
Laplacian score and genetic algorithm based automatic feature selection for Markov State Models in adaptive sampling based molecular dynamics
- Published
- Accepted
- Received
- Academic Editor
- Ho Leung Ng
- Subject Areas
- Theoretical and Computational Chemistry, Biophysical Chemistry
- Keywords
- Molecular Dynamics, Markov State Models, Laplacian scores, Computational Chemistry, Accelerated Molecular Dynamics
- Copyright
- © 2020 George et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Physical Chemistry) and either DOI or URL of the article must be cited.
- Cite this article
- 2020. Laplacian score and genetic algorithm based automatic feature selection for Markov State Models in adaptive sampling based molecular dynamics. PeerJ Physical Chemistry 2:e9 https://doi.org/10.7717/peerj-pchem.9
Abstract
Adaptive sampling molecular dynamics based on Markov State Models use short parallel MD simulations to accelerate simulations, and are proven to identify hidden conformers. The accuracy of the predictions provided by it depends on the features extracted from the simulated data that is used to construct it. The identification of the most important features in the trajectories of the simulated system has a considerable effect on the results.
Methods
In this study, we use a combination of Laplacian scoring and genetic algorithms to obtain an optimized feature subset for the construction of the MSM. The approach is validated on simulations of three protein folding complexes, and two protein ligand binding complexes.
Results
Our experiments show that this approach produces better results when the number of samples is significantly lesser than the number of features extracted. We also observed that this method mitigates over fitting that occurs due to high dimensionality of large biosystems with shorter simulation times.
Introduction
The study of biomolecular dynamics and interactions play a crucial role in understanding cell functions, and development of novel drugs (De Vivo et al., 2016; Śledź & Caflisch, 2018) Molecular dynamics (MD) simulations have the unique capability to track conformational changes, which in turn allows the establishment of target-specific and conformation-specific drug discovery research (Lecina, Gilabert & Guallar, 2017). The cost of bringing a drug to the market is still estimated to be 2.6 billion dollars (USD) (DiMasi, Grabowski & Hansen, 2016). Techniques like MD, and other computer-aided drug discovery (CADD) tools, reduce the burden of exorbitant drug discovery and development costs to healthcare. Various experimental methodologies such as NMR, fluorescence, X-ray crystallography and high-throughput screening are routinely used in drug discovery and development. While they are still the core to conducting drug discovery research, CADD tools can contribute to making the process rationally driven, reduce trial and error costs and allow accurate high throughput explorations that are otherwise too expensive to conduct. CADD tools have proven to complement experimental techniques by making the discovery process more systematic and rational, and even opening up new search spaces previously unexplored. The past decade has even seen CADD tools contribute to upstream discovery research that resulted in full life cycle drug development programs, effectively translating research from the bench to the bed-side (Leelananda & Lindert, 2016).
MD is a low-throughput CADD technology that has vast applications (Hollingsworth & Dror, 2018), and central to that is the ability of MDs to provide time-continuous structural variation data of residues, co-factors, small molecules, etc., alongside the ability to clearly understand the modalities of interaction with the biomolecule in question in 3D. Experimental structures are rigid representations of the structure. MDs use Newtonian physics, chemical property parametrizations (Alder & Wainwright, 1957) to transform this static information to dynamic trajectories, which in turn provide advanced analytic and predictive capabilities to researchers. Many applications of MD simulations exist. For example, studies that explore conformational changes (Flocco & Mowbray, 1995; Grant, Gorfe & McCammon, 2010; Lindorff-Larsen et al., 2011; Schwantes, Shukla & Pande, 2016), binding-unbinding (Fabritiis et al., 2008; Buch, Giorgino & De Fabritiis, 2011; Kohlhoff et al., 2014; Meyer et al., 2014), and others that influence fundamental drug design in conjunction with experimental work (Martinez et al., 2003; Namboori et al, 2010; Mohan et al., 2010; De Vivo et al., 2016; Childers & Daggett, 2017). In this study, we explore the usage of Laplacian scoring based enhanced feature selection that can be employed in adaptive sampling MD. Below, we introduce each of the components briefly before we move to a detailed Methods section.
MDs provide a time-continuous trajectory, which comprises movement of atoms in microscopic, spatial and temporal detail. Frequencies of oscillation of atoms are in the range of femtoseconds, which is a limitation when the biological process being investigated require longer timescales. This is especially true in the context of all-atom calculations of MD (Vanatta et al., 2015; Kohlhoff et al., 2014). In recent times, all-atom, single, long simulations of few micro- to milliseconds are able to run on highly specialized hardware (Shaw et al., 2014), and supercomputers (Stone et al., 2007; Dakka et al., 2018). Microsecond range calculations are now possible even on workstations or server machines via GPU computing (Salomon-Ferrer et al., 2013) at costs that are orders of magnitude lower, as compared to specialized hardware or large compute clusters. However, the timescales of biological processes for molecules with millions of atoms, such as RNA Polymerase, are still out of reach (Da et al., 2016; Wang et al., 2018).
Apart from the above hardware-assisted acceleration, enhanced sampling techniques such as Replica-Exchange (Sugita & Okamoto, 1999; Zhang et al., 2016), Meta-Dynamics (Zheng & Pfaendtner, 2015), Transition path sampling (Bolhuis et al., 2002), Adaptive sampling MD (Noé et al., 2009; Plattner & Noé, 2015; Pande, 2014), high-throughput simulations (Buch et al., 2010; Harvey & De Fabritiis, 2012) and other methods (Laio & Parrinello, 2002; Namboori et al, 2010; Vargiu et al., 2008; Tiwary & van de Walle, 2016) have also proven to accelerate MD calculations. Adaptive sampling MD initiates many short parallel simulations from different starting conformers of the biomolecule, and repeats this in sequential iterations. Each iteration of the set of simulations that run in tandem, constitute an epoch. The starting structures in successive epochs are determined based on the analysis of MSM constructed, which is in turn calculated using features obtained from the previous epochs (Doerr et al., 2016). The final integrated trajectory is an aggregation of many epochs. Adaptive sampling MD, based on Markov State Models (MSM), is shown to explore under-sampled regions of the conformational space. This allows it to effectively overcome energetic barriers, and eventually sample optimal conformations (Husic & Pande, 2018; Mittal & Shukla, 2017) through MSM analysis. Identification of meta-stable states in the bio-dynamics and acceleration of simulation, depends on the efficiency of the MSM constructed. It is characteristic of MSMs based on optimal features to better identify hidden conformers (Chen et al., 2018). At the end of all the epochs, the MSM constructed is used to predict the characteristics of interactions. These may include protein-ligand binding free energy (Noé et al., 2009), transition rate between different protein conformations with possible binding sites (Plattner & Noé, 2015), hidden conformer states in protein folding (Pande, 2014), etc.
Examples of statistical learning techniques include Laplacian eigenmaps, spectral clustering, and Laplacian scores. In the context of applications to MD trajectory analysis, they have been successfully used to select features that can identify hidden conformers (Sgourakis et al., 2011). Also, Genetic algorithms (GAs), a class of meta-heuristics, are a good fit to automate the selection of optimal set of features to construct efficient MSMs (Chen et al., 2018; Mittal & Shukla, 2017). In our study, we explore a method for improving MSM efficiency, as measured by the established Generalized Matrix Rayleigh Quotient (GMRQ) scoring system (Noé & Nuske, 2013), by combining both Laplacian scoring (to provide a heuristic initialization) and GAs. As a result of this study, we identify features using this technique with a goal to construct better MSMs, as quantified by GMRQ scores. Various features can be used to discriminate between conformers, and features we have collected are detailed later. Time-structure Independent Component Analysis (TICA) (Molgedey & Schuster, 1994) is a dimensionality reduction method that is used in the MSM construction. The use of lesser number of features as compared to data samples, assures that the co-variance matrix used by TICA is a positive definite. This can be achieved by feature selection. This in turn avoids redundancy, and ensures a more accurate representation of the features by the TICA components. In summary, we propose the use of Laplacian scores in conjunction with GA for feature selection to produce improved MSMs. The goal of this study was to verify if the use of these two selection criteria, in conjunction, can lead to an efficient MSM. We worked on the hypothesis that this would lead to a more efficient selection of meta-stable states or hidden conformers for the next epoch of the adaptive sampling MD, and result in the acceleration of the overall simulation since low energy conformers are found rapidly.
Feature selection is an important step that avoids increased sampling and over-fitting. Automatic feature selection is being widely researched to construct efficient MSMs (Tiwary & Berne, 2016; Shamsi, Cheng & Shukla, 2018). Feature selection techniques, towards better MSMs, endeavor to select optimal sets of Collective Variables (CVs) (Noé & Clementi, 2017), or in other words, those that identify the most important residues with respect to the slowest processes. If all features are used, it results in poor clustering performance, which will adversely affect the MSM. Further, when the number of frames in simulation data is less than the number of features, it results in poor generalization of the MSM due to over-fitting (Dy & Brodley, 2004). This will necessitate the need for increased sampling at a higher computational cost (Malmstrom et al., 2014). However, the increased sampling is in direct opposition to one of the primary goals of accelerating the single long simulation through the use of short parallel simulations. Manual selection of CVs based on prior information about the system and human intuition have also been used to construct MSMs (Lovera et al., 2012; Ahalawat & Mondal, 2018). Nonetheless, this can lead to loss of information and very slow convergence in thermodynamic and kinetic property calculations. The automatic feature selection methods of highly-discriminant non-redundant features, like the one proposed in this study, provide a more generalized model with reduced computational complexity (García & García-Pedrajas, 2018).
In summary, and to highlight some of the principal points of the discussions above, in our approach, the initial feature selection is performed by Laplacian scoring (He, Cai & Niyogi, 2006; Sgourakis et al., 2011), and the model is optimized using a GA (Chen et al., 2018) on this subset. The time-structure independent components (tiCs) in TICA is used to identify the states in the system by grouping kinetically similar structures (Schwantes & Pande, 2013; Pérez-Hernández et al., 2013). During the MSM construction step, if the number of features is larger than the number of samples in the dataset, the co-variance matrix used for TICA is not guaranteed to be a positive definite. It means that some of the features can be expressed as a linear combination of the other features. This emphasizes the need for use of feature selection before dimensionality reduction to avoid over-fitting. Hence, in this study we use Laplacian scoring to pre-rank and select the features based on its importance. The ability of Laplacian score to identify only the significant features (Sgourakis et al., 2011), avoids the need for large amount of simulation data and over-fitting of data in the MSM constructed. Hence, these Laplacian scored feature subsets are used as a heuristic initialization of the zeroth population in the GA. The GA further improves the MSM efficiency, by adopting natural evolution strategies such as crossover and mutation (Chen et al., 2018; Mittal & Shukla, 2017). It is important to note that our method only provides an extra tool to perform effective GA-based searches for MSM based studies. We have shown that this improves the results in specific datasets used in this work. It can be seen as an adjunct tool that could be opportunistically used alongside previous techniques mentioned above.
Materials and Methods
We have applied Laplacian scores driven winnowing, and the subsequent, GA based feature selection to improve the MSM models of the following complexes (previously studied, further detailed in subsection ‘Datasets to evaluate the approach’) Villin (PDB ID:2F4K), Fs Peptide (Ace-A_5(AAARA)_3A-NME), WW domain (PDB ID: 2F21), Piperidine-Thrombin with Thrombin taken from PDB ID: 3D49 and Benzamidine-Trypsin (PDB ID:3PTB). This work was carried out on the Intel vLab Knights Landing cluster that consists of 252 Intel Xeon-Phi nodes connected with Omni-Path (Intel OPA) 100 Series interconnect and academic desktop with Intel Core i7-7500U @2.70 GHz with Nvidia GPU GTX TITAN Black @889 GHz. An outline of MSMs (see Husic & Pande, 2018) for a detailed review of MSM), and the application of MSM in adaptive sampling MD follows. We have inherited this part of the workflow from previous studies. Finally, we also provide details about the additional components of our workflow.
