
Unveiling the synthesis and spectral characterizations of novel (E)-furan-2-yl acrylohydrazides: an exploration in molecular design
- Published
- Accepted
- Received
- Academic Editor
- Giovanna Bosica
- Subject Areas
- Spectroscopy, Synthetic Organic Chemistry
- Keywords
- (E)-3-(furan-2-yl)acrylohydrazide, Acylhydrazone, Conformational analysis, NMR spectroscopy
- Copyright
- © 2024 Coulibaly et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Organic Chemistry) and either DOI or URL of the article must be cited.
- Cite this article
- 2024. Unveiling the synthesis and spectral characterizations of novel (E)-furan-2-yl acrylohydrazides: an exploration in molecular design. PeerJ Organic Chemistry 6:e11 https://doi.org/10.7717/peerj-ochem.11
Abstract
In this study, we present the synthesis of novel derivatives of 3-furan-2-yl acrylohydrazide using a meticulous three-step reaction sequence. The synthesis ends up in the condensation of (E)-3-(furan-2-yl) acrylohydrazide (3) with diverse benzaldehyde and acetophenone derivatives. Comprehensive characterization of the synthesized compounds was achieved through 1D nuclear magnetic resonance spectroscopic analyses (1H and 13C NMR), 2D nuclear magnetic resonance spectroscopy (HSQC, NOESY), and high-resolution mass spectrometry (HRMS). The investigation of 1H NMR data at room temperature in deuterated dimethyl sulfoxide (DMSO-d6 ) unveiled the existence of (E)-3-(furan-2-yl) acrylohydrazide derivatives (4a–h) in a conformational equilibrium, manifesting as a mixture of synperiplanar E (sp E) and antiperiplanar E (ap E), Notably, compounds 4a and 4b predominantly adopted the sp E conformer (Ec=c sp EC=N), while the remaining compounds (4c–h), favored the antiperiplanar conformation (Ec=c ap EC=N) even if for the 4g compound it was challenging to determine the EC=N conformer. These findings contribute valuable insights into the conformational dynamics of this class of compounds, holding significance for applications in diverse scientific domains.
Introduction
N-acylhydrazones (NAH) are distinguished by the presence of an azomethine fragment (CO-NH-N=CH), which, when integrated or connected into a heterocyclic scaffold, frequently yields compounds exhibiting noteworthy biological activities, (Popiołek et al., 2020; Coulibaly et al., 2023; Popiołek, 2021) including anticancer, (Zheng et al., 2009; Terzioglu & Gürsoy, 2003) antibacterial, (Cardoso et al., 2014) and antidepressant properties (Salgin-Goksen et al., 2021). Acrylohydrazides, a distinct class within these organic compounds, have recently garnered attention due to their multifunctional properties (Yan & Mihail, 2016). Among them, (E)-3-(furan-2-yl) acrylohydrazides stand out, featuring the conjugated furan-2-yl scaffold linked to a hydrazide function. This unique combination of structures holds considerable potential for diverse applications, encompassing bioactive functionalities (Ge et al., 2014).
Building upon our prior studies, (Achi et al., 2022; Ablo et al., 2022) which consistently established the stable (E) conformation of N-acylhydrazones in various solvents, particularly the prevalence of the syn conformation in solvents like DMSO-d6, we identified conditions favoring the anti conformation, including less polar solvents (e.g., CDCl3), intramolecular hydrogen bonding, or steric hindrance. The persistent dominance of the syn conformation, where crucial substituents adopt specific spatial orientations, suggests a structural regularity with promising implications for the design of novel molecules tailored for specific purposes.
Aligned with our previous investigations characterizing benzimidazoles N-acylhydrazones and acylhydrazones derived from nitroimidazo[1,2-a]pyridine, intimately linked to (E)-3-(furan-2-yl) acrylohydrazides by their fundamental structure, this study endeavors to comprehensively explore the preferential conformation of these newly synthesized molecular entities. Employing a rigorous synthetic approach coupled with structural analyses, encompassing NMR, and HRMS, our objective is to delve into the conformational properties of (E)-3-(furan-2-yl) acrylohydrazides. Ultimately, this research aspires to make a significant contribution to the understanding of molecular conformation, thereby laying the foundation for potential applications in the field of materials science.
Materials and Methods
Material
Portions of this text were previously published as part of a preprint in ChemRxiv. 2024; doi:10.26434/chemrxiv-2024-f8gtf. Unless otherwise stated, 1H et 13C NMR spectra were recorded at 500 and 126 MHz, at 400 MHz for 1H and 101 MHz for 13C, respectively, in CDCl3, Methanol-d4, and DMSO-d6 solutions provided by Eurisotop, using TMS as an internal reference. Chemical shifts are reported in ppm on the δ scale. Multiplicities are denoted as s (singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartet), m (multiplet), and coupling constants (J) are reported in Hz. High-resolution mass spectra (HRMS) were acquired in electrospray ionization (ESI) mode using an LC-MSD TOF mass analyzer. The infrared analyses were conducted using a SHIMADZU FTIR spectrometer equipped with an ATR stage featuring a diamond crystal. Data acquisition involved performing 20 scans in the range of 4000 to 400 cm−1. Reaction monitoring and compound purity were assessed by thin-layer chromatography (TLC) on silica gel plates (Merck Kisselgel 60F254 nm, silica thickness 0.2 mm) revealed under UV-Vis light (λ = 254 and 365 nm). Product purification was carried out using silica gel column chromatography (Kisselgel SI60, 40–63).
Solid compound melting points were determined using a Köpfler melting point apparatus with a temperature range of 44–266 °C.
The required reagents for the synthesis protocols were procured from suppliers. Triphenylphosphine, 3-fluoroacetophenone, 4-fluoroacetophenone, furan-2-carbaldehyde, thiophene-2-carbaldehyde, and pyridine-2-carbaldehyde were sourced from Sigma Aldrich. Hydrazine monohydrate and pyrrole carboxaldehyde were obtained from Thermo Scientific Chemicals. Additionally, sodium hydrogenocarbonate was sourced from Chem Lab. All solvents utilized in the experiments were provided by Polychimie Côte d’Ivoire, ensuring high quality and consistency throughout the experimental procedures.
Methods
Synthesis procedure for (E)-ethyl 3-(furan-2-yl) acrylate (2)
In a 100 mL round-bottom flask (rbf) fitted with a magnetic stirrer, a reaction mixture comprising triphenylphosphine (PPh3, 1.5 eq., 36.46 mmol, 9,48 g), a saturated solution of sodium bicarbonate (NaHCO3) (50 mL), ethyl 2-chloroacetate (1.8 eq, 43.40 mmol, 4.80 mL), and furan-2-carbaldehyde (1 eq, 24.10 mmol) was stirred at ambient temperature for a duration of 24 h. The reaction was monitored using thin-layer chromatography (TLC), and the completion of the reaction is indicated by TLC analysis. At the end of the reaction, the reaction mixture was extracted with dichloromethane (DCM), and the resulting organic layer was dried using magnesium sulfate (MgSO4) before undergoing concentration under reduced pressure. Subsequently, diethyl ether (Et2O) was introduced to induce the precipitation of triphenylphosphine oxide (O=PPh3), which was subsequently separated via filtration. The obtained crude product undergoes purification via column chromatography on silica gel, using cyclohexane or hexane as the eluent, resulting in the isolation of a brown oil with a purity of 77% (3.07 g).
(E)-Ethyl-3-(furan -2-yl) acrylate (2); brown oil, yield = 77% (3.07 g) (E/Z-ratio:88/12)
1H NMR (500 MHz, CDCl3) δ: E (major): 7.42 (d, J = 1.9 Hz, 1H, HAr), 7.38 (d, J = 15.8 Hz, 1H, HC=), 6.55 (d, J = 3.4 Hz, 1H, HAr), 6.41 (dd, J = 3.4, 1.9 Hz, 1H, HAr), 6.27 (d, J = 15.8 Hz, 1H, =CH), 4.18 (q, J = 7.1 Hz, 2H, -CH2-), 1.26 (t, J = 7.1, Hz, 3H, -CH3).
13C NMR (126 MHz, CDCl3) δ 167.00, 150.95, 144.69, 130.95, 115.94, 114.62, 112.24, 60.40, 14.28.
1H NMR (500 MHz, CDCl3) δ: Z (minor): 7.64 (d, J = 3.6 Hz, 1H, HAr), 6.73 (d, J = 13.0 Hz, 1H), 7.47 (d, J = 1.9 Hz, 1H, HAr), 6.45 (ddd, J = 3.6, 1.9, 0.8 Hz, 1H, HAr), 5.69 (d, J = 13.0 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H).
13C NMR (126 MHz, CDCl3) δ 165.96, 150.83, 143.89, 130.35, 117.01, 114.39, 112.58, 60.11, 14.25.
Synthesis procedure for (E)-3-(furan-2-yl) acrylohydrazide (3)
In a 50 mL round bottom flask equipped with a magnetic stirrer, 1 g (1 eq, 6.00 mmol) of (E)-ethyl-3-( furan-2-yl) acrylate was dissolved in 2 mL of ethanol, followed by the addition of 1.5 mL (5 eq, 30.00 mmol) of hydrazine monohydrate (H2N-NH2.H2O). The mixture was stirred for 3 h at room temperature. TLC, indicative of reaction completion, was performed. Upon cessation of the reaction, the reaction medium was extracted using DCM. The organic layer was washed multiple times with water (H2O), then dried with MgSO4 and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel, using cyclohexane/ethyl acetate 80/20 as the eluent, yielding a beige powder (0.48 g, 52%).
(E)-3-(furan-2-yl) acrylohydrazide (3), beige powder, yield = 52% (480 mg), m.p = 72–74 °C
1H NMR (500 MHz, CDCl3) δ 7.45 (d, J = 15.5 Hz, 1H, HC=), 7.43 (d, J = 1.9 Hz, 1H, HAr), 7.19 (s, 1H, NH), 6.55 (d, J = 3.6 Hz, 1H, HAr), 6.44 (dd, J = 3.6, 1.9 Hz, 1H, HAr), 6.27 (d, J = 15.5 Hz, 1H, =CH), 4.05 (s, 2H, NH2).
