PeerJ Section
Bioinformatics and Genomics
Welcome to your community’s home at PeerJ. Sections are community led and exemplify a research community’s shared values, norms and interests.
The citation average is 6.1 (view impact metrics).
67,241 Followers
Section Highlights
View all Bioinformatics and Genomics articles

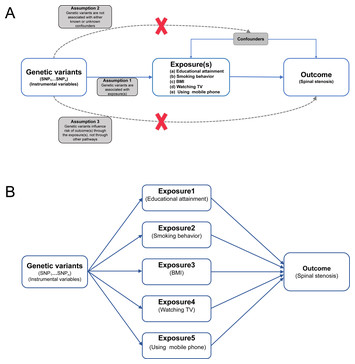
21 March 2023
Evaluating the causal relationship between five modifiable factors and the risk of spinal stenosis: a multivariable Mendelian randomization analysis
"This is an interesting manuscript that contributes to increasing our understanding of spinal stenosis."
 Renato Polimanti, Handling Editor
Renato Polimanti, Handling Editor
Renato Polimanti, Handling Editor
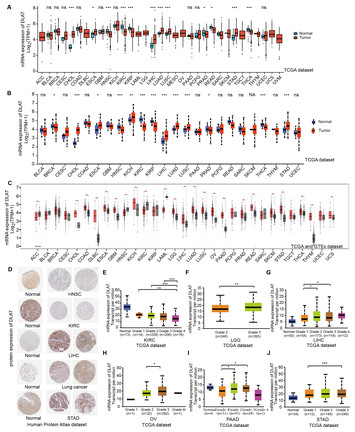
17 March 2023
Roles of cuproptosis-related gene DLAT in various cancers: a bioinformatic analysis and preliminary verification on pro-survival autophagy
"Studies on the correlation between DLAT and hepatocellular carcinoma will help to better understand the immune response mechanism in tumors and guide clinical treatment."
 Yuzhen Xu, Handling Editor
Yuzhen Xu, Handling Editor
Yuzhen Xu, Handling Editor
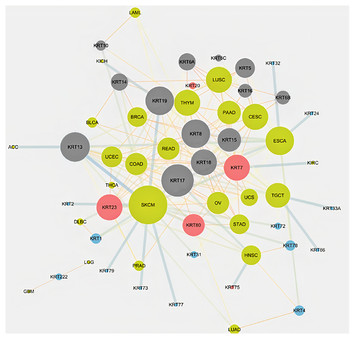
17 March 2023
“In the light of evolution:” keratins as exceptional tumor biomarkers
"Keratins have a key role in several aspects of cancer pathophysiology, including cancer cell invasion and metastasis, and several members of the Keratin family serve as diagnostic or prognostic markers, such model use in a non-mammalian species model organisms in functional studies to uncover Keratin genes which are associated with human cancers."
 Abdul Hafeez Kandhro, Handling Editor
Abdul Hafeez Kandhro, Handling Editor
Abdul Hafeez Kandhro, Handling Editor