Markov State Models (MSM)
MSMs are used to model randomly changing systems. They have been used to study time-series events in which the current state depends only on the previous state and not on any other states before it (Tang, Bevan & Grover, 2017). It has a wide variety of applications in the field of physics, chemistry, medicine, finance, management etc. It has been used for biological modelling such as the simulation of brain function (George & Hawkins, 2009), viral infection of single cells (Gupta & Rawlings, 2014), analysis of bacterial genome (Skewes & Welch, 2013), as well as MD simulations using adaptive sampling techniques (Doerr et al., 2016; Doerr & De Fabritiis, 2014) etc.
Figure 1 represents the workflow adopted towards the application of MSM to analyze MD trajectories of bio-molecules. The goal is to find meta-stable conformers, with a few variations as explained below. Prediction accuracy of MSM, as indicative by GMRQ score, can be improved by selecting optimal features from the simulation trajectory data.
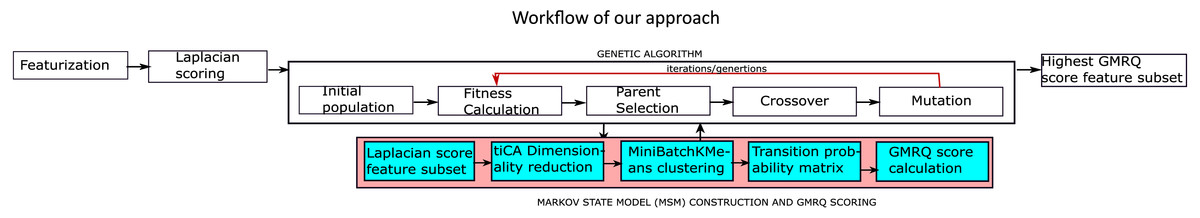
Figure 1: The proposed methodology for the identification of the optimal features.
{kind=link}
MSM is represented by an Nx N matrix which gives information about the probability of transition between N states (Singhal, Snow & Pande, 2004; Noé & Fischer, 2008). In Fig. 2, xij is the average of transition count matrix and its transpose. The probability of each row in the average transition count matrix is calculated to obtain the MSM. From the MSM, the eigen-flux is calculated. Eigen flux (-n1 for s4, n2 for s3, n3 for s2 and n4 for s1) of each state shows the transition from source state to the sink state, and this is used to identify the slowest process (Beauchamp et al., 2011). The N states in the MSM represents the state decomposition into which the simulation data is clustered. The ideal MSM, or in other words, the ideal state decomposition reveals even the slowest dynamical process. In this specific use case, it is the conformational change occurring in the simulated system. The probability with which transition to a state j occurs is calculated as (1) It can be generalized as (2) where pj(t + τ) is a population vector after time τ; τ is lag time; pi(t) is a population vector at time t; Tij(τ) is the probability of transition from state i to state j; T(τ) is the probability transition matrix that represents the MSM.
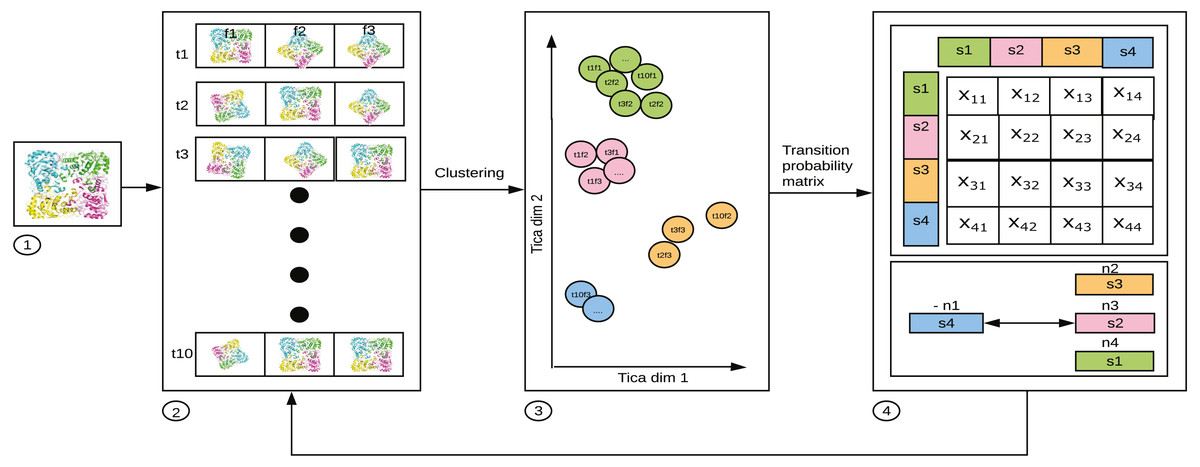
Figure 2: 1. Starting structure (frames f) of the bio-molecule used for parallel simulation. 2. 10 (arbitrary number for illustration) parallel trajectories (t1 through t10) produced by MD software, such as ACEMD, GROMACS,AMBER, etc. 3. The frames per structure, having reduced to 2 dimensions using TICA. 4. MSM constructed with each of the values in a cell of the matrix, xij, where i is the source state, and j is the destination state showing the probability of transition. from i to j.
The figure has been drawn based on Fig. 1 in Husic & Pande (2018).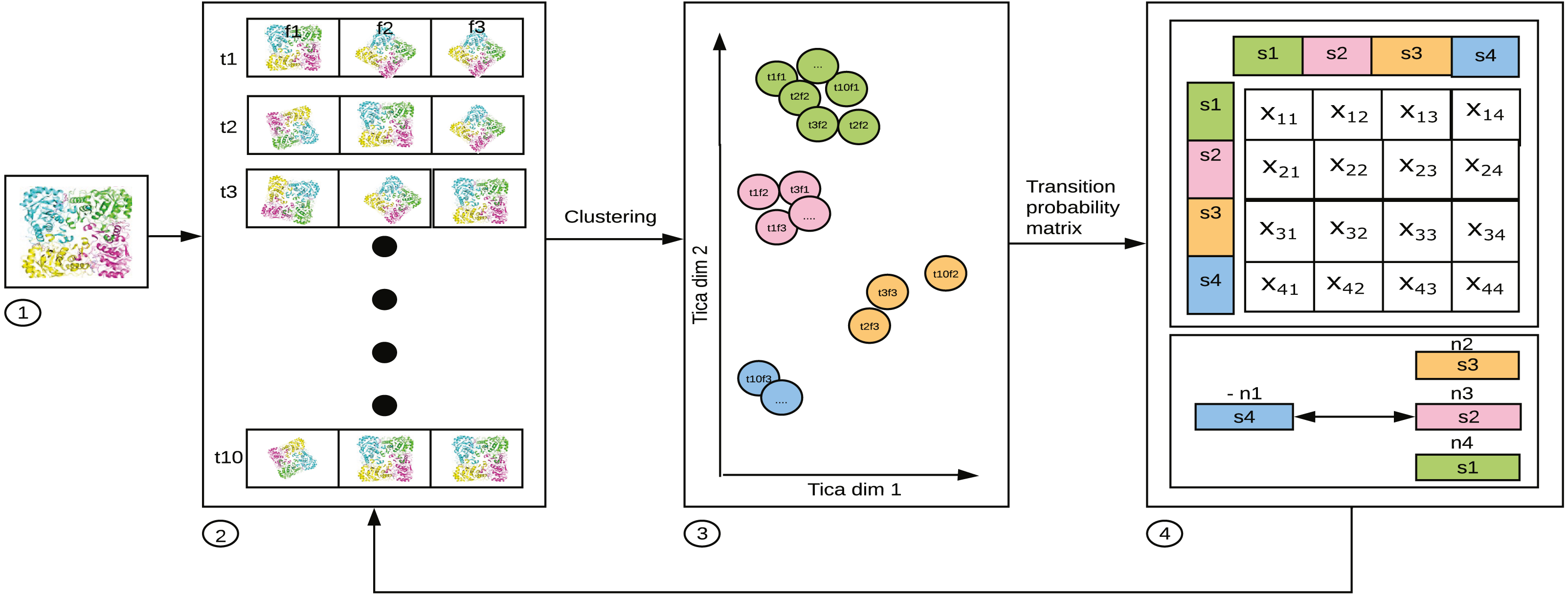{kind=link}
The eigenvalues λi and eigenfunctions ψi of T(τ) can decompose the transition probability matrix (Shukla et al., 2015) as in Eq. (3). The eigenfunctions represents the slowest processes that occurs in the simulated system. (3)
Generalized Matrix Rayleigh Quotient (GMRQ) derived from the variational principle of conformation dynamics (Noé & Nuske, 2013) is a metric used to evaluate the efficiency of the MSM (McGibbon & Pande, 2015). MSMs with higher GMRQ scores provide higher discriminatory abilities towards identifying the slowest dynamical process occurring in the system (McGibbon & Pande, 2015). Variational principle is used in the calculus of variations, or changes to develop a function that maximizes the variables dependent on it. Based on variational principle, the eigen values and eigen functions are estimated for the MSM constructed using the simulated data. GMRQ score of the MSM is the sum of the first p eigenvalues of T(τ). They also identify the slow processes, p. The upper boundary for the GMRQ score is set by variational principle as shown below (Husic & Pande, 2017; Noé & Nuske, 2013): (4) where ; λi are estimated and real i-th eigenvalues respectively. The efficiency of MSM is improved by trying to reach the upper boundary set by the sum of real eigenvalues (λi). MSMs with higher GMRQ scores are better able to identify the slowest conformational change in the system (Noé & Nuske, 2013; McGibbon & Pande, 2015).
Adaptive sampling MD based on MSM and Optimized feature selection for MSM
We have used simulation trajectories that are produced by adaptive sampling MD, based on the MSM. In this protocol, the first step in the construction of MSM involves the extraction of feature, namely, dihedral angles, distance between amino-acid residues, root mean square deviation (RMSD) from the raw Cartesian coordination, etc. This step is called featurization. The selection of optimal features or collective variables have a critical impact on the thermodynamic and kinetic property calculations predicted by the model, and is explored in many studies (Shamsi, Cheng & Shukla, 2018; Mittal & Shukla, 2017; Sultan et al., 2014). Consequently, using the optimal feature set for the construction of MSM affects its prediction accuracy. Identification of the most critical dihedral or contact features assist in identifying the optimum conformers. These conformers are then used as starting structures for the next epoch of adaptive sampling in MSM based adaptive sampling MD. Appropriate feature selection circumvents issues due to energy barriers leading to convergence of thermodynamic properties (such as free energy of binding in the case of protein-ligand binding simulation), or folding of the unfolded protein (Doerr & De Fabritiis, 2014).
Feature selection is an unsupervised machine learning task. One method of categorizing them identifies three sub-classes, namely, filters (Devakumari & Thangavel, 2010; Mitra, Murthy & Pal, 2002; Tabakhi & Moradi, 2015), wrappers (Dy & Brodley, 2004; Dutta, Dutta & Sil, 2014; Breaban & Luchian, 2011), and hybrids (Solorio-Fernández, Carrasco-Ochoa & Martínez-Trinidad, 2016; Li, Lu & Wu, 2006). In filter methods, the importance of a feature is studied based on the intrinsic properties of the data. They are not selected by training on the model on which it is to be used. In wrapper-based methods, the features are first used to train the model. Then their impact on the performance of the model is evaluated. Filter methods are computationally less expensive than wrapper methods. Hybrid methods combine the advantages of both filter and wrapper methods (Li et al., 2008). In this method, the feature subset works best when the same model used for feature selection is used subsequently (Solorio-Fernández, Carrasco-Ochoa & Martínez-Trinidad, 2020). Laplacian-score based feature selection is a filter-based method. This method finds the features that have the power to preserve the clusters in the data. Features that can maintain the structure of the plotted nearest neighbor graph are selected.
