes and heart-related diseases. This might be a new and significant biomarker for coronary heart disease. Research on ITPK1 remains scarce, with only one study labeling ITPK1 as being associated with migraine and thyroid dysfunction (Tasnim & Nyholt, 2023). In future studies, we may be able to obtain different macrophage phenotypic characteristics by reprogramming KCNQ1 as well as ITPK1 expression in adult macrophages, which may provide new ideas for the treatment of CAD.
In order to uncover potential molecular mechanisms and targets for diabetes-related CAD modifications, we have attached various concerns regarding the enrichment and analysis of pathways and molecular variances between different CAD phenotypes. Our data imply that phenotype -associated genes lead to changes in different pathways, including the Hippo system and NRF2 pathway. Interestingly, of the 32 biomarkers, both YAP and BAG3 were found to be associated with the Hippo and NRF2 pathways, which could be a potential cause of their changes (Moya & Halder, 2019; Wackerhage et al., 2019). Additionally, NRF2 was also found to be correlated with MT1G, the potential biomarker in CAD (Tang & Kang, 2024). we discovered that changed genes are primarily associated with pathway enrichment in pathways related to immunological, metabolic, and cardiac damage. Additional differentiation analysis of metabolic pathway enrichment revealed that protein, carbohydrate, and lipid metabolism are the primary metabolic reprogramming changes. Patients with CAD tend to experience changes in metabolic patterns, including alterations in glucose and fatty acid oxidative phosphorylation (Jiang et al., 2021). Previous studies also found that MAP4K1 may be involved in mitochondrial injury to alter energy metabolism. The upregulated MAP4K1 may be involved in the metabolic reprogramming process of CAD. These findings imply that metabolic reprogramming frequently occurs in tandem with changes in the diabetes-related phenotype. In general, synergistic investigation of the immune microenvironment and metabolism may lead to new discoveries regarding CAD treatment targets.
This study has limitations that must be acknowledged. The sample size was restricted, and some critical indices for CAD and DM were not consistent, such as probe cleaning, correction, annotation details, and sample clinical information. Although we have provided some additional information regarding functional assays for KCNQ1 and ITPK1, the experimental validation remains quite limited in scope. our conclusions need validation through more mechanism experiments.
Our study has some potential future applications in clinical flow. Our identified genetic markers are significantly upregulated in patients with coronary artery disease and have a high co-diagnostic value, they may be used for early CAD diagnosis in diabetic patients. We can integrate the diagnostic value of these genes into the clinical flow by constructing relevant gene probes, fitting them in patient blood samples, and detecting the relative expression of these biomarkers.
Conclusion
To conclude, we constructed a diabetes related CAD phenotype and an optimal diagnostic model. We found that different CAD phenotypes have large differences in inflammatory and immune microenvironment, and found that KCNQ1 and ITK1 are potential pathogenic genes and immune regulators of CAD.