PeerJ Section
Bioinformatics and Genomics
Welcome to your community’s home at PeerJ. Sections are community led and exemplify a research community’s shared values, norms and interests.
The citation average is 5.9 (view impact metrics).
65,861 Followers
Section Highlights
View all Bioinformatics and Genomics articles 
13 September 2024
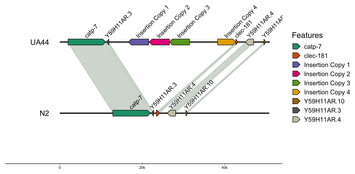
Identifying transgene insertions in Caenorhabditis elegans genomes with Oxford Nanopore sequencing
"The ability to use Oxford Nanopore kits to improve genomes is important in the field. As such the paper will appeal beyond the community interested in C. elegans to the wider community doing new or revised genome sequencing and using experimental transgene manipulations."
 Clement Kent, Handling Editor
Clement Kent, Handling Editor
Clement Kent, Handling Editor
5 August 2024
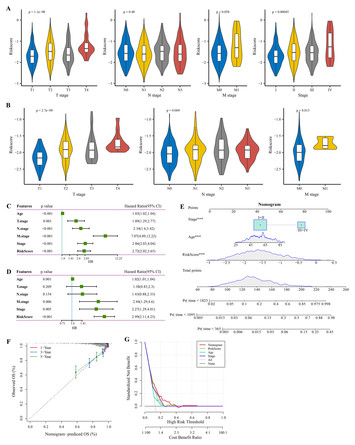
A glycolysis-related signature to improve the current treatment and prognostic evaluation for breast cancer
"This manuscript presents a study on breast cancer, focusing on the development of a glycolysis-related gene signature for prognostic evaluation. The research utilizes glycolysis-related genes to create a prognostic model, offering an alternative approach to breast cancer risk stratification. The authors employ various analytical methods, including GSVA, LASSO regression, and immune infiltration analysis, to develop their model. The proposed Riskscore and nomogram are designed to predict breast cancer patient outcomes, though their clinical utility remains to be fully established. The study examines potential relationships between glycolysis, tumor immune microenvironment, and breast cancer progression. The findings are supported by both computational analysis and some laboratory experiments. This work introduces a potential tool for assessing breast cancer prognosis and explores the role of metabolic factors in cancer progression. While the research contributes to the current understanding of glycolysis in cancer biology, further validation and investigation may be needed to determine its full impact on breast cancer patient care and prognostic evaluation. The study provides a foundation for future research in this area, but additional work will be required to confirm its clinical significance."
 Fanglin Guan, Handling Editor
Fanglin Guan, Handling Editor
Fanglin Guan, Handling Editor
23 July 2024
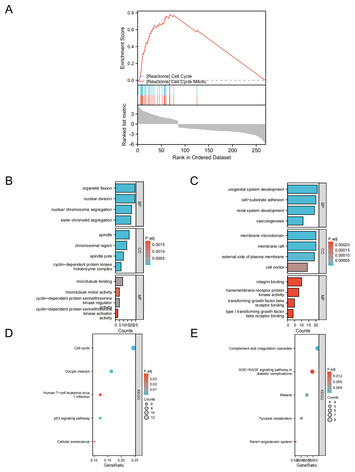
Machine learning and bioinformatics analysis of diagnostic biomarkers associated with the occurrence and development of lung adenocarcinoma
"Authors conducted machine learning and multi-omics data analysis using standard Bioinformatics pipelines to identify potential biomarkers BUB1B, CENPF, and PLK1. which are playing key roles
in LUAD development of lung adenocarcinoma."
 Praveen Korla, Handling Editor
Praveen Korla, Handling Editor
Praveen Korla, Handling Editor
27 June 2024
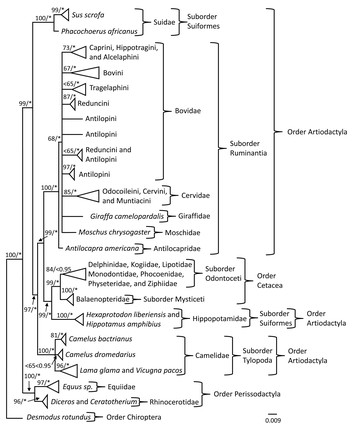
Genetic characterization of the prion protein gene in camels (Camelus) with comments on the evolutionary history of prion disease in Cetartiodactyla
"In my view, this work makes very important contribution to the field and brings attention to the prion disease in camels"
 Vladimir Uversky, Handling Editor
Vladimir Uversky, Handling Editor
Vladimir Uversky, Handling Editor
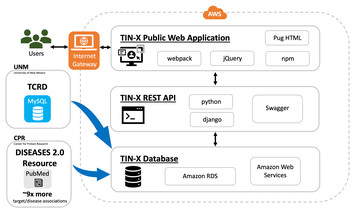
25 June 2024
TIN-X version 3: update with expanded dataset and modernized architecture for enhanced illumination of understudied targets
"The papers presents improvements to an algorithm that aids AI in identifying diseases and gene associations, This approach could improve research in a number of areas, such pest control products and insecticide resistance."