Laplacian scores have been used to reveal hidden structures in the complex conformational space of the intrinsically disordered peptide, Aβ(1-42) (Sgourakis et al., 2011). Laplacian scores identified crucial interactions in these conformers, which provided new drug design hypothesis that can, potentially, be used to discover drugs that inhibit the oligomers and fibril formation critical to the progression of Alzheimer’s disease. In this study, inspired by the success of the study mentioned above, the initial population of a GA’s chromosomes have genes pre-filtered through Laplacian scores-based ordering. In bio-molecules, conformational changes happen in localized regions (Fan et al., 2015). Laplacian score ranks the features based on its ability to preserve the local structure of the graph constructed based on the k-nearest neighbor algorithm (He, Cai & Niyogi, 2006). Due to this property of the Laplacian score, it is used to identify a subset of the features. The feature subset selection is further optimized through the GA.
Datasets to evaluate the approach
Molecular dynamics trajectories of five systems are used to validate our approach by analyzing the kinetic and thermodynamic properties predicted by the MSMs. The features selected were derived from the GA run, whose initial population was based on our Laplacian score ranking scheme. The simulation dataset of the two protein-ligand complexes are Piperidine-Thrombin Doerr et al. (2016) and Benzamidine-Trypsin Scherer et al. (2015). The protein folding trajectories of Fs Peptide (Beauchamp et al., 2011), WW domain (Lindorff-Larsen et al., 2011) (generously shared by D.E Shaw Research), and Villin simulation Doerr et al. (2016) were also analyzed as a part of this study.
Our approach
Figure 1 provides a detailed view of the protocol we have implemented, while the principal choices for each component of the workflow are shown in Table 1. The hyperparameters for the protocol were chosen based on the published research, HTMD and Msmbuilder tutorial (Chen et al., 2018; Doerr et al., 2016; Beauchamp et al., 2011). Scikit-learn (Pedregosa et al., 2011) was used for feature preprocessing, and to calculate the Laplacian scores, IPython (Pérez & Granger, 2007) for the execution of the python scripts in the form of a pipeline. MDTraj (McGibbon et al., 2015) was used to analyze the simulation trajectories, and HTMD (Doerr et al., 2016) was used for the construction of free energy heatmaps, standard free energy plots and cktest graphs for the MSMs. These graphs were constructed for selected residues to implement our approach, and for all residues to re-implement previous work (for comparison). MSMBuilder (Beauchamp et al., 2011) was used for MSM construction and GMRQ scoring.
| Featurization: Types |
| • Dihedrals- measured as phi and psi angles of respective amino acid residues |
| • Contacts- measured as distance between CA atoms of amino acid residues |
| Feature Selection Method |
| Genetic algorithm with initial population selected using Laplacian scored features |
| Dimensionality Reduction Method |
| TICA algorithm with 4 components and lag time of 2 ns |
| Clustering Method |
| Mini-Batch K-means with 200 clusters |
| MSM Model- Hyperparameters |
| • N_timescales = 5 Lag time (ns) = 50 |
| • Scoring using GMRQ |
| Genetic Algorithm- Hyperparameters |
| • Crossover Probability = 0.8 |
| • Mutation Probability = 0.2 |
-
Featurization: Featurization is performed to transform rotational and transitional motion from the simulation data to a vector. Dihedral angles and distance between CA atoms of amino-acid residues, from the Cartesian coordinates were extracted as features from the simulation data (for the 5 bio-systems). Dihedral featurization comprises backbone and side chain dihedral angles, along with sine and cosine of these angles (Beauchamp et al., 2011). All of the above are calculated for each frame in the simulation trajectory. In our approach, we used the sine and cosine of back bone dihedral angles for dihedral features. It has been proved to be one of the important metrics that is able to identify different conformers in an MD trajectory (Cossio, Laio & Pietrucci, 2011). Contact featurization calculates the distance between each pair of amino acid residue, or the distance between the specified pair of residues for each frame (Beauchamp et al., 2011). In this study, we implemented contact featurization as a vector of distances between CA atom of each amino acid residue and ligand for the protein-ligand systems, and the distance between the CA atoms of amino acid residues for the protein-folding simulation data.
-
Laplacian scoring: An average, across all trajectories, of the Laplacian scores obtained for each feature is calculated. The features were then sorted based on this average Laplacian score (of each feature). This ordered list is used to build the initial population for the GA. Laplacian Score (LS) is fundamentally based on Laplacian Eigenmaps and Locality Preserving Projection. The algorithm for calculating the Laplacians (He, Cai & Niyogi, 2006) is given below. Let Lr denote the Laplacian score of the rth feature. Let fri denote the ith sample of the rth feature; where i = 1, …, m.
-
Construct a nearest neighbor graph—G with m nodes—. The ith node corresponds to xi. We connect the edge between nodes i and j, if xi and xj are close, i.e., xi is among k nearest neighbors of xj, or xj is among k nearest neighbors of xi.
-
The weight matrix S of the graph, models the local structure of the data space. , when nodes i and j are connected, and Sij = 0 otherwise.
-
For the rth feature, we define: fr = [fr1, fr2, …, frm]T, D = diag(S1), 1 = [1, …, 1]T, L = DS where the matrix L is called graph Laplacian (Chung, 1997). Let (5)
-
The Laplacian score of the rth feature is: (6)
-
-
Genetic Algorithm (GA):
-
Initial population - Features are shortlisted based on Laplacian score. To avoid over-fitting or under-fitting, we use the thumb rule of selecting th of the number of samples (Sánchez & García, 2018) as the target number of features. This subset is then used as the initial population for GA. Under-fitting occurs when the number of features that represent the model are significantly lesser than the number of data points available. In MD centered protein systems, the number of features or the dimensionality is high due to the nature of the problem, and thus probability of under-fitting is negligible.
-
Fitness calculation - The fitness of the feature subset is scored based on GMRQ score of the respective MSM. Dimensionality reduction is performed using TICA (Pérez-Hernández et al., 2013). Dimensionality reduction produces a linear combination of the features, and the top components (called time structure-based independent components or tICs) capture the most prominent processes in the simulation. Further, clustering of frames is performed using MiniBatchKmeans clustering (McGibbon & Pande, 2015). The MSM is constructed using the clusters identified and the transition probability between these clusters. The number of clusters, lag time of TICA and MSM, number of components used for TICA and n_timescales which represents the timescale of the n slow processes is presented in Table 1. K-fold cross-validation (K = 5) is used to avoid over-fitting. The value of GMRQ score for the MSM is calculated based on the k-fold cross validation.
-
Selection - Selection of parents is performed for the production of off-springs for the next generation. This is performed based on the fitness of the feature subset. This helps in the selection of ideal traits, which in this case is the important CVs.
-
Crossover - The crossover probability decides the number of parents taking part and, in effect, the number of off-springs generated. We have used a single point crossover.
-
Mutation - It is performed to increase the gene variations in each of the generations. Single point mutation is carried out, in which a randomly selected gene is replaced with the highest ranked feature based on its Laplacian score, which is not already present in the chromosome.
-
Repeat the above four steps for the specified number of generations
-
-
MSM construction and GMRQ scoring: The MSM is constructed using the most optimal CVs obtained after the GA. This is then compared with MSM constructed using all the features in terms of the GMRQ score, and also compared using the implied timescale. The methodology is summarized in the Fig. 1
Details on Bio-systems studied
Five systems were analyzed in this work (The original complexes have been uploaded in figshare).
-
Benzamidine-Trypsin (Doerr et al., 2016) : This work studies the binding of serine protease beta-trypsin to inhibitor benzamidine. It comprises 10 microseconds of simulation data, with component trajectories of 200 nanoseconds each.
-
Piperidine-Thrombin (Scherer et al., 2015) : Thrombin is a serine protease that acts in the coagulation pathway to convert factor XI. The simulation dataset of Piperidine-Thrombin comprises 810 trajectories of 200 nanoseconds each, resulting in a cumulative simulation length of 162 microseconds.
-
Fs Peptide (Beauchamp et al., 2011) : Fs peptide is a system widely used to study intricacies of protein folding. The simulations were carried out using the AMBER99SB-ILDN force field with OpenMM 6.0.1. The Fs peptide dataset includes 28 trajectories of 500 nanoseconds each, totalling to 14 microseconds of data.
-
Villin(Doerr et al., 2016) : Villin is an actin binding protein. The Villin dataset consists of 1,374 trajectories of 100 nanoseconds each and total simulation data of length 137.4 microseconds.
-
WW domain (Lindorff-Larsen et al., 2011) : WW domain is a protein domain that plays an important role in the interaction between protein ligands. The WW-domain dataset consists of 325 trajectories with a total of 1,137 microseconds.
Results
The effectiveness of MSMs that use features selected through GA with Laplacian-based initialization is compared to MSMs that used all features. The metrics used are GMRQ scores, and the implied timescales obtained for the slowest process for the two different sets of features. Recall that dihedrals and contacts are used to generate the feature set. The features selected by our approach in systems where we saw an improvement MSM metrics are shown in Fig. 3.
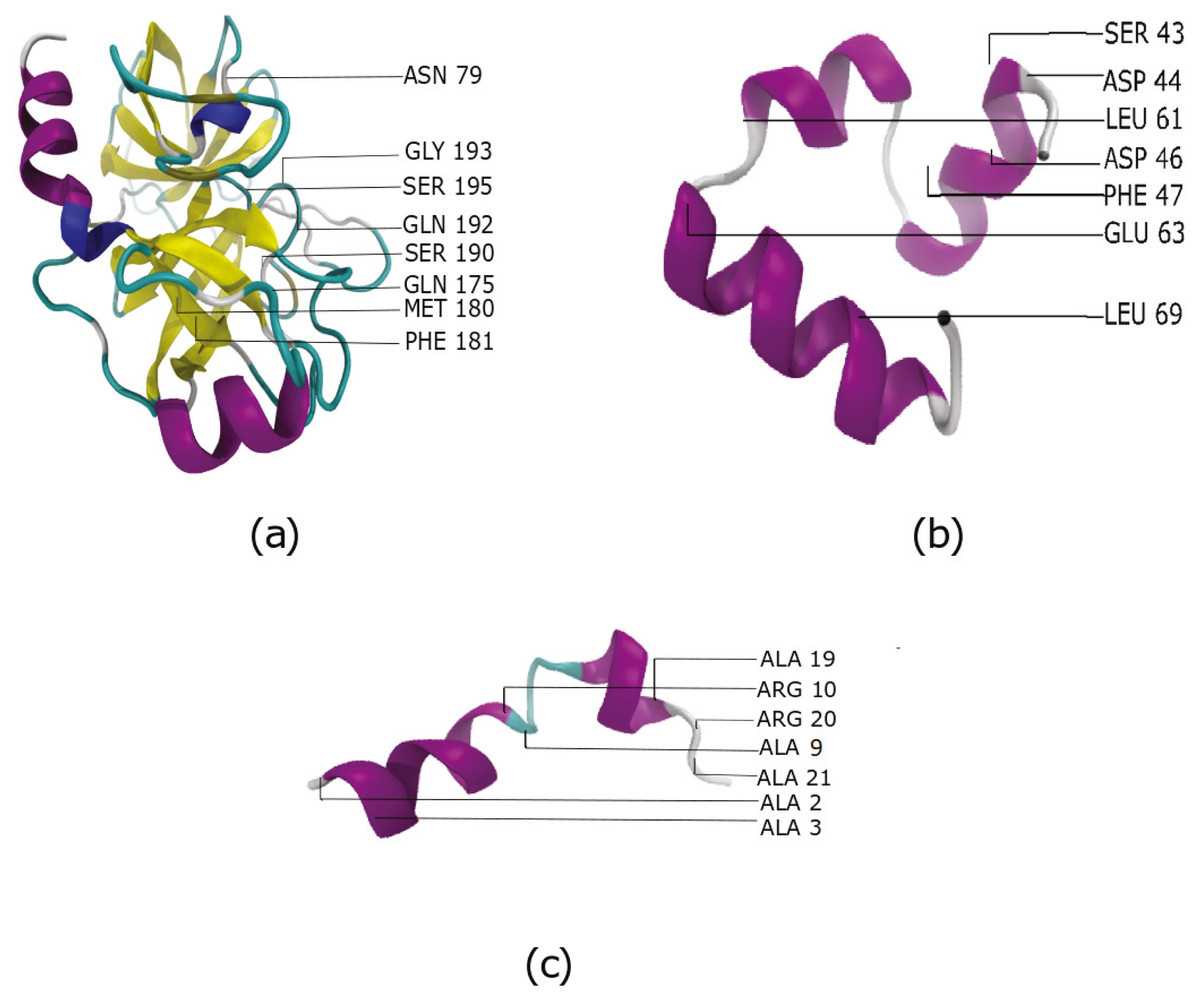
Figure 3: The most important features identified by our approach for three systems (A) Benzamidine -Trypsin (B) Villin and (C) Fs Peptide.