13C NMR (126 MHz, CDCl3) δ 167.16, 151.23, 144.31, 128.60, 115.69, 114.33, 112.33.
General procedure for the synthesis of hydrazide-hydrazone derivatives (4a–h)
In a 10 mL Schlenk equipped with a magnetic stirrer, a mixture was prepared by combining 0.21 mmol (1 eq) of 3-(furan-2-yl) acrylohydrazide, 0.23 mmol (1.1 eq) of acetophenone or aldehyde derivatives, 1.5 mL of ethanol, and two drops of concentrated hydrochloric acid (HCl). The resulting mixture was stirred at room temperature (r.t) for 3 h. Subsequently, the reaction medium was concentrated under reduced pressure and subjected to purification via silica gel chromatography, using a DCM/MeOH mixture (95/5) as eluent. The isolated product was obtained from this process.
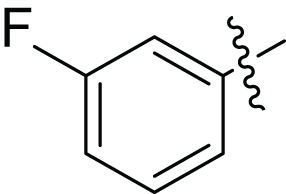
(2E, N’E)-N′-(1-(3-fluorophenyl) ethylidene)-3-(furan-2-yl) acrylohydrazide (4a), gold yellow powder, yield = 54% (23.90 mg), m.p = 172-174 °C
3-(furan-2-yl) acrylohydrazide (1 eq, 31.80 mg), 3-fluoroacetophenone (1.1 eq, 28 μL) were used in the procedure.
1H NMR (500 MHz, Methanol-d4) δ 7.74 (dt, J = 10.8, 1.9 Hz, 1H, HAr), 7.69sp, 7.65ap (dd, J = 7.9, 1.9 Hz, 1H, HAr), 7.67ap, 7.63sp (d, J = 1.9 Hz, 1H, HAr), 7.56sp, 7.55ap (d, J = 15.4 Hz, 1H, HC=), 7.40 (td, J = 7.9, 5.9 Hz, 1H, HAr), 7.13 (td, J = 7.9, 2.6 Hz, 1H, HAr), 6.78 (d, J = 15.4 Hz, 1H, =CH), 6.77ap, 6.75sp (d, J = 3.5 Hz, 1H, HAr), 6.56 (dd, J = 3.5, 1.9 Hz, 1H, HAr), 2.36sp, 2.30ap (s, 3H, -CH3).
13C NMR (126 MHz, Methanol-d4) δ 168.38ap, 164.43sp (C=O), 162.94ap, 162.90sp (d, J = 243.9 Hz, =C-F), 152.47 (d, J = 3.1 Hz, C), 151.62ap, 151.44sp (CAr), 145.01ap, 144.78sp (CH), 140.53 (d, J = 7.7 Hz, CAr), 129.87ap, 129.69sp (d, J = 8.4 Hz, CH Ar), 129.63ap, 129.04sp (CH), 122.43sp, 121.83ap (d, J = 3.0 Hz, CH Ar), 115.92sp, 115.58ap (d, J = 21.8 Hz, CH Ar), 115.94sp, 114.86ap (CH), 114.50sp, 113.84ap (CH), 113.12sp, 112.40ap (d, J = 23.4 Hz, CH Ar), 112.08 (CH), 12.90sp, 12.08ap (s, CH3).
1H NMR (400 MHz, DMSO-d6) δ 10.74ap, 10.69sp (s, 1H, NH), 7.86ap, 7.83sp (s, 1H, HAr), 7.61 (m, 2H, HAr), 7.50 (m, 1H, HAr), 7.43sp, 7.35ap (d, J = 15.7 Hz, 1H, HAr), 7.24(sp+ap) (t, J = 8.5 Hz, 1H, HAr), 6.93ap, 6.86sp (d, J = 3.5 Hz, 1H, HAr), 6.83 (d, J = 15.7 Hz, 1H, =CH), 6.62(sp+ap) (d, J = 2.0 Hz, 1H), 2.31sp, 2.29ap (s, 3H, N=C(CH3)).
FT-IR (ATR) cm−1: 3,186.40 (NH), 2,978.09 (N=CH), 2,885.51 (CH), 1,651.07 (C=O), 1,620.21 (C=N), 1,325.10 (C-O-C), 1,014.56 (N-N), 966.34 (HC=CH).
(2E, N’E)-N′-(1-(4-fluorophenyl) ethylidene)-3-(furan-2-yl) acrylohydrazide (4b), gold yellow powder, yield = 41% (21.70 mg), m.p = 178–180 °C
3-(furan-2-yl) acrylohydrazide (1 eq, 30.00 mg), 4-fluoroacetophenone (1.1 eq, 26 μL) were used in the procedure.
1H NMR (500 MHz, Methanol-d4) δ 7.95sp, 7.86ap (dd, J = 9.0, 5.4 Hz, 2H, HAr), 7.65ap, 7.62sp (d, J = 2.1 Hz, 1H, HAr), 7.54sp, 7.49ap (d, J = 15.9 Hz, 1H, =CH), 7.15ap, 7.11sp (t, J = 9.0 Hz, 2H, HAr), 6.76sp (d, J = 15.9 Hz, 1H, HC=), 6.76ap, 6.74sp (d, J = 3.3 Hz, HAr), 6.55 (dd, J = 3.3, 2.1 Hz, 1H, HAr), 2.36sp, 2.30ap (s, 3H, -CH3).
13C NMR (126 MHz, Methanol-d4) δ 169.73ap, 165.76sp (C=O), 165.44ap, 165.18sp (d, J = 248.7 Hz, =C-F), 154.48sp, 153.05ap ( C Ar), 152.85ap, 149.38sp (C Ar), 146.31ap 146.13sp (CH Ar), 135.94ap, 135.77sp (d, J = 3.4 Hz, C Ar), 130.89ap, 130.27sp (HC=), 130.16sp, 129.42ap (d, J = 8.4 Hz, 2CH Ar), 117.42ap, 116.16ap (=CH), 116.19sp, 115.38ap (CH Ar), 116.16ap, 113.46sp (CH Ar), 115.90 (d, J = 28.1 Hz, 2 CH Ar), 14.36sp, 13.51ap (CH3).
FT-IR (ATR) cm−1: 3,161.33 (NH), 3,020 (N=CH), 2,960 (CH), 1,658.78 (C=O), 1,625 (C=N), 1,384.99 (C-O-C), 1,016.49 (N-N), 972.12 (HC=CH)
HRMS (ESI): Calc. C15H14N2O2F [M+H] = 273.1039 found = 273.1033.
Calc. C15H13N2O2F [M−H] = 271.0883 found = 271.0895.
1H NMR (500 MHz, DMSO-d6) δ 10.67ap, 10.63sp (s, 1H, NH), 7.85 (m, 3H), 7.50ap, 7.42sp (d, J = 15.8 Hz, =CH), 7.34ap, 6.83sp (d, J = 15.8 Hz, HC=), 7.27 (m, 2H), 6.93ap, 6.85sp (d, J = 3.6 Hz, 1H, HAr), 6.64ap, 6.62sp (dd, J = 3.6, 1.7 Hz, 1H), 2.31sp, 2.28ap (s, 3H, -CH3).
13C NMR (126 MHz, DMSO-d6) δ 165.25sp, 162.73ap (d, J = 363.8 Hz, =C-F), 161.88sp, 161.85ap (C=O), 151.16 (C Ar), 150.55sp, 147.02ap (C Ar), 145.53ap, 145.17sp (CH Ar), 134.78 (C Ar), 129.06ap, 127.49sp (HC=), 128.58sp, 128.21ap (d, J = 8.6 Hz, 2 CH Ar), 117.92ap, 117.87sp (=CH), 115.44ap, 115.24sp (d, J = 21.3 Hz, 2CH Ar), 114.66sp, 114.46ap (CH Ar), 112.70ap, 112.62sp (CH Ar), 14.16sp, 13.87ap (CH3).
(2E, N’E)′-3-(furan-2-yl)-N’-(thiophen-2-ylmethylene) acrylohydrazide (4c), Brown solid, yield = 23% (11.47 mg), m.p = 216–218 °C
3-(furan-2-yl) acrylohydrazide (1 eq, 31.10 mg), thiophene-2-carbaldehyde (1.1 eq, 21 μL) were used in the procedure.
1H NMR (500 MHz, DMSO-d6) δ 11.60ap, 11.47sp (s, 1H, NH), 8.45ap, 8.23sp (s, 1H, N=CH), 7.87sp, 7.82ap (d, J = 1.9 Hz, 1H, HAr), 7.66ap, 7.63sp (d, J = 5.0 Hz, 1H, HAr), 7.47ap, 7.41sp (m, 1H), 7.43sp, 7.41ap (d, J = 15.7 Hz, 1H, =CH), 7.21sp, 6.46ap (d, J = 15.7 Hz, 1H, HC=), 7.13(sp + ap) (m, 1H, HAr), 6.91sp, 6.86ap (d, J = 3.5 Hz, 1H, HAr), 6.64sp, 6.62ap (dd, J = 3.5, 1.9 Hz, 1H, HAr).
13C NMR (126 MHz, DMSO-d6) δ 165.49sp, 161.18ap (C=O), 151.02sp, 150.85ap (C-O), 145.49sp, 145.19ap (CH Ar), 141.79sp, 138.17ap (N=CH), 139.10ap, 138.92sp (C -S), 130.87ap, 130.19sp (CH Ar), 128.89ap, 128.21sp (CHAr), 128.85sp, 127.59ap (HC=), 127.90sp, 127.82ap (CH Ar), 117.21ap, 113.79sp (=CH), 115.39sp, 114.74ap (CH Ar), 112.60sp, 112.55ap (CH Ar).