27 February 2023
PrismEXP: gene annotation prediction from stratified gene-gene co-expression matrices
"With so many genes in many different organisms lacking annotation or associated functions, PrismEXP has the potential to greatly expand our understanding of gene function that will lead to improvements in many different downstream applications."
 Brenda Oppert, Section Editor
Brenda Oppert, Section Editor
Brenda Oppert, Section Editor
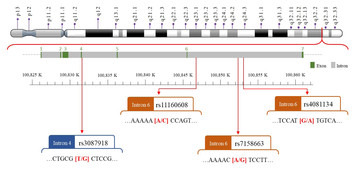
27 January 2023
Novel genetic variants in long non-coding RNA MEG3 are associated with the risk of asthma
"Novel genetic variants in long non-coding RNA MEG3 are associated with the risk of asthma."
 Ramcés Falfán-Valencia, Handling Editor
Ramcés Falfán-Valencia, Handling Editor
Ramcés Falfán-Valencia, Handling Editor
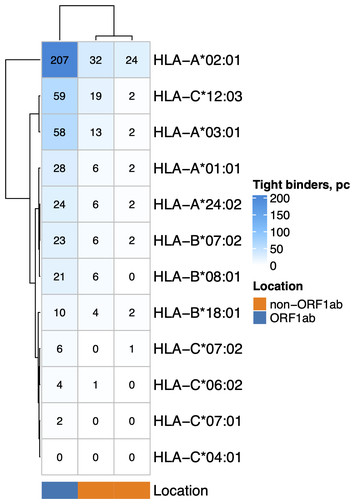
18 January 2023
HLA-A*01:01 allele diminishing in COVID-19 patients population associated with non-structural epitope abundance in CD8+ T-cell repertoire
"The manuscript describes underlying research into the evolving mutations in SARS-covid2 and how this can relate to immune responses. As reviewer states, this approach can be applied to other disease outbreaks."
Brenda Oppert, Section Editor
Brenda Oppert, Section Editor
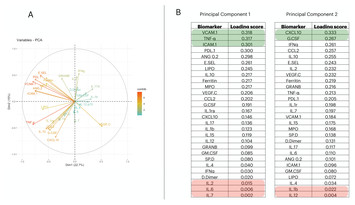
12 December 2022
An artificial neural network classification method employing longitudinally monitored immune biomarkers to predict the clinical outcome of critically ill COVID-19 patients
"Research contributes to knowledge about COVID-19. Both the role of specific biomarkers on the severity of COVID-19 and the use of Artificial Neural Network makes this study novel."
 Aslı Suner, Handling Editor
Aslı Suner, Handling Editor
Aslı Suner, Handling Editor
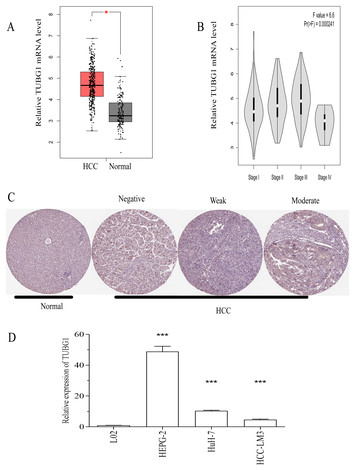
5 December 2022
Upregulated TUBG1 expression is correlated with poor prognosis in hepatocellular carcinoma
"Identification of novel theranostic biomarkers is a very hot topic for all cancer types. Thus this study will be of interest not only for scientists working in the field of HCC but also in different cancer types. Though the results need to be validated in an independent study."
 Hilal Ozdag, Handling Editor
Hilal Ozdag, Handling Editor
Hilal Ozdag, Handling Editor
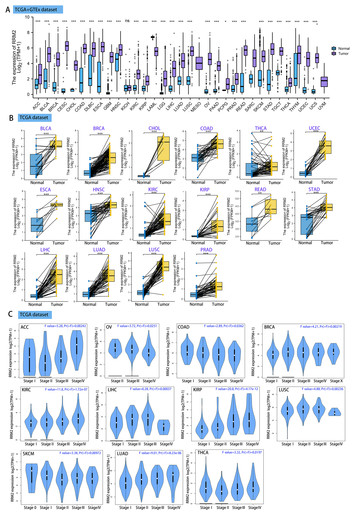
28 November 2022
A pan-cancer analysis of the oncogenic role of ribonucleotide reductase subunit M2 in human tumors
"The authors used pan-cancer analysis to demonstrate the promising important roles of ribonucleotide reductase subunit M2 in human cancers."
 Zhijie Xu, Handling Editor
Zhijie Xu, Handling Editor
Zhijie Xu, Handling Editor
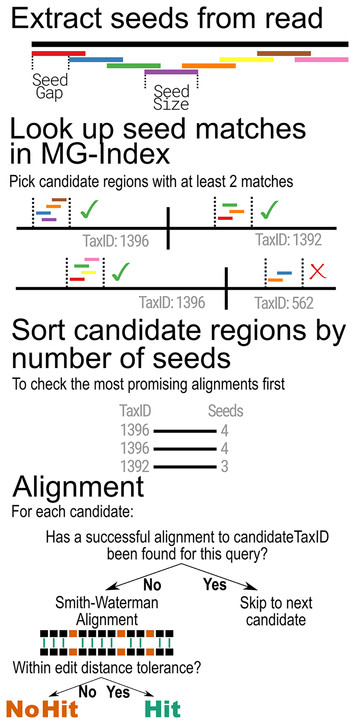
8 November 2022
MTSv: rapid alignment-based taxonomic classification and high-confidence metagenomic analysis
"MTSv classifies metagenomic reads with a good balance of speed and accuracy."
 Michael LaMontagne, Handling Editor
Michael LaMontagne, Handling Editor
Michael LaMontagne, Handling Editor




67,241 Followers