{kind=link}
Among the important residues identified for Villin, are 47 and 69. These are two of the most important hydrophobic residues that play an important role in the folding of Villin (Frank et al., 2002). In the Benzamidine-Trypsin system, residues 79, 175, 180, 190 are observed to play important roles in ligand-binding process (Buch, Giorgino & De Fabritiis, 2011) and calcium binding loop (Plattner & Noé, 2015). Indeed, these were also identified as important residues using our workflow. This validates that the proposed approach is able to identify residues important to the biological process being studied.
Comparison of GMRQ scores
In this section, we have compared the GMRQ scores of MSM models obtained when the full set of dihedral angle-based features and the full set of features calculated using all contacts are considered, against the ones where we use only selected features using our approach. The summary of these results are shown in Table 2. For the Benzamidine-Trypsin system, the model with select features has a higher GMRQ score of 3.21 and 2.59. The GMRQ scores of MSMs constructed for Villin and Fs Peptide systems with reduced set of dihedral and contact features are both higher by 19.15%, 19.95%, 17.44%, 11.25% respectively, as compared to when constructed with all the features. This indicates that the features identified by our method is able to capture the most important features and reduce the noise by avoiding unwanted features in certain cases. However, in the case of Piperidine-Thrombin and WW-domain systems, the GMRQ score of the MSM constructed with reduced set of selected features is less than the score obtained with all the features selected. This may be due the fact that the interactions captured by the reduced set of features is not enough to capture the slowest process that occurs in the respective simulation data.
| System | Feature | GMRQ score (all) used as Benchmark | GMRQ score (selected with Laplacian score and GA) |
|---|---|---|---|
| Benzamidine-Trypsin | Dihedral | 3.54 | 3.21 |
| Contacts | 1.87 | 2.59 | |