FT-IR (ATR) cm−1: 3,200 (NH), 2,989 (N=CH), 1,670 (C=O), 1,610 (C=N), 1,328.95 (C-O-C), 1,016.49 (N-N), 961 (HC=CH).
(2E, N’E)′-N’-((1H pyrrol-2-yl) methylene)-3-(furan-2-yl) acrylohydrazide (4d), green powder, yield = 48% (36.00 mg), m.p = 236–238 °C
3-(furan-2-yl) acrylohydrazide (1 eq, 50.00 mg), pyrrole-2-carbaldehyde (1.1 eq, 34.39 mg) were used in the procedure.
1H NMR (500 MHz, DMSO-d6) δ 11.50ap, 11.44sp (s, 1H, NH pyrrole), 11.32ap, 11.16sp (s, 1H, NH), 8.07ap, 7.88sp (s, 1H, N=CH), 7.84sp, 7.80ap (d, J = 1.9 Hz, 1H, HAr), 7.42 (d, J = 1.9 Hz, 1H, HAr) 7.37 (d, J = 15.5 Hz, 1H, =CH), 6.93sp, 6.90ap (m 1H), 6.90ap, 6.82sp (d, J = 3.5 Hz, 1H, HAr), 6.64sp, 6.61ap (dd, J = 3.5, 1.9 Hz, 1H, HAr), 6.47 (d, J = 15.5 Hz, 1H, HC=), 6.42sp, 6.13ap (m, 1H).
13C NMR (126 MHz, DMSO-d6) δ 165.36sp, 160.75ap (C=O), 151.40sp, 150.99ap (C-O), 145.11sp, 144.99ap (CH Ar), 139.72ap, 136.09sp (N=CH), 128.35ap, 115.11sp (CH Ar), 127.11sp, 127.02ap (C-N, C Ar), 126.94ap, 127.03sp (=CH), 122.52ap, 121.86sp (CH Ar), 117.74, 113.30 (HC=), 114.56sp, 114.26ap (CH Ar), 112.58, 112.50sp, 112.48ap (CH Ar), 109.25ap, 109.14sp (CH Ar).
FT-IR (ATR) cm−1: 3,415.93 (NH), 3,211.48 (NH), 2,987.74 (N=CH), 2,889.37 (CH), 1,656.85 (C=O), 1,600.92 (C=N), 1,328.95 (C-O-C), 1,016.49 (N-N), 968.27 (HC=CH).
(2E, N’E)′-3-(furan-2-yl)-N’-(furan-2-ylmethylene) acrylohydrazide (4e), beige powder, yield = 56% (44.00 mg), m.p = 224–226 °C
3-(furan-2-yl) acrylohydrazide (1 eq, 50.00 mg), furane-2-carbaldehyde (1.1 eq, 30 μL) were used in the procedure.
1H NMR (500 MHz, DMSO-d6) δ 11.59ap, 11.44sp (s, 1H, NH), 8.13ap, 7.94sp (s, 1H, N=CH), 7.84(ap + sp) (td, J = 8.35, 2.1 Hz, 2H, HAr), 7.45sp, 7.43ap (d, J = 15.8 Hz, 1H, =CH), 7.24sp, 6.45ap (d, J = 15.8 Hz, 1H, HC=), 6.92sp, 6.62ap (m, 3H) 6.89sp, 6.86ap (d, J = 3.5 Hz, 1H, HAr).
13C NMR (126 MHz, DMSO-d6) δ 165.68sp, 161.27ap (C=O), 151.05sp, 150.85ap (C-O), 149.41ap, 149.16sp (C-O), 145.44ap, 144.90sp (CH Ar), 145.21ap, 145.14sp (CH Ar), 136.41ap, 133.14sp (N=CH), 128.80sp, 127.65ap (HC=), 117.17ap, 113.95sp (=CH), 115.32sp, 114.74ap (CH Ar), 113.51ap, 113.36ap (CH Ar), 112.60sp, 112.55ap (CH Ar), 112.17ap, 112.05sp (CH Ar).
FT-IR (ATR) cm−1: 3,126.61 (NH), 2,989.66 (N=CH), 2,887.44 (CH), 1,651.07 (C=O), 1,606.70 (C=N), 1,338.60 (C-O-C), 1,014.56 (N-N), 968.27 (HC=CH).
(2E, N’E)′-3-(furan-2-yl)-N’-(pyridin-2-ylmethylene) acrylohydrazide (4f), yellow powder, yield = 96% (47.17 mg), m.p = 104–106 °C
3-(furan-2-yl) acrylohydrazide (1 eq, 31.00 mg), pyridine-2-carbaldehyde (1.1 eq, 21 μL) were used in the procedure.
1H NMR (500 MHz, Methanol-d4) δ 8.56 (m, 1H), 8.27ap, 8.05sp (d, J = 7.8 Hz, 1H, HAr), 8.19ap, 8.05sp (s, 1H, N=CH), 7.90sp, 7.88ap (t, J = 7.8 Hz, 1H, HAr), 7.67sp, 7.63ap (d, J = 1.8 Hz, 1H, HAr), 7.58ap, 7.52sp (d, J = 15.4 Hz, 1H, HC=), 7.42 (dd, J = 7.4, 4.8 Hz, 1H, HAr), 6.81sp, 6.77ap (d, J = 3.3 Hz, 1H, HAr), 6.56ap, 6.39sp (dd, J = 3.3, 1.8 Hz, 1H, HAr), 6.52 (d, J = 15.4 Hz, 1H, =CH).
13C NMR (126 MHz, Methanol-d4) δ 169.37sp, 165.46ap (C=O), 154.47ap, 154.39sp (C-N), 153.02sp, 152.59ap (C-O), 150.26sp, 150.15ap (CH), 148.29ap, 143.96sp (CH), 146.39ap, 144.62sp (N=CH), 138.89sp, 138.60ap (CH), 131.32sp, 130.93ap (CH), 125.97ap, 125.71sp (CH), 122.30ap, 121.92sp (CH), 116.68sp, 116.31ap (CH), 114.77sp, 113.53ap (CH).
HRMS (ESI): Calc. C13H12N3O2 [M+H] = 242.0929 found = 242.0924.
Calc. C13H10N3O2 [M−H] =240.0773 found = 240.0781.
1H NMR (400 MHz, DMSO-d6) δ 11.89ap, 11.72sp (s, 1H, NH), 8.61(sp + ap) (d, J = 4.9 Hz, 1H, HAr), 8.23ap, 8.10sp (s, 1H, N=CH), 7.96sp, 7.94ap (d, J = 3.5 Hz, 1H, HAr), 7.89 (sp + ap) (td, J = 7.5, 1.9 Hz, 1H, HAr), 7.88ap, 7.84sp (d, J = 1.9 Hz, 1H, HAr), 7.51sp, 7.47ap (d, J = 15.5 Hz, =CH), 7.42(sp + ap) (dd, J = 7.5, 4.9 Hz, 1H, HAr), 7.31sp, 6.50ap (d, J = 15.5 Hz, 1H, HC=), 6.98sp, 6.89ap (d, J = 3.5 Hz, 1H, HAr), 6.65sp, 6.64ap (dd, J = 3.5, 1.9 Hz, 1H, HAr).
13C NMR (101 MHz, DMSO-d6) δ 166.01sp, 161.55ap (C=O), 153.17ap, 152.97sp (C-N), 151.05sp, 150.79ap (C-O), 149.51ap, 146.82sp (CHAr), 145.57ap, 145.40sp (CH), 143.68sp, 136.91ap (CH), 129.30sp, 128.19ap (CH), 124.35ap, 124.19sp (CH), 119.93ap, 119.61sp (CH), 116.90ap, 115.51sp (CH), 115.13ap, 113.73sp (CH), 112.68ap, 112.65sp (CH).
FT-IR (ATR) cm−1: 3,180.62 (NH), 2,990 (N=CH), 2,866.22 (CH), 1,660.71 (C=O), 1,624.09 (C=N), 1,556.55 (C=N), 1,323.17 (C-O-C), 1,014.56 (N-N), 968.27 (HC=CH).
(2E, N’E)-N′-(1-(4chloro-3-nitrobenzylidene)-3-(furan-2-yl) acrylohydrazide (4g), yellow powder, yield = 70% (44.80 mg), m.p = 98–100 °C
3-(furan-2-yl) acrylohydrazide (1 eq, 30.60 mg), 4-chloro-3-nitrobenzaldehyde (1.1 eq, 41.00 mg) were used in the procedure.
1H NMR (500 MHz, Methanol-d4) δ 8.43ap, 8.23sp (d, J = 2.0 Hz, 1H, HAr), 8.17ap, 8.03sp (s, 1H, N=CH), 8.03ap, 8.00sp (dd, J = 8.4, 2.0 Hz, 1H, HAr), 7.74sp, 7.71ap (d, J = 8.4 Hz, 1H, HAr), 7.69sp, 7.65ap (d, J = 2.0 Hz, 1H, HAr), 7.57ap, 7.53sp (d, J = 15.5 Hz, =CH), 6.82sp, 6.78ap (d, J = 3.5 Hz, 1H, HAr), 6.59sp, 6.58ap (dd, J = 3.5, 2.0 Hz, 1 H, HAr), 6.50 (d, J = 15.5 Hz, HC=).
13C NMR (126 MHz, Methanol-d4) δ 169.20sp, 165.40ap (C=O), 152.93sp, 152.56ap (C-O), 149.99ap (C-NO2), 146.45sp, 146.39ap (CH), 145.60ap, 142.08sp (CH), 136.28 (C Ar), 133.35sp, 133.19ap (CH), 132.71ap, 131.87sp (CH), 131.35sp, 130.90ap (CH), 128.26ap, 127.99sp (C-Cl), 124.86ap, 124.52sp (CH), 116.59ap, 116.44sp (CH), 116.34ap, 114.55sp (CH), 113.53 (CH).