 Brenda Oppert, Section Editor
Brenda Oppert, Section Editor
Brenda Oppert, Section Editor
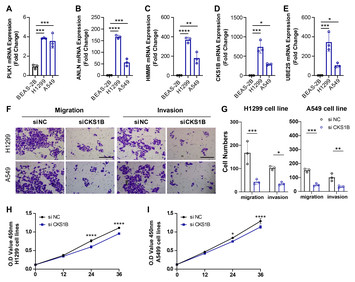
24 June 2024
Integrated bulk and single-cell RNA sequencing identifies an aneuploidy-based gene signature to predict sensitivity of lung adenocarcinoma to traditional chemotherapy drugs and patients’ prognosis
"The study introduces a novel aneuploidy-related risk score (ARS) model, which could significantly enhance the ability to predict patient prognosis in lung adenocarcinoma (LUAD). This is crucial for tailoring personalized treatment strategies and improving patient outcomes. By identifying an aneuploidy-based gene signature, the research provides insights into the sensitivity of LUAD to traditional chemotherapy drugs such as Erlotinib and Roscovitine. This could potentially guide more effective treatment regimens and improve response rates in patients. The use of both bulk RNA sequencing and single-cell RNA sequencing (scRNA-seq) allows for a comprehensive analysis of gene expression and cellular heterogeneity in LUAD. This integrative approach enhances the understanding of tumor biology and the tumor immune.
The study also identifies specific genes (e.g., CKS1B) and signaling pathways (e.g., SPP1, MK) that are involved in LUAD progression and immune cell communication. These findings could lead to the development of novel therapeutic targets and biomarkers. Moreover, the research highlights the potential for patients with low ARS to benefit more from immunotherapy. This underscores the importance of the ARS model in identifying patients who are likely to respond to immunotherapeutic approaches, thereby optimizing treatment efficacy.
These findings add to the growing body of knowledge on the role of aneuploidy in cancer progression and treatment resistance. This could stimulate further research into aneuploidy-related mechanisms across various cancer types."
Fanglin Guan, Handling Editor
Fanglin Guan, Handling Editor
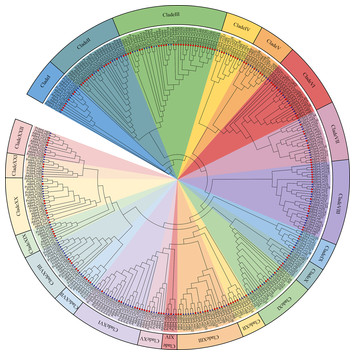
27 May 2024
Genome-wide identification of bHLH gene family and its response to cadmium stress in Populus × canescens
"Important for plant biologists who want to improve certain traits under specific stress conditions!"
 Muhammad Aamer Mehmood, Handling Editor
Muhammad Aamer Mehmood, Handling Editor
Muhammad Aamer Mehmood, Handling Editor
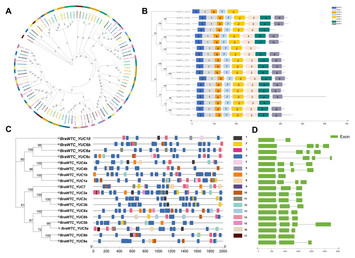
20 May 2024
Identification of YUC genes associated with leaf wrinkling trait in Tacai variety of Chinese cabbage
"The leaf wrinkle trait forms a very important phenotypic trait in Chinese cabbage. Therefore, elucidation of the molecular basis of this phenotypic trait is very important for use in future breeding programs of Chinese cabbage."
 Andrew Eamens, Handling Editor
Andrew Eamens, Handling Editor
Andrew Eamens, Handling Editor
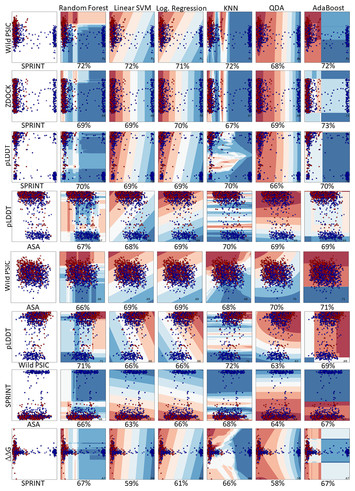
14 May 2024
Var3PPred: variant prediction based on 3-D structure and sequence analyses of protein-protein interactions on autoinflammatory diseases
"Interdisciplinary study addressing variant prediction based on protein sequence and 3D structural data."
 Efe Sezgin, Handling Editor
Efe Sezgin, Handling Editor
Efe Sezgin, Handling Editor

13 May 2024
Prognostic and chemotherapeutic implications of a novel four-gene pyroptosis model in head and neck squamous cell carcinoma
"This article extends the clinical value of assessing pyroptosis in squamous cell carcinomas, with potentail application to other cancer types."
 Philip Coates, Handling Editor
Philip Coates, Handling Editor




65,861 Followers