| Piperidine-Thrombin | Dihedral | 5.68 | 5.65 |
| Contacts | 5.25 | 5.09 | |
| Villin | Dihedral | 3.76 | 4.48 |
| Contacts | 3.71 | 4.45 | |
| WW-domain | Dihedral | 4.66 | 4.46 |
| Contacts | 5.29 | 5.23 | |
| Fs Peptide | Dihedral | 4.53 | 5.32 |
| Contacts | 4.8 | 5.34 |
The Table 3 shows that our approach is able to mitigate model inaccuracies caused by the over-fitting that occurs when the length of simulation (and hence, number of observations) is relatively shorter, while the number of features are higher. The variance in the GMRQ score for the model with selected residues is 5 to 10 times lesser than when all residues are used for MSM construction. This shows that the MSM constructed using selected residues is able to generalize better, and overcome over-fitting issues. The WW-domain is not given in the Table 3 as simulation length of 1,137 microseconds is adequate, and hence, over-fitting does not occur.
| System | Variance in GMRQ score (all features) | Variance in GMRQ score (selected with Laplacian score and GA) |
|---|---|---|
| Benzamidine-Trypsin | 0.27 | 0.04 |
| Piperidine-Thrombin | 0.06 | 0.005 |
| Villin | 0.42 | 0.08 |
| Fs Peptide | 0.92 | 0.18 |
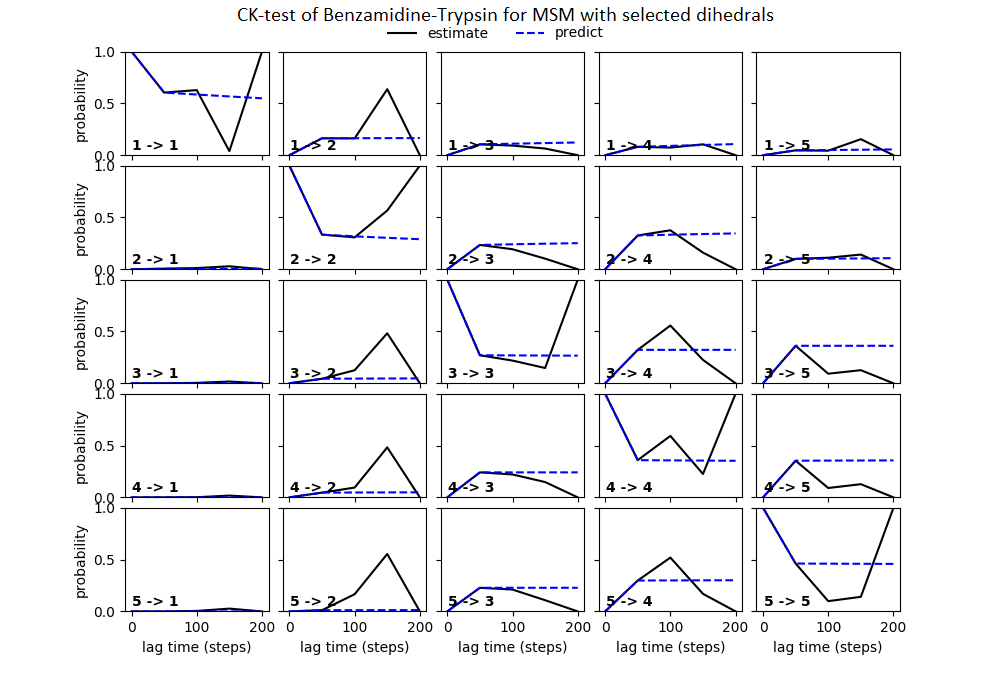
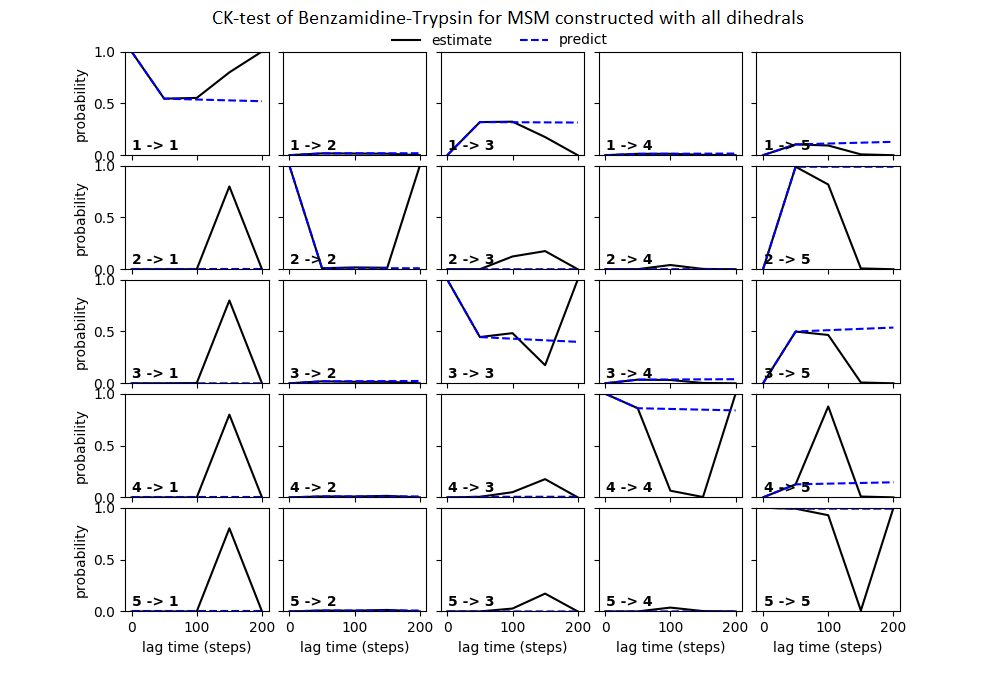
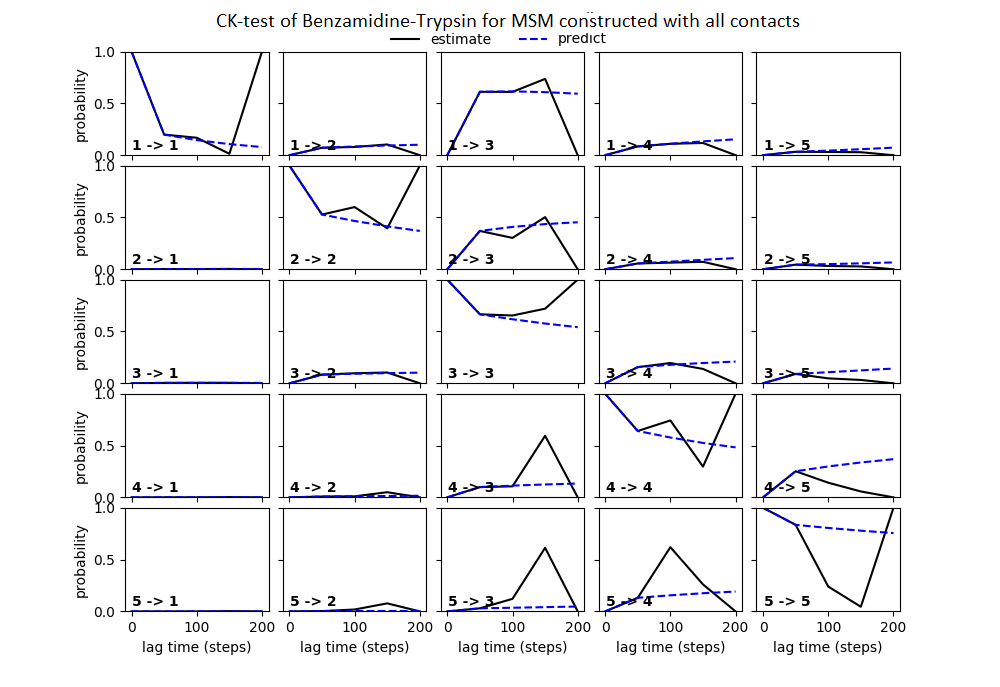
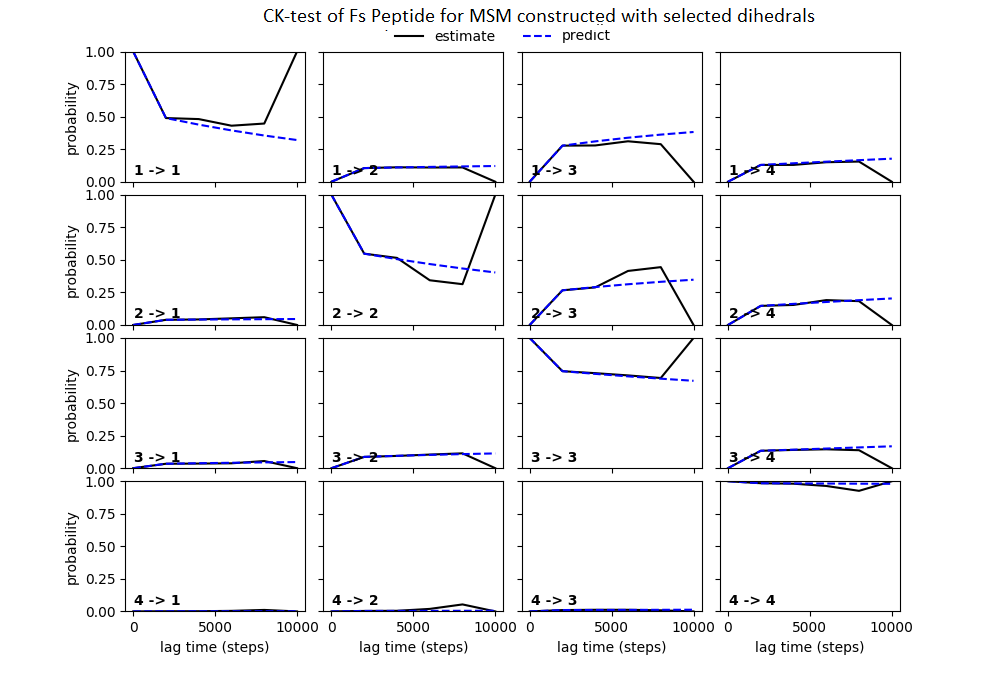
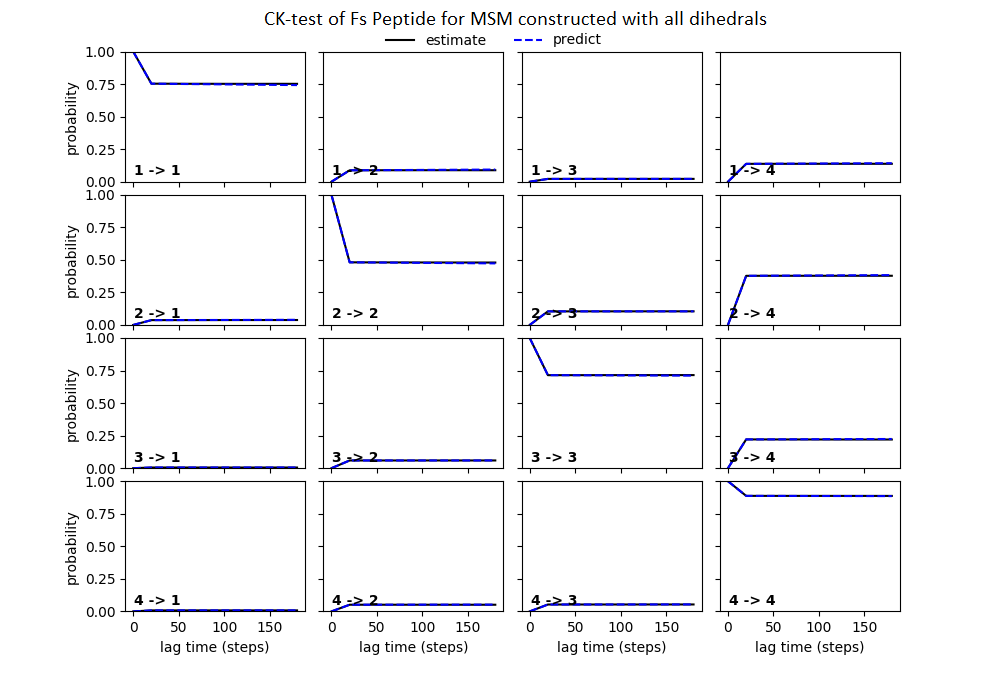
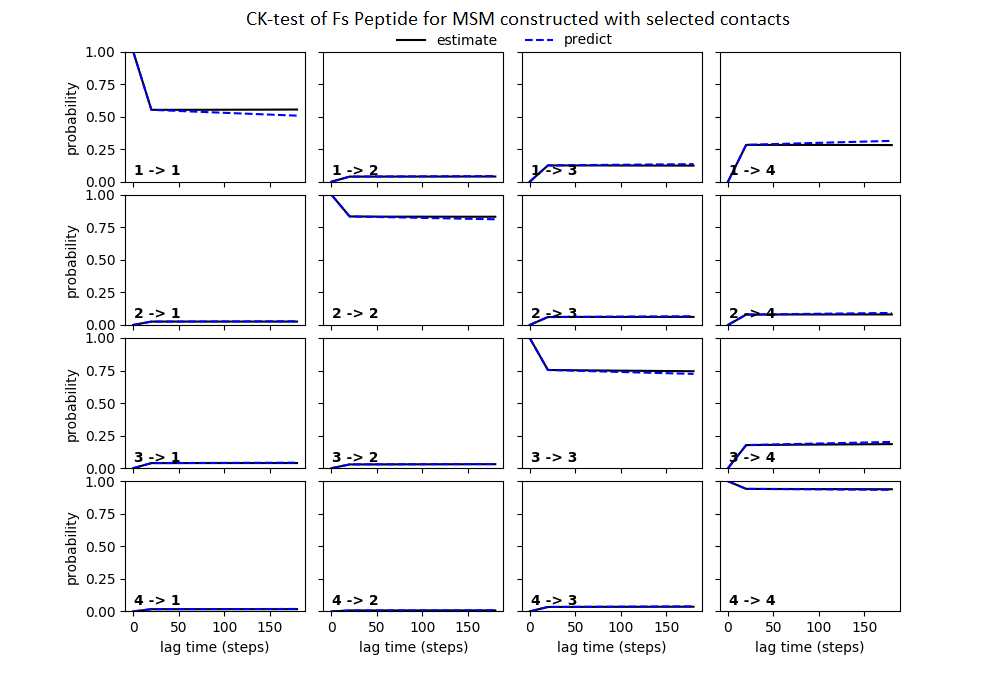
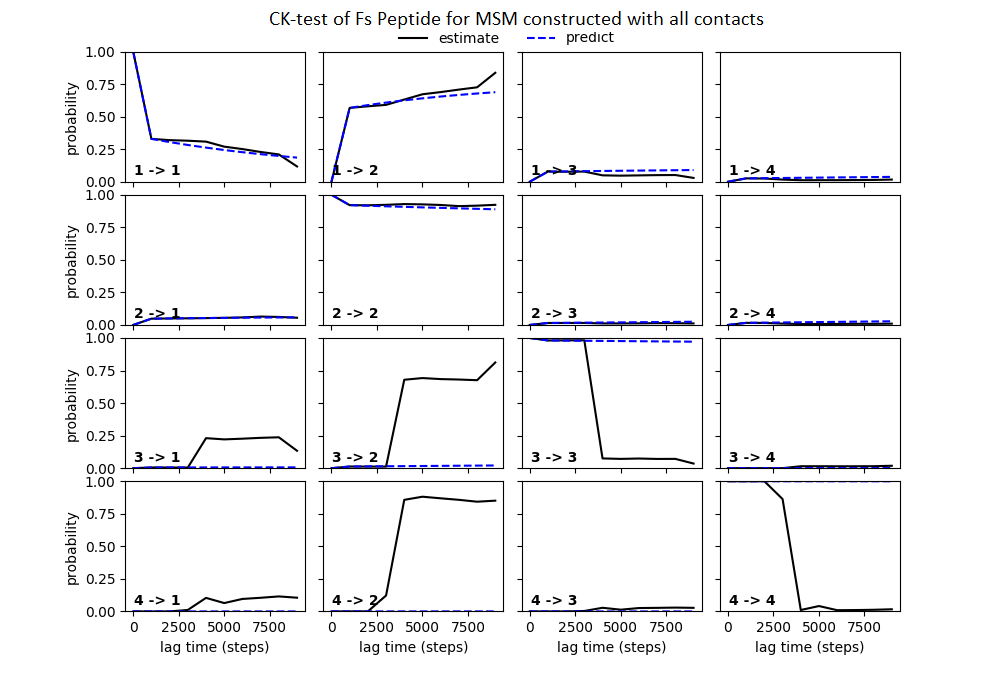
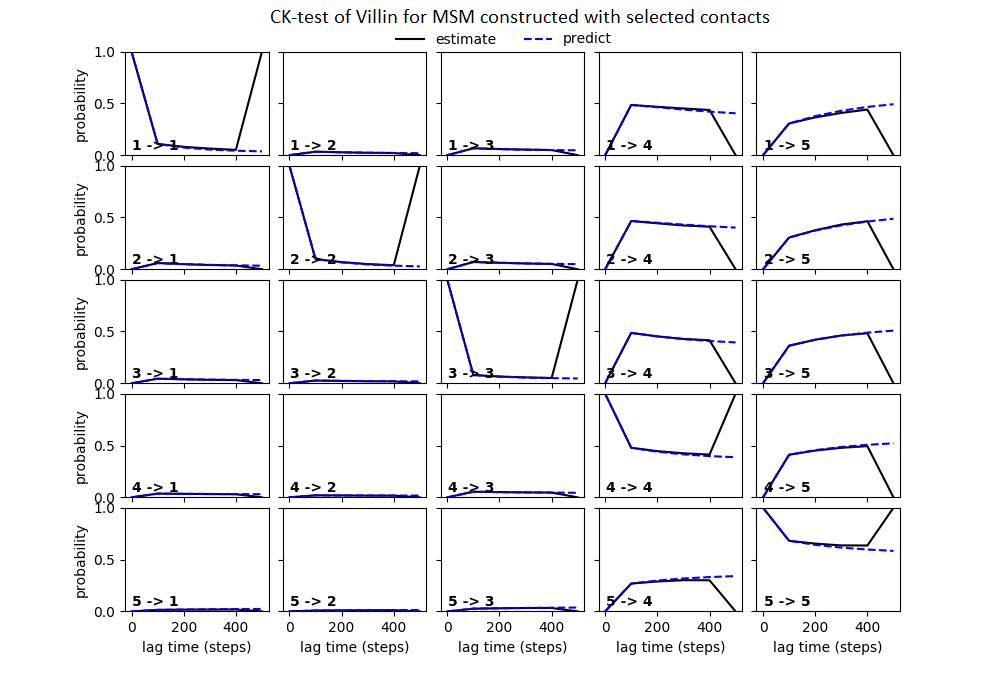
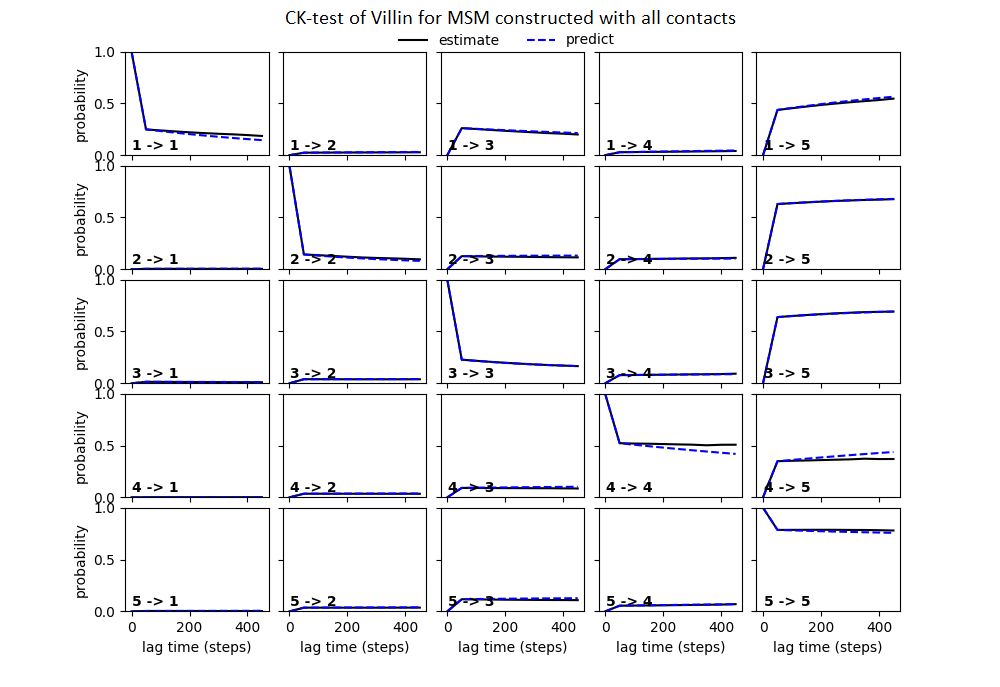
Some studies use the Chapman–Kolmogorov property (Noé et al., 2009; Prinz et al., 2011; Bowman, Pande & Noé, 2013) as a measure of self-consistency of individual MSMs. Nonetheless, since our goal was to find the optimum MSM that identifies the slowest implied timescale, we have used GMRQ scores for the primary analysis. The analysis of Chapman–Kolmogorov test is given for the three systems that produced better results (see Table 2 for the ranking) through our approach is provided in form of figures in the (Figs. S1 through S3).