HRMS (ESI) Calc. C14H11ClN3O4 [M+H] = 320.0438 found = 320.0427
Calc. C14H9ClN3O4 [M−H] = 318.0281 found = 318.0284
1H NMR (500 MHz, DMSO-d6) δ 11.93ap, 11.75sp (s, 1H, NH), 8.38ap, 8.37sp (d, J = 2.1 Hz, HAr), 8.29ap, 8.11sp (s, 1H, N=CH), 8.03(ap + sp) (m, 1H), 7.88sp, 7.83ap (s, 1H, HAr), 7.86ap, 7.84sp (s, 1H, HAr), 7.50sp, 7.46ap (d, J = 15.5 Hz, 1H, =CH), 7.31sp, 6.51ap (d, J = 15.5 Hz, HC=), 6.97sp, 6.89ap (d, J = 3.5 Hz, 1H, HAr), 6.65sp, 6.64ap (dd, J = 3.5, 2.1 Hz, HAr).
13C NMR (126 MHz, DMSO-d6) δ 166.09sp, 161.63ap (C=O), 151.03sp, 150.80ap (C-O), 148.05sp, 147.86ap (C-NO2), 145.61sp, 145.41ap (CH Ar), 143.16ap, 140.00sp (N=CH), 135.04ap, 134.82sp (C Ar), 132.15ap, 131.46sp (CH Ar), 130.98 (CH Ar), 129.44sp, 128.24ap (HC=), 125.66ap, 125.29sp (C-Cl), 123.58ap, 123.37sp (CH Ar), 116.85ap, 113.63sp (=CH), 115.56sp, 115.17ap (CH Ar), 112.74sp, 112.65ap (CH Ar).
FT-IR (ATR) cm−1: 3,429.43 (NH), 3,000 (N=CH), 2,889.37 (CH), 1,658.78 (C=O), 1,606.70 (C=N), 1,531.48 (NO2), 1,328.95 (C-O-C), 1,016.49 (N-N), 968.27 (HC=CH).
(2E, N’E)-N′-(3-(benzyloxy) benzylidene)-3-(furan-2-yl) acrylohydrazide (4h), yellow powder, yield = 40% (28.10 mg), m.p = 116–118 °C
3-(furan-2-yl) acrylohydrazide (1 eq, 30.06 mg), 3-benzyloxybenzaldehyde (1.1 eq, 47.00 mg) were used in the procedure.
1H NMR (500 MHz, CDCl3) δ 9.33 (s, NH), 7.78 (s, 1 H, N=CH), 7.62 (d, J = 15.8 Hz, 1H, =CH), 7.51 (d, J = 15.8 Hz, 1H, HC=), 7.46 (d, J = 7.3 Hz, 2H, HAr), 7.40 (t, J = 7.3 Hz, 4H, HAr), 7.34 (t, J = 7.7 Hz, 2H, HAr), 7.28 (d, J = 1.4 Hz, 1H, HAr), 7.03 (dd, J = 7.5, 1.9 Hz, 1H, HAr), 6.67 (d, J = 3.5 Hz, 1H, HAr), 6.50ap, 6.47sp (dd, J = 3.5, 1.9 Hz, 1H, HAr), 5.15 (s, 2H, -CH2-).
13C NMR (126 MHz, CDCl3) δ 167.88 (C=O), 159.21 (C-O), 151.93 (C-O), 144.71 (CH Ar), 143.77 (N=CH), 136.93 (C Ar), 135.42 (C Ar), 130.28 (HC=), 129.94 (=CH), 128.81 (2C, CH Ar), 128.26 (CH Ar), 127.77 (2C, CH Ar), 120.75 (CH Ar), 117.11 (CH Ar), 115.00 (CH Ar), 114.35 (CH Ar), 112.87 (CH Ar), 112.47 (CH Ar), 70.30 (CH2).
HRMS (ESI) Calc. C21H19N2O [M+H] = 347, 13,957 found = 347, 13,864
Calc. C21H17N2O [M−H] = 345.12391 found = 345.12466
1H NMR (400 MHz, DMSO-d6) δ 11.70ap, 11.53sp (s, 1H, NH), 8.19ap, 8.01sp (s, 1H, N=CH), 7.85 (d, J = 15.4 Hz, 1H, =CH), 7.48 (m, 3H), 7.37 (m, 6H), 7.27ap, 7.08sp (d, J = 7.7 Hz, 1H, HAr), 6.95sp, 6.85ap (d, J = 3.5 Hz, 1H, HAr), 6.65sp, 6.63ap (dd, J = 3.5, 1.9, 1H, HAr), 6.50 (d, J = 15.4 Hz, 1H, HC=), 5.18sp, 5.15ap (s, 2H, -CH2-).
13C NMR (101 MHz, DMSO-d6) δ 165.84sp, 161.34ap (C=O), 158.64ap, 158.61sp (C-O), 151.10sp, 150.87ap (C-O), 146.45ap, 145.48sp (CH Ar), 145.24ap, 142.98sp (N=CH), 136.96sp, 136.90ap (CAr), 135.77ap, 135.57sp (CAr), 130.02sp, 129.93ap (CH), 128.97 (CH), 128.45sp, 128.42ap (CH Ar, 2C), 127.85 (CH), 127.73sp, 127.68ap (CH Ar, 2C), 120.02ap, 119.50sp (CH), 117.27ap, 116.81sp (CH), 116.48sp, 115.27ap (CH), 114.81ap, 114.06sp (CH), 112.64sp, 112.59ap (CH), 112.47 (CH), 69.27 (CH2).
FT-IR (ATR) cm−1: 3,200 (NH), 2,947.23 (N=CH), 2,931.80 (CH), 1,660.71 (C=O), 1,620.21 (C=N), 1,328.49 (C-O-C), 1,016.49 (N-N), 972.12 (HC=CH).
Results and discussion
Chemical synthesis
The N-acylhydrazone derivatives (4a–h) were obtained through a three-step synthetic pathway starting from furan-2-carbaldehyde (1). The initial step involved the addition of ethyl chloroacetate to aldehyde (1) in the presence of triphenylphosphine (PPh3) under basic conditions, employing the Wittig reaction (El-Batta et al., 2007). Consequently, the ester (2) was obtained with a yield of 77%. Subsequently, the ester (2) reacted with hydrazine monohydrate (H2N-NH2.H2O) in ethanol at room temperature to yield the corresponding hydrazide (3) with a yield of 52%. Finally, a series of acrylohydrazide derivatives (4a–h) was synthesized with yields ranging from 23% to 96% (Table 1). This was achieved by condensing hydrazide (3) with various derivatives of benzaldehyde and acetophenone in ethanol at room temperature for 3 h, in the presence of a catalytic amount of concentrated hydrochloric acid (HCl) (Scheme 1).
| Compounds | Structure | Ar | Yield (%) |
|---|---|---|---|
| 4a | 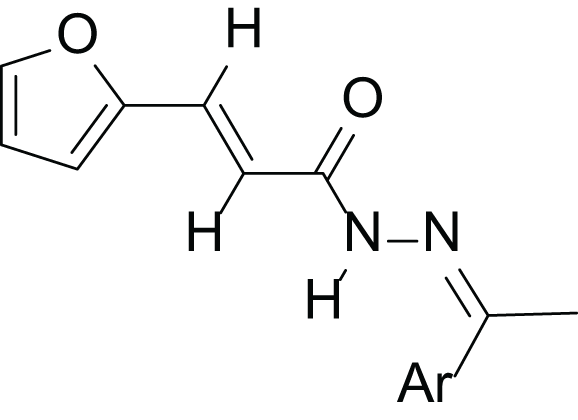 |
 |
54 |
| 4b | 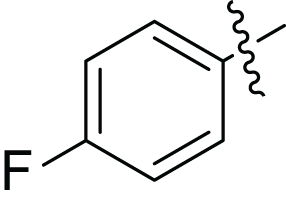 |
41 | |
| 4c | 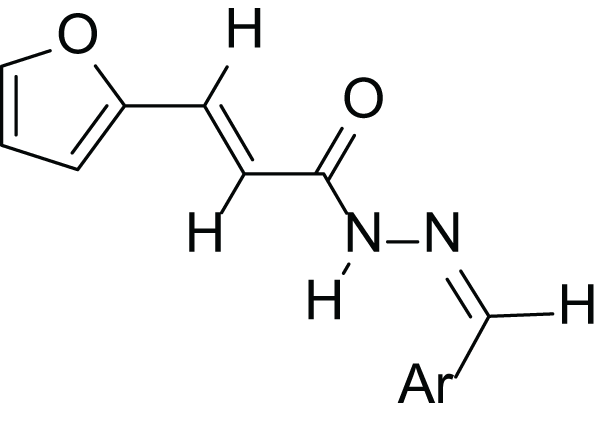 |
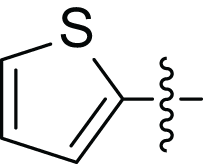 |
23 |
| 4d | 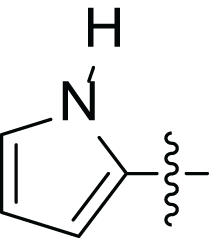 |
48 | |
| 4e | 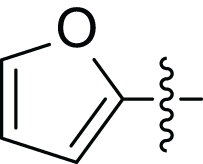 |
56 | |
| 4f | 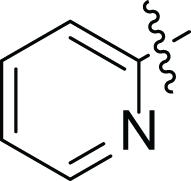 |
96 | |
| 4g | 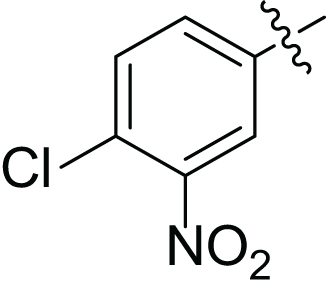 |
70 | |
| 4h | 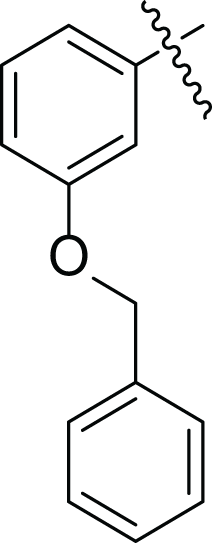 |
40 |
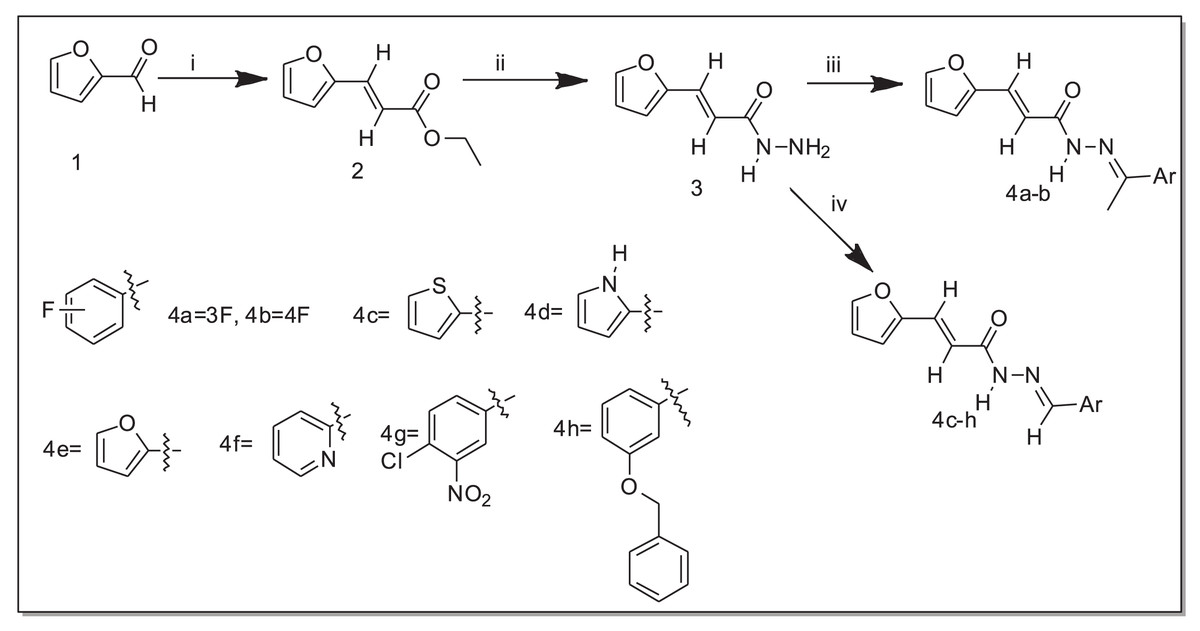
Scheme 1: Synthesis of acrylohydrazide derivatives.