Standard free energy prediction
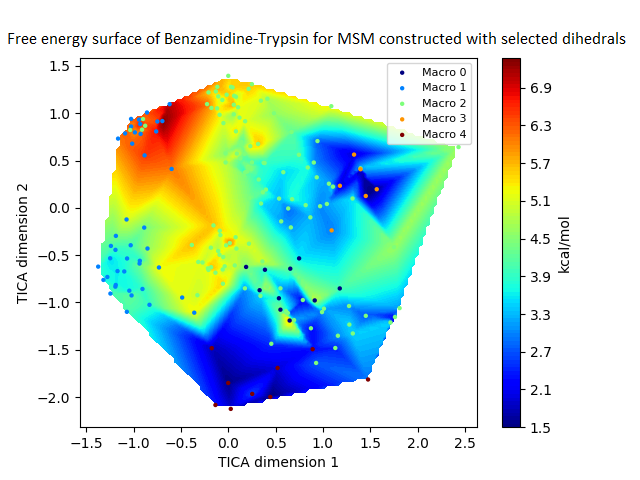
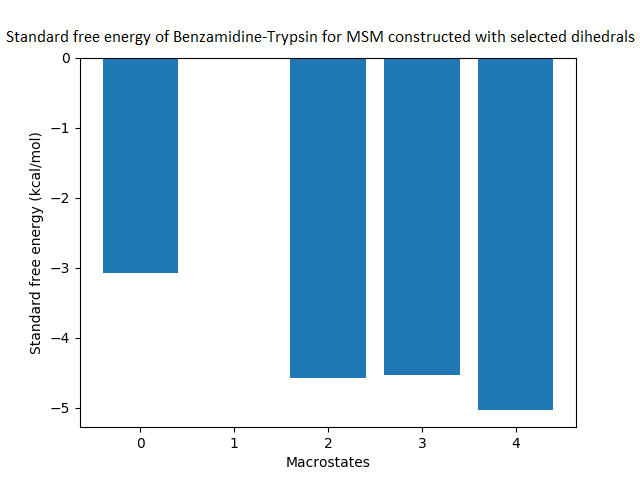
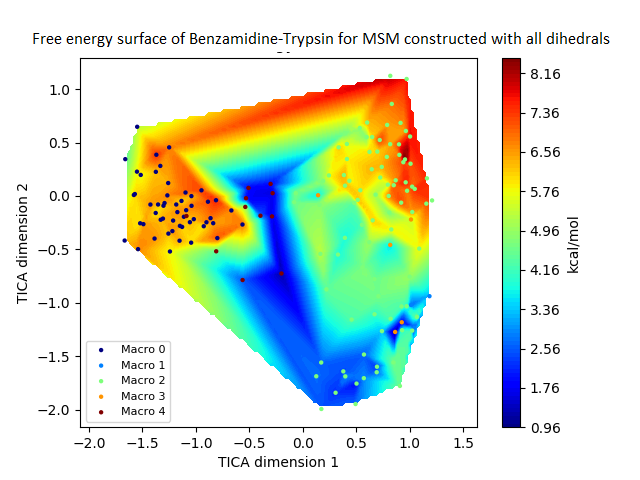
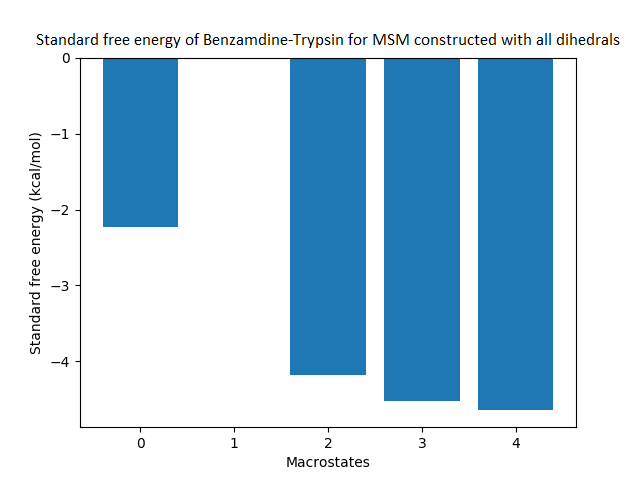
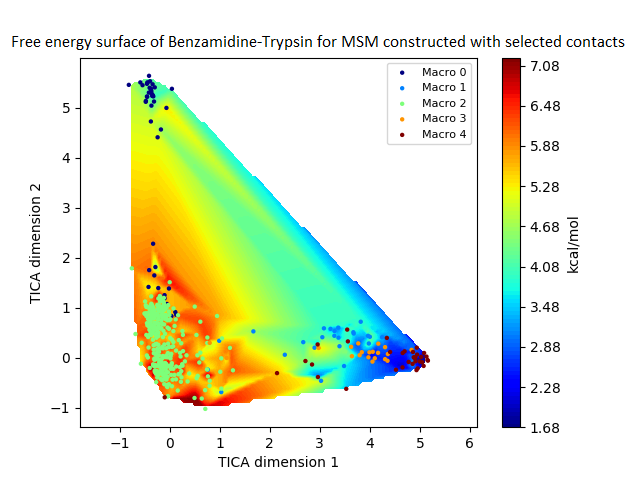
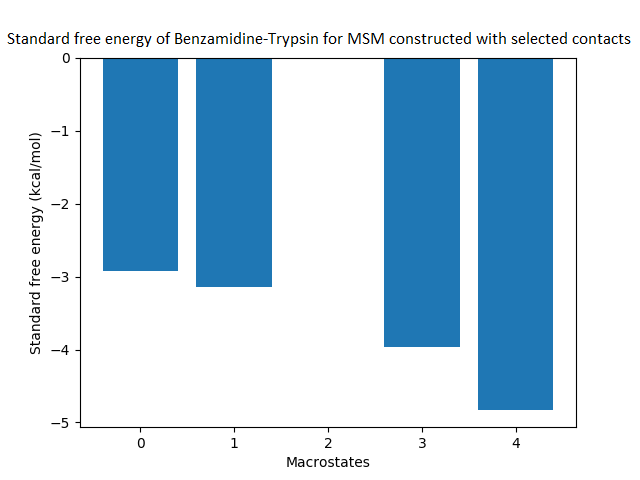
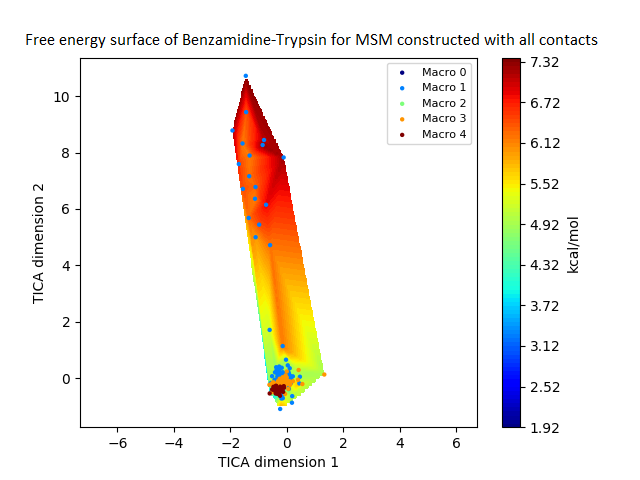
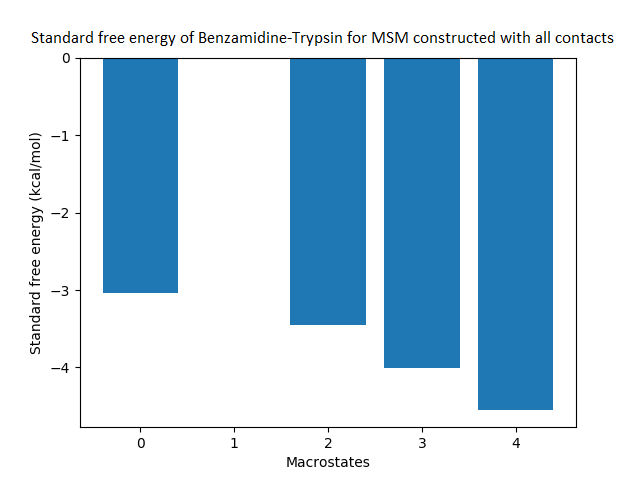
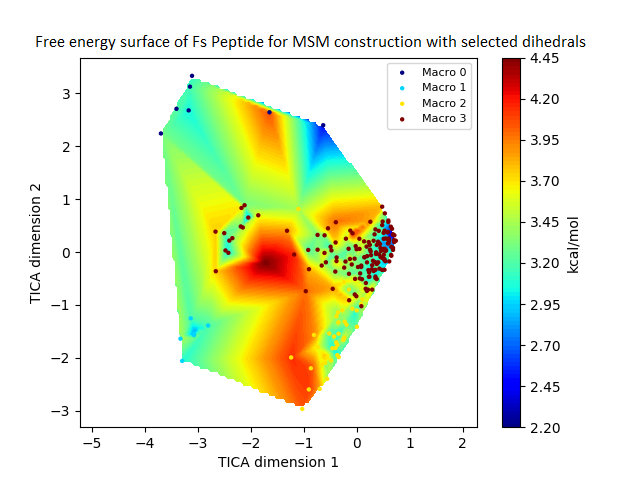
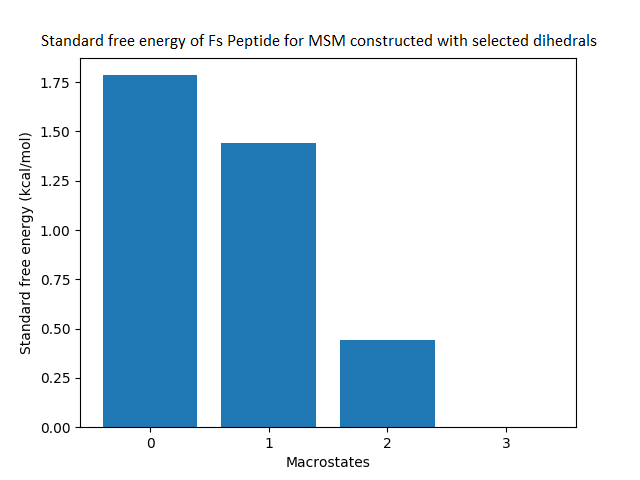
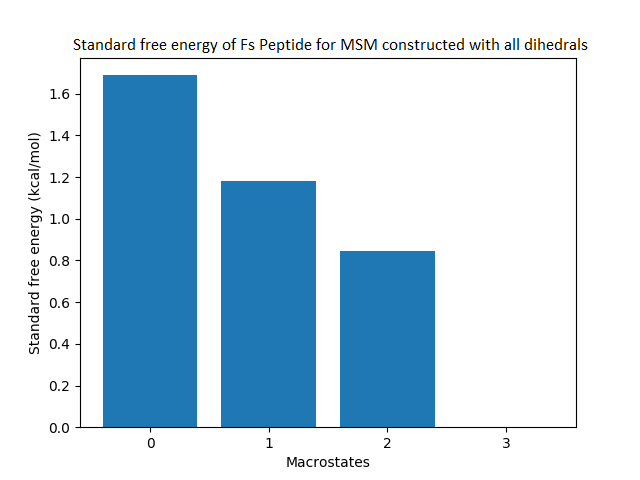
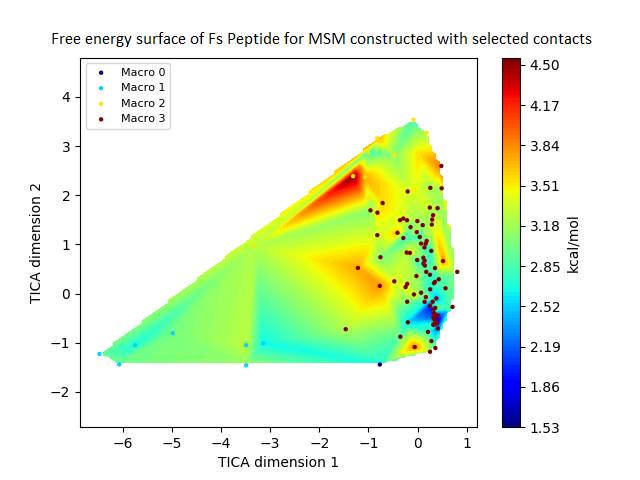
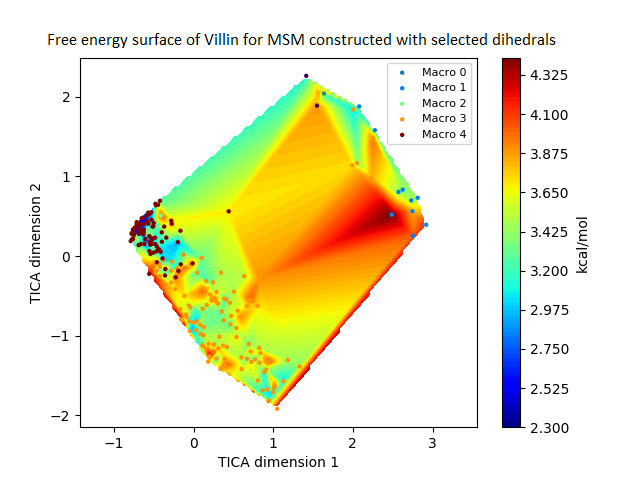
The free energy surface is indicative of the thermodynamic and kinetic properties of the system. The free energy surface and the standard free energy plots of the meta-stable states, identified as macro-states in MSMs, of the Villin system are given in Fig. 4. One of the main advantages of an MSM is its ability to predict the thermodynamic and kinetic properties of the system. The meta-stable states identified by the MSM model map to different conformers of the bio-molecule in the simulation. These are identified as different points on the free energy surface. The stable, lowest energy state is the bonded state in protein-ligand binding simulation, and folded state in protein folding simulation. For the sake of brevity, the same for the other two high scoring MSM models, namely, Benzamidine-Trypsin system, Fs Peptide and Villin (dihedral features only) are given in Figs. S4 to S6.

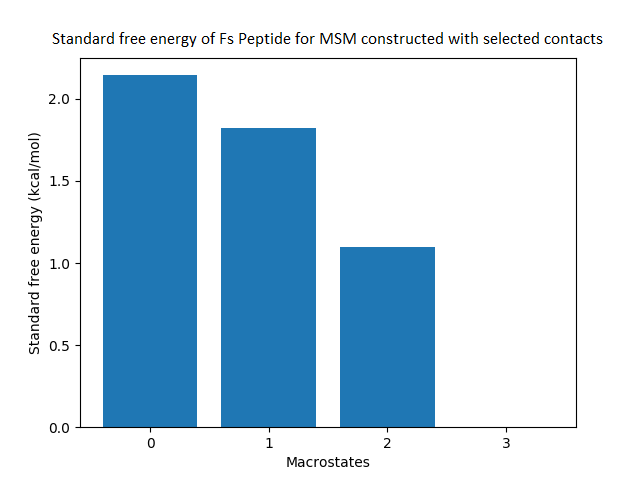
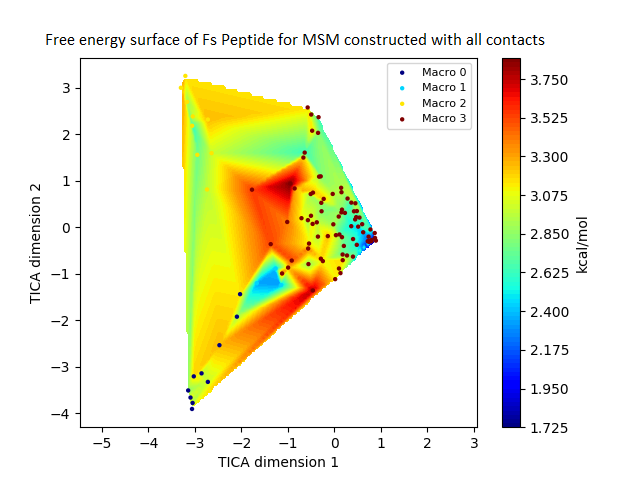
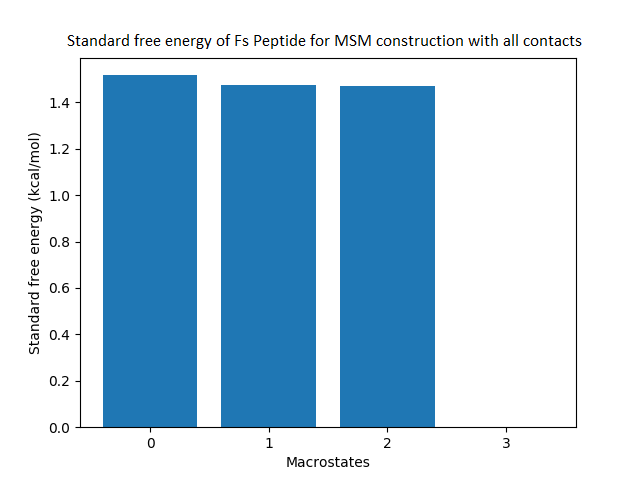
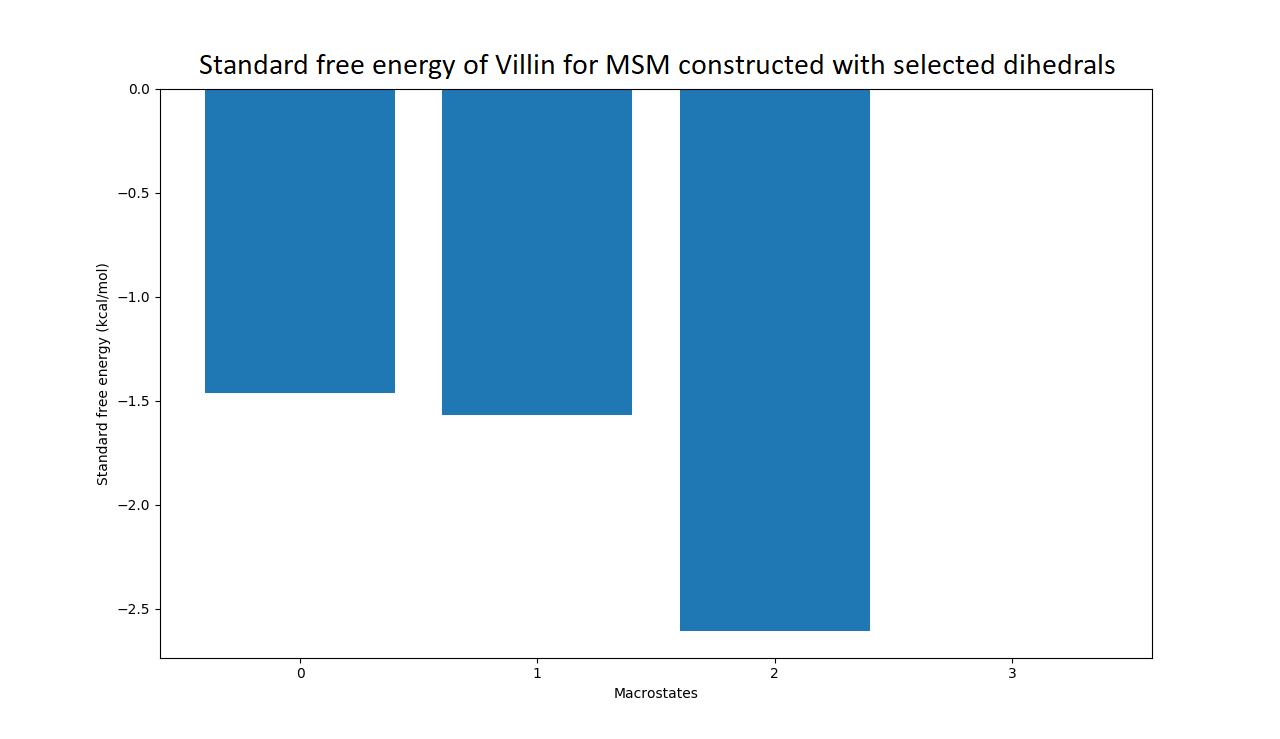
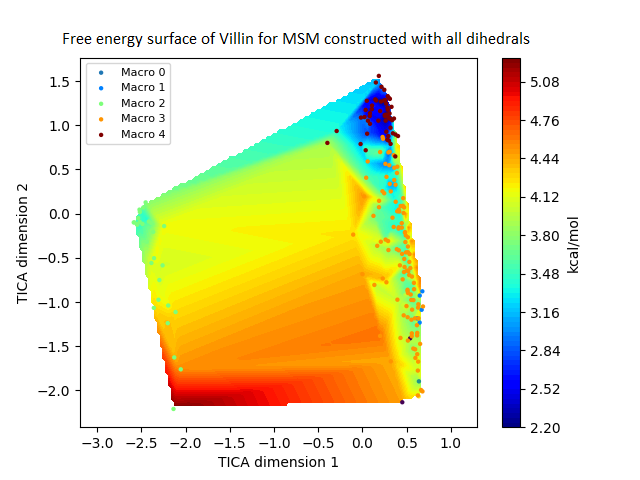
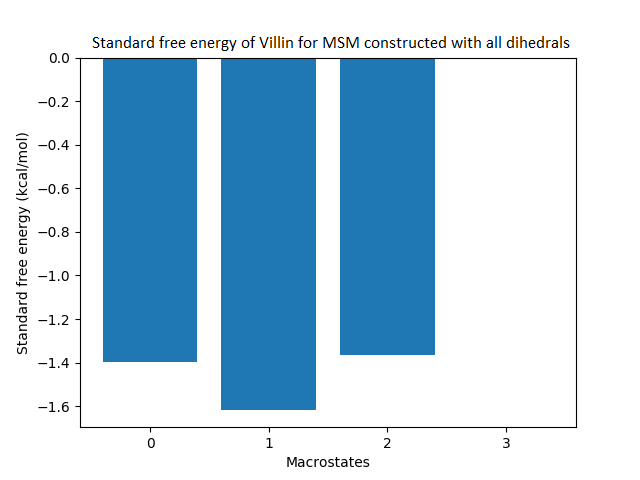
Figure 4: Free energy surface projected on the first two TICA dimensions and the corresponding standard free energy of meta-stable states of Villin for the MSM models.