i. PPh3, ethyl chloroacetate, NaHCO3, ii. H2NNH2, EtOH, iii. EtOH, HCl, acetophenone derivatives, iv. EtOH, HCl, aldehydes.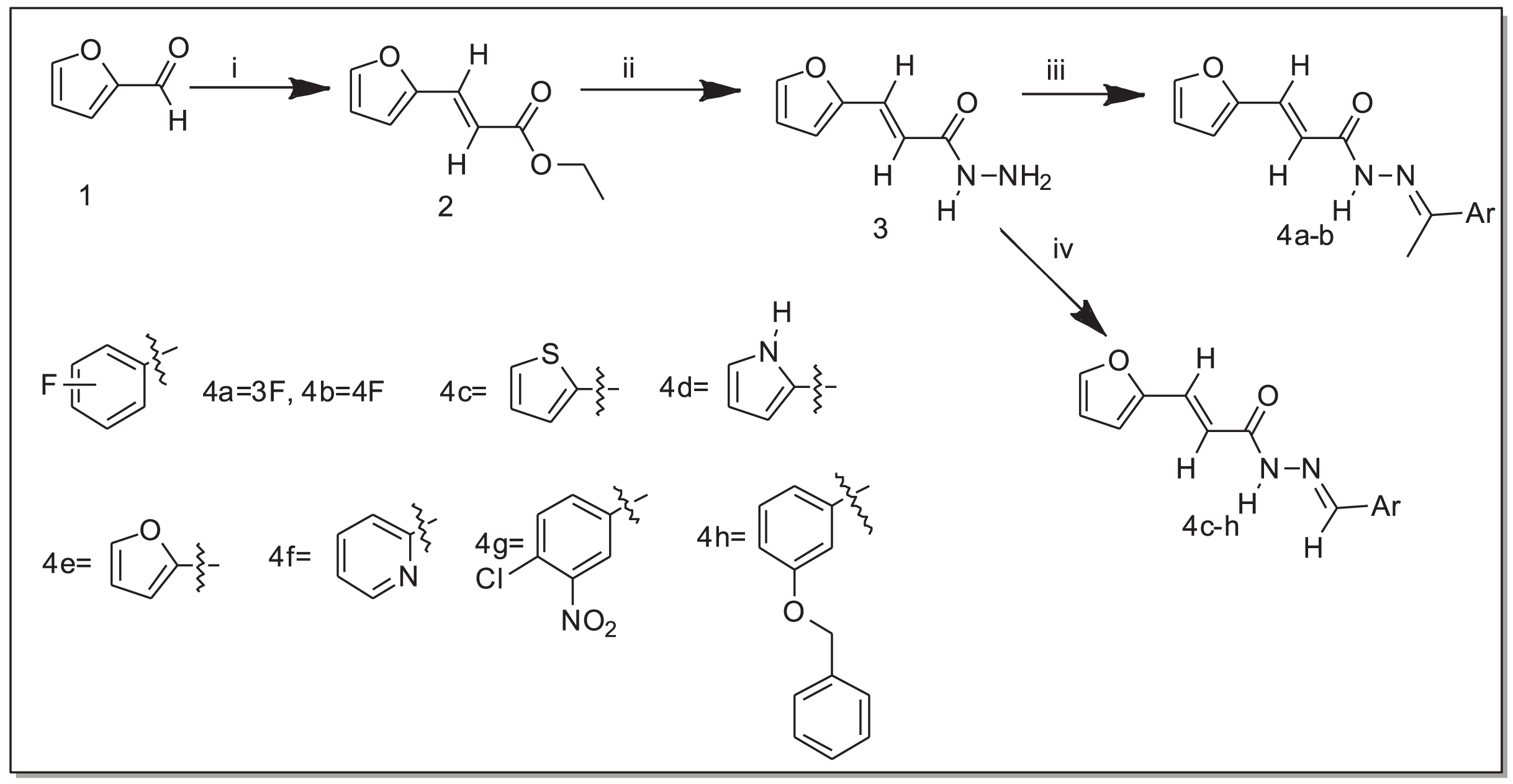{kind=link}
The newly synthesized compounds (4a–h) were fully characterized using various analytical methods, notably NMR, which revealed the presence of a dynamic equilibrium between two isomers for each compound in the N-acrylohydrazone series (4a–h). The results of this stereochemical study will be discussed in the following section.
Spectral analysis
The examination of the 1H NMR spectrum for compound (2) reveals the presence of two distinct E and Z isomers, with an approximate ratio of 88/12 (E/Z). Each isomer exhibits characteristic doublet signals. In the predominant E isomer, a doublet is observed at 6.27 ppm, corresponding to the alpha (α) proton adjacent to the carbonyl CO, while another doublet is detected at 7.38 ppm, indicative of the beta (β) proton. Conversely, in the minor Z isomer, the alpha (α) proton resonates at 5.69 ppm, while the beta (β) proton is discerned at 7.73 ppm.
The pronounced mesomeric attractive effect between the alkene and the carbonyl bonds results in notable deshielding of the beta (β) proton in the vicinity of the carbonyl. Furthermore, a quadruplet, integrating for two protons around 4.18 ppm, and a triplet, integrating for three protons at 1.26 ppm, are evident, corroborating the presence of the methyl group of the ester function. In the 13C NMR spectrum, discernible peaks at 167 ppm (E isomer) and 165.96 ppm (Z isomer) confirm the presence of the carbonyl carbon in both forms.
The scrutiny of the 1H NMR spectrum for compound (3) reveals the disappearance of signals attributed to the ethoxy group (OCH2CH3) and the emergence of broad singlets at 4.05 and 7.19 ppm. These singlets correspond to the NH protons of the NH2 and C(O)-NH groups, respectively. Even if compound (2) was initially utilized, consisting of a mixture of E and Z isomers in an 88/12 ratio, only the E isomer was successfully isolated for compound (3).