(A) Free energy surface of MSM constructed with all contacts, (B) Standard free energy of meta-stable states plotted in (A), (C) Free energy surface of MSM constructed with selected with selected contacts, (D) Standard free energy of meta-stable states plotted in (C).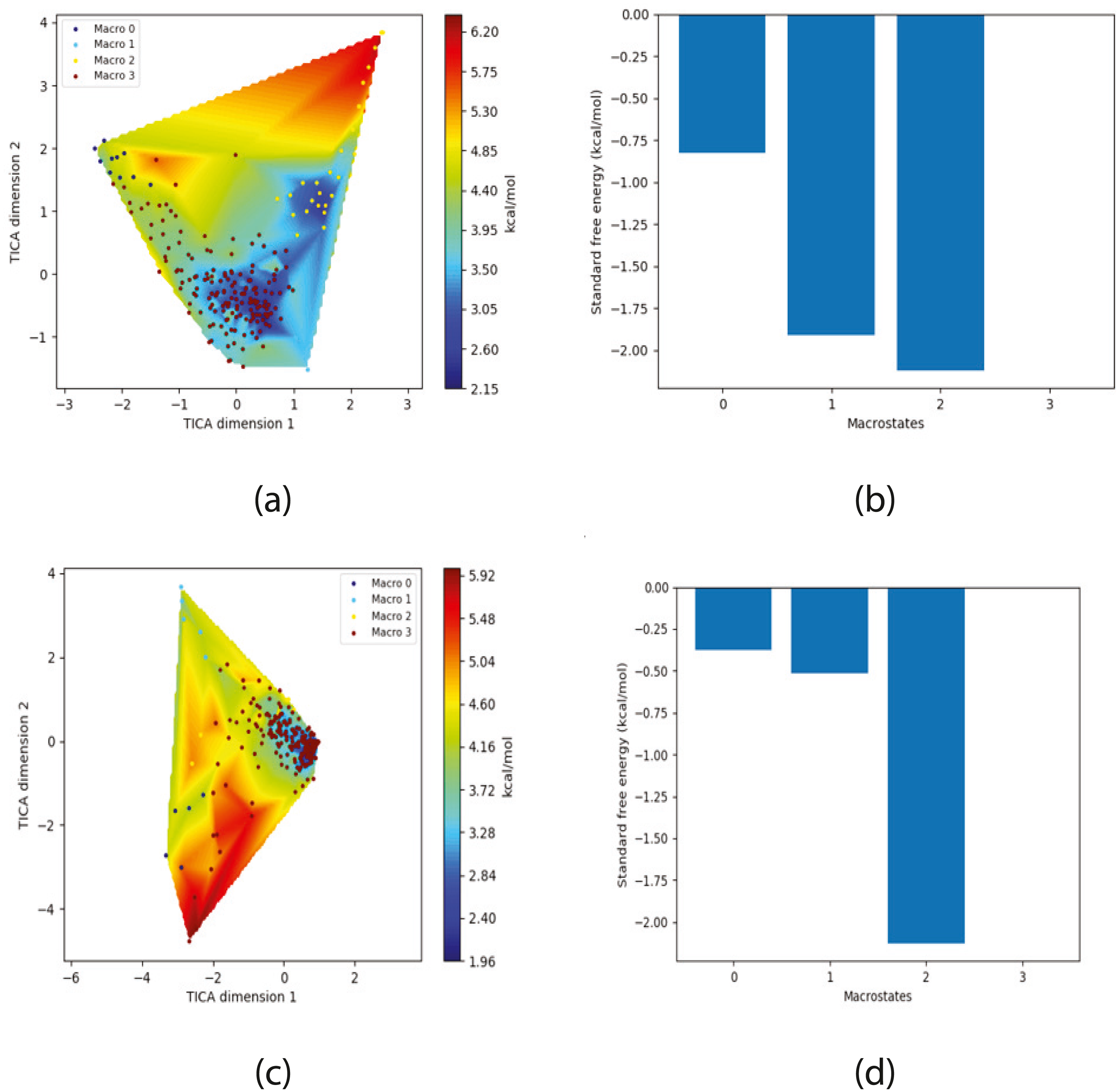{kind=link}
The free energy surfaces of the Villin system for full-set and selected of features are given in Figs. 4A and 4C respectively. The standard free energy of each of the meta-stable states identified in the corresponding free energy heat map are shown for full-set and selected of features in Figs. 4A and 4D. Figure 4A shows four meta-stable states on the free energy surface that was plotted using all the contact features. The standard free energy of each of the meta-stable states identified in Fig. 4A) is shown in Fig. 4B). The lowest energy of −2.29 kcal/mol, observed for the meta-stable state 2, is identified as the sink state by the MSM model. Similarly, four meta-stable states are identified (Fig. 4C) for the MSM model that used contact features between selected residues that were identified by our approach. The standard free energy of each of the meta-stable states is plotted in Fig. 4D, and lowest energy of −2.31 kcal/mol is observed for the meta-stable state 2, identified as the sink state by the MSM model. The lower standard energy stable state (−2.31 kcal/mol) is identified by the MSM model constructed using selected residues identified by our approach.
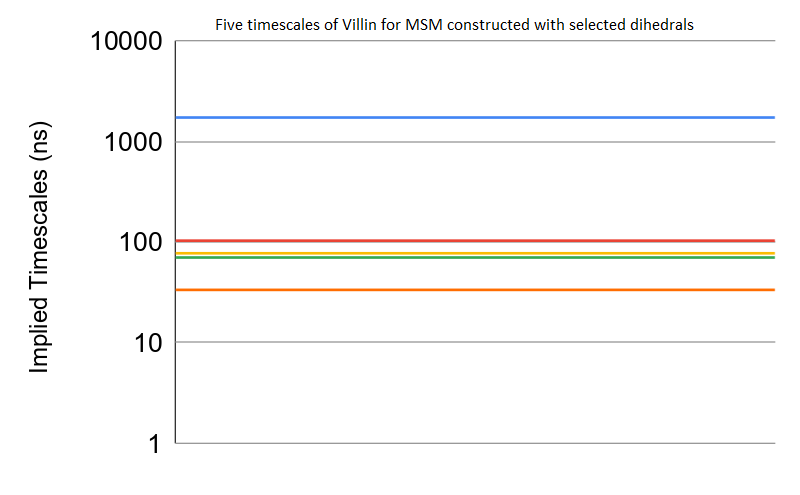
Implied timescale comparisons
Implied timescale values refer to the time taken by the slowest processes captured by MSMs. Table 4 shows that the MSMs constructed using the selected dihedral and contact features of Benzamidine-Trypsin, Villin and Fs Peptide are able to capture slowest processes. Since our goal is to capture the slowest timescale that represents the folded or the protein-ligand bonded state, the other four slower timescales is given in the Fig. S7, for the interested reader. Five timescales are compared for systems with better MSMs as identified by our approach, namely, Benzamidine-Trypsin, Fs Peptide and Villin.
| System | Feature | Implied timescale in ns (all features) Benchmark | Implied timescale in ns (selected with Laplacian score and GA) |
|---|---|---|---|
| Benzamidine-Trypsin | Dihedral | 3,980.45 | 1,069.21 |
| Contacts | 5,571.51 | 14,082.11 | |
| Piperidine-Thrombin | Dihedral | 2,453.09 | 1,530.06 |
| Contacts | 5,017.45 | 4,055.23 | |
| Villin | Dihedral | 1003.65 | 1,725.33 |
| Contacts | 788.59 | 1,169.06 | |
| WW-domain | Dihedral | 7,934.28 | 7,101.21 |
| Contacts | 8,679.26 | 8,535.81 | |
| Fs Peptide | Dihedral | 1,515.48 | 2,034.63 |
| Contacts | 1,943.93 | 2,198.82 |
The MSM constructed using the full-set of all the contacts with the ligand, in the Piperidine-Thrombin system, is able to capture the slower process. Nonetheless, it can be seen that, in this case, GMRQ score for the model from the reduced set is not significantly higher against the one with the full-set of contact-based features.
Discussion
Optimal feature selection to identify an optimal set of collective variables, in other words, residues of the simulated bio-molecules, ensures that the co-variance matrix calculated during dimensionality reduction phase is a positive definite matrix. Additionally, avoiding over fitting, due to the lack of sampling data relative to a large number of features in each frame of the samples is highly desirable. In this work, we show that enhanced feature selection using Laplacian scoring addresses both these issues i.e., the requirement of increased sampling and avoiding over fitting.
In the protein-ligand system, Benzamidine-Trypsin, the contact features between selected residues (Fig. 3A) are able to identify the slowest processes correlated to the binding of Benzamidine to Trypsin, better than when all the features are selected. This may be because using all the contact features adds noise to the model when the MSM is constructed with all contacts. Results also show that contact features are relatively more discriminant towards identifying the slowest processes, as compared to dihedral features. This suggests that researchers should empirically check the set of features that best fit the system in hand, when using Laplacian scoring. In this system, the number of features (for example, 888 dihedrals) are very large as compared to the number of frames (200). Our proposed methodology, allows the use of selected features for construction of efficient MSM, and thus shows that this approach is useful when the size of the molecules are larger, and have smaller length simulations.
In the protein folding systems used, when the amount of simulation data is more than the number of dihedral and contact features extracted, the GMRQ score of MSM constructed with all the features is higher than when using a reduced set of features selected by our approach. The GMRQ score of MSM constructed for WW-domain with all features is higher. Whereas, in the case of Villin and Fs Peptide, MSM with the reduced set of features has higher GMRQ score, and is able to identify the slowest process.
Conclusions
Identification of CVs, that is feature selection, is one of the the critical steps in the construction of MSM. The features determines the ability of MSM to capture the slowest processes and thus the hidden conformers in the molecular dynamics simulation data. The GMRQ score of the MSM provides a metric to calculate the accuracy of the prediction made by the MSM. In this study, we have shown that, in some systems, more efficient MSMs are constructed using Laplacian score with GA using GMRQ score as fitness score. This method helps to identify which of the features, between dihedrals or contacts, is to be chosen for the construction of an efficient MSM. The most significant advantage of this method is that it helps to reduce the amount of sampling and overcomes the bottleneck of long simulation. It has the potential to circumvent over fitting caused by large dimensionality of the simulated data. This approach has been applied to simulation data involving folding of protein and protein-ligand binding. In this approach, MSM building had the goal of finding out the slowest process in the simulation. In protein folding, the movement from the unfolded state to the folded state, and in protein-ligand binding the binding of ligand to the protein are the slowest process in the simulation data identified by MSM.
The time required to find out the optimal subset of features is significant due to use of wrapper method, along with filter based feature selection. The GMRQ score of MSM constructed with all the features is higher for longer simulations. Nonetheless, this approach significantly reduces the amount of sampling required to construct an efficient MSM as shown in the case of Benzamdine-Trypsin, Villin and Fs Peptide systems, and hence helps to take advantage of adaptive sampling MD which involves a large number of short simulations. This method helps to construct MSM with higher GMRQ score and find out the optimum features that maximally affects the slowest process in the simulation data. In summary, our study shows that heuristic initialization of the GA population can automatically select the essential features that contribute to the construction of MSM with improved GMRQ score. To our knowledge, this is the first approach that uses Laplacian score along with GA to automatically select features to construct MSMs with a reduced set of features. The code used is available at GitHub: https://github.com/anuginu/MSM_latest.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}