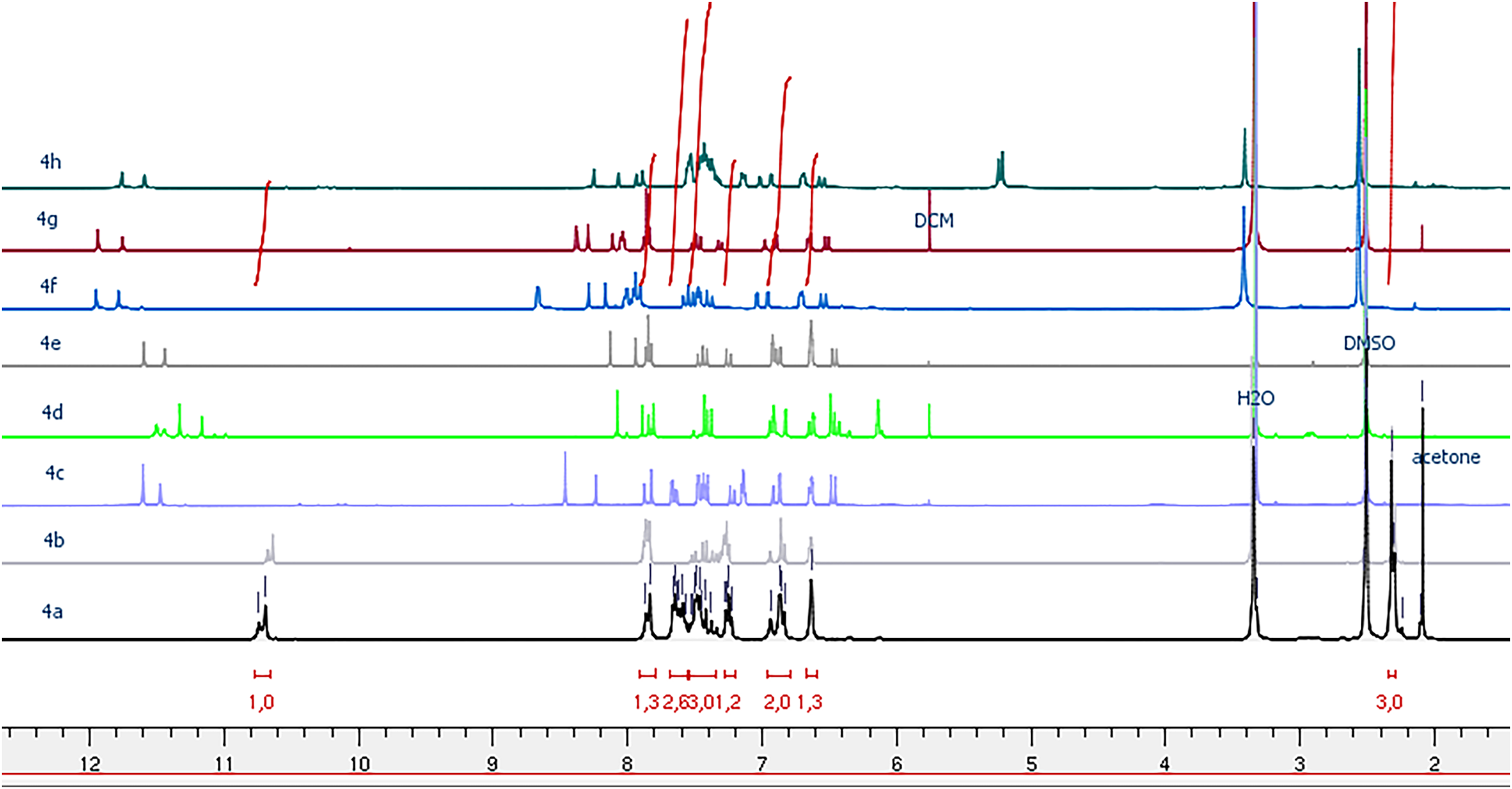
The 1H NMR spectra of the ultimate compounds (4a–h), acquired in DMSO-d6, exhibit a marked duplication of signals pertaining to three neighboring atom groups within the amide fragment: NH, N=CH or N=C(CH3), and CH=CH(CO) (Figs. 1–4). Table 2 provides the chemical shift data for the duplicated proton signals of the amide function, as well as the azomethine (N=CH) and azoethylidine (N=C(CH3)) fragments. Additionally, a systematic duplication of signals is discernible in the 13C NMR spectra for all compounds (4a–h). Table 3 compiles the chemical shift values for the duplicated carbons of the amide carbonyl function, and the N=CH and N=C(CH3) fragments.
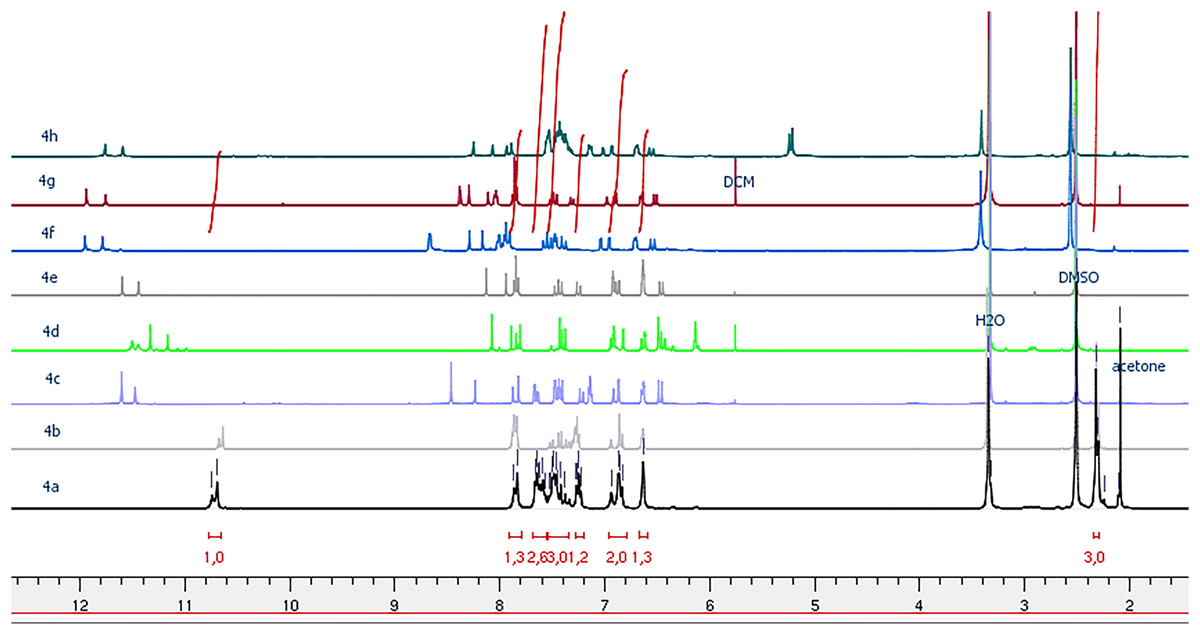
Figure 1: 1H NMR spectra of compounds 4a–h.
{kind=link}
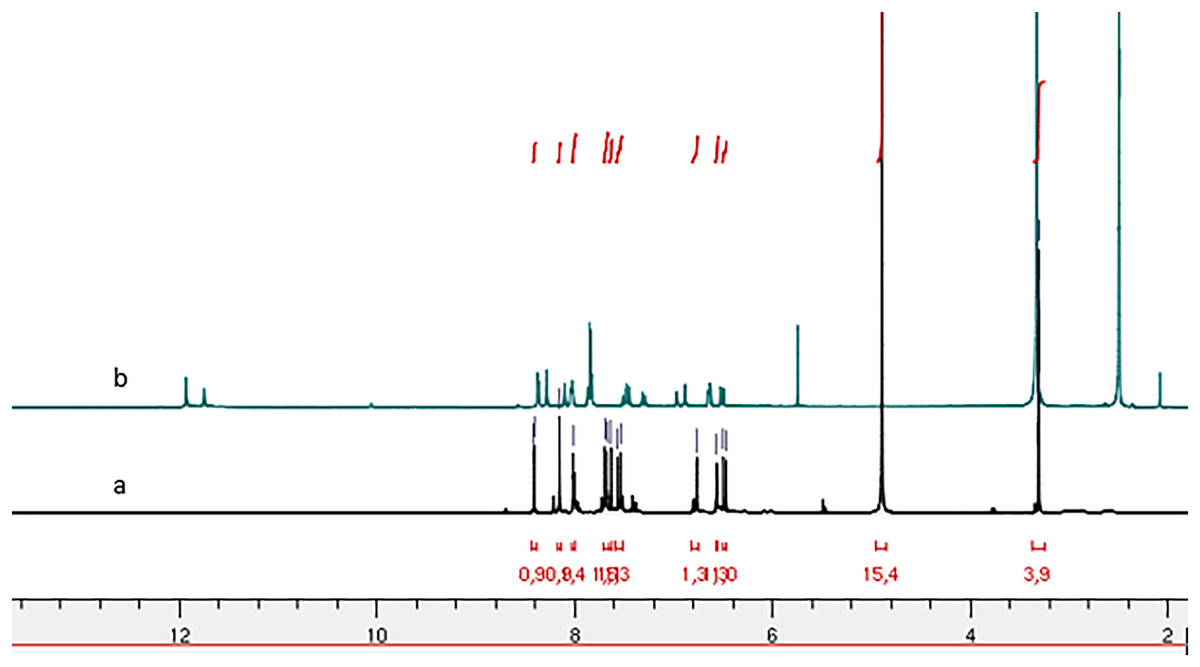
Figure 2: 1H NMR spectrum of compound 4g: (A) in Methanol-d4 and (B) in DMSO-d6.
{kind=link}
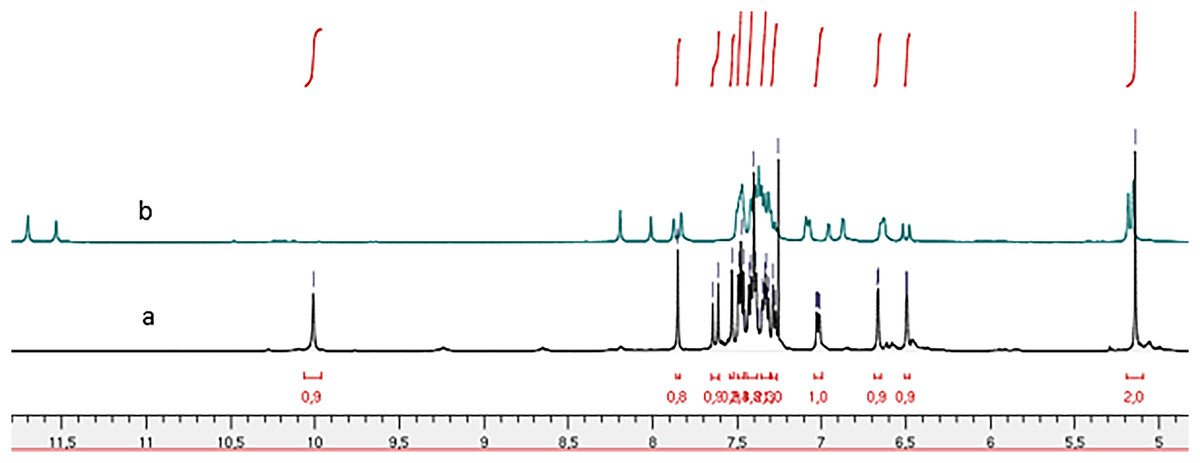
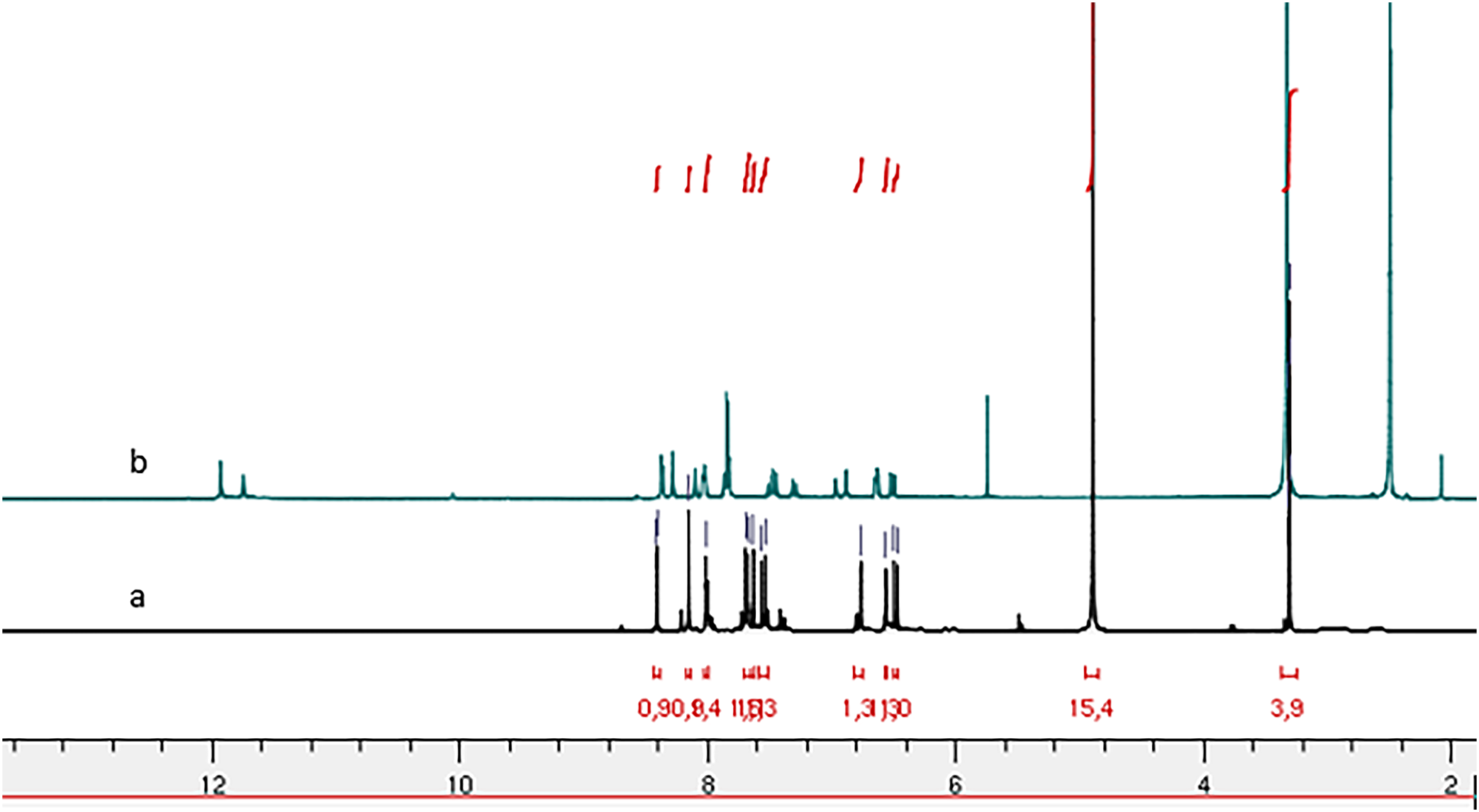
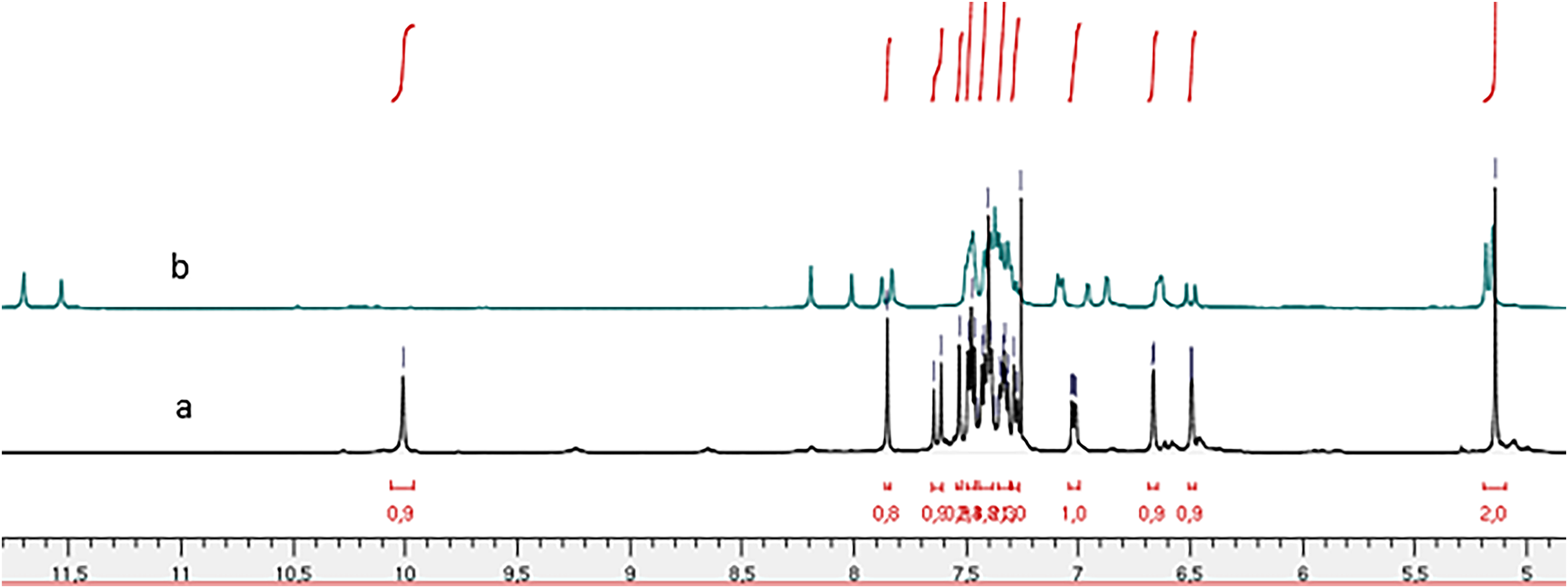
Figure 3: 1H NMR spectrum of compound 4h: (A) in CDCl3 and (B) in DMSO-d6.
{kind=link}
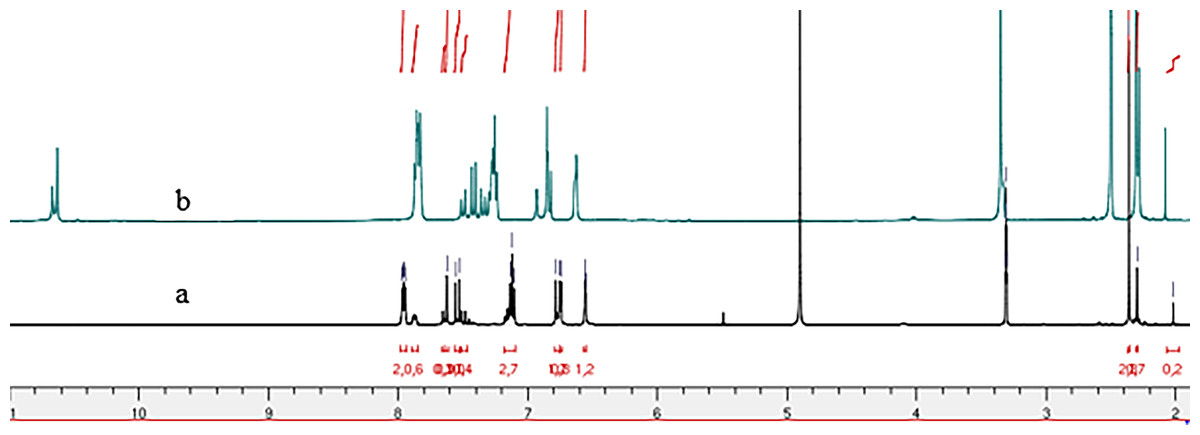
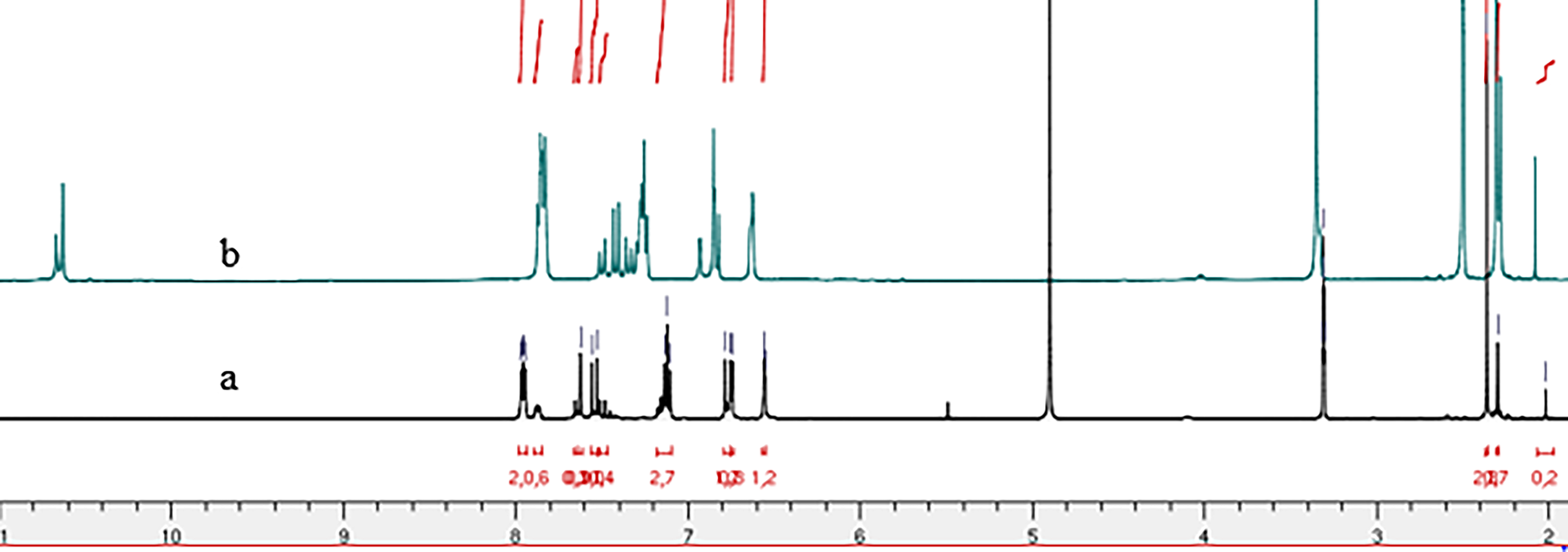
Figure 4: 1H NMR spectrum of compound 4b: (A) in methanol-d3 et (B) in DMSO-d6.
{kind=link}
| Chemical shift δ (ppm) | |||||||
|---|---|---|---|---|---|---|---|
| C(O)-NH | N=C(CH3) or N=CH | Ar | Ratio (%) | ||||
| Compounds | Anti | Syn | Anti | Syn | Anti | Syn | |
| 4a | 10.74 | 10.69 | 2.29 | 2.31 | 3-F-C6H4 | 38 | 62 |
| 4b | 10.67 | 10.63 | 2.28 | 2.30 | 4-F- C6H4 | 38 | 62 |
| 4c | 11.60 | 11.47 | 8.46 | 8.23 | C4H3S | 62 | 38 |
| 4d | 11.33 | 11.16 | 8.07 | 7.88 | C4H4N | 62 | 38 |
| 4e | 11.59 | 11.43 | 8.12 | 7.93 | C4H3O | 53 | 47 |
| 4f | 11.89 | 11.72 | 8.22 | 8.10 | C5H4N | 53 | 47 |
| 4g | 11.93 | 11.74 | 8.29 | 8.10 | 3-Cl-4-NO2- C6H3 | 56 | 44 |
| 4h | 11.70 | 11.53 | 8.19 | 8.01 | 3-O-CH2-C6H5 | 56 | 44 |
| Compounds | Ratio (%) relative to C(O)-NH | Proportion | |
|---|---|---|---|
| Anti | Syn | ||
| 4a | 38 | 62 | 1:1.6 |
| 4b | 38 | 62 | 1:1.6 |
| 4c | 62 | 38 | 1.6:1 |
| 4d | 62 | 38 | 1.6:1 |
| 4e | 53 | 47 | 1.1:1 |
| 4f | 53 | 47 | 1.1:1 |
| 4g | 56 | 44 | 1.3:1 |
| 4h | 56 | 44 | 1.3:1 |
Comparison of the 1H NMR spectra (Fig. 3) for compound (4h) recorded in deuterated chloroform (CDCl3) and deuterated dimethyl sulfoxide (DMSO-d6) reveals the presence of two conformers characterized by the splitting of the NH function signal, one at 11.70 ppm and the other at 11.53 ppm in the DMSO-d6 spectrum. In contrast, the CDCl3 spectrum indicates a singular conformer with the NH function signal appearing at 9.33 ppm. Notably, the chemical shift of NH is reduced in the CDCl3 spectrum compared to the DMSO-d6 spectrum. This observation aligns with the findings reported by Palla et al. (1986), suggesting that the marginal chemical shifts observed in the CDCl3 spectra may be attributed to intramolecular hydrogen bonding.
Comparative analysis of the 1H NMR spectra for compounds 4b and 4g, recorded in deuterated methanol (Methanol-d4) and DMSO-d6 (Figs. 2 and 4) indicates signal duplication for various protons. However, the NH signal is absent in the spectrum recorded in Methanol-d4, and the duplicated signals exhibit lower intensity compared to the 1H NMR spectrum recorded in DMSO-d6. The duplicated NH signals manifest at 11.75 ppm and 11.93 ppm for compound (4g) and 10.63 ppm and 10.67 ppm for compound (4b). The absence of the NH signal in the spectra recorded in Methanol-d4 may be ascribed to proton exchange with deuterium from the solvent.
Stereochemical analysis of Acrylohydrazide derivatives (4a–h)
The duplication of characteristic signals for NH and N=CH groups observed in both 1H and 13C NMR spectra (Tables 2 and 4) of the acrylohydrazides (compounds 4a–h) indicates the presence of two distinct isomers. Considering the structure of acrylohydrazide, this signal duplication could be attributed to the geometric stereoisomerism (Z or E) of the azomethine (N=CH) or azoethylidine (N=C(CH3)) fragment, or the ethylenic (CH=CH) group, or to a slow rotation around the N-N or N-C=O bond (Scheme 2).
| Chemical shift δ ppm (13C) | |||||
|---|---|---|---|---|---|
| C(O)-NH | N=C(CH3) or N=CH | Ar | |||
| Compounds | Anti | Syn | Anti | Syn | |
| 4a | 164.43 | 168.38 | 13.62 | 14.45 | 3-F-C6H4 |
| 4b | 165.77 | 169.74 | 13.66 | 14.51 | 4-F- C6H4 |
| 4c | 161.18 | 165.49 | 141.79 | 138.92 | C4H3S |
| 4d | 160.75 | 165.36 | 139.72 | 136.09 | C4H4N |
| 4e | 161.27 | 165.68 | 136.41 | 133.14 | C4H3O |
| 4f | 165.42 | 169.30 | – | – | C5H4N |
| 4g | 161.63 | 166.09 | 143.16 | 140.00 | 3-Cl-4-NO2- C6H3 |
| 4h | 161.39 | 165.84 | – | – | 3-O-CH2-C6H5 |
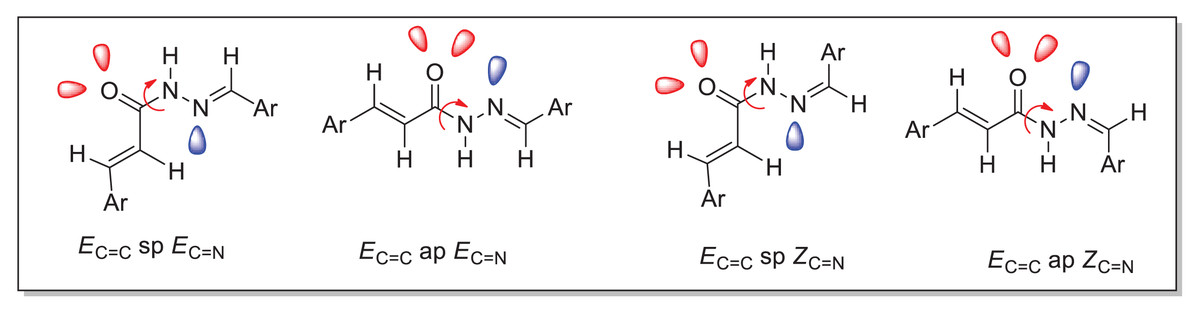
Scheme 2: The various possible conformers of the acrylohydrazides derivatives.
{kind=link}
Concerning the geometric stereoisomerism of the ethylenic fragment in acrylohydrazides, they all exhibit the Ec=c configuration confirmed by the coupling constant (J = 15.40–15.90 Hz) of the ethylenic proton signals ranging between 6.46 ppm and 7.62 ppm.
Due to the assembly of amide and imine functionalities, acrylohydrazides can exist as stereoisomers with a C=N double bond (geometric configurations Z or E) and as syn/antiperiplanar conformers around the amide (C(O)-NH) bond (Scheme 2). In the 1H NMR spectra, the maximum intensity difference for compounds (4a–h) confirms that both conformers were obtained with different ratios (Table 2).
Analysis of the various ratios among all possible conformers reveals that the synperiplanar (sp) conformer is predominant for compounds 4a–b, while the antiperiplanar (ap) conformer is favored for compounds 4c–d. For compounds 4e–h, the syn/anti-periplanar conformers are nearly in equal proportions.
Palla et al.’s (1986) previous work has demonstrated that the Z isomer of acrylohydrazides is characterized by a singlet around 14 ppm corresponding to the NH proton. The absence of a duplicated singlet around this chemical shift in the proton spectra (Fig. 1) suggests the absence of the Z(C=N) isomer. However, the correlation between the NH proton and the azomethine fragment in the NOESY spectrum of compounds 4h and 4f (Fig. 5) confirms the presence of the E(C=N) isomer, according to the findings of Palla et al. (1986). Regarding compounds 4a–f and 4h, the emergence of a weak correlation between the NH proton and azomethine (N=CH) and azoethylidine (N=C(CH3)) fragments in the NOESY spectrum (Fig. 2) confirms also the presence of the E(C=N) isomer. Table 3 summarizes the calculated ratios relative to the duplicated NH signal. These results indicate that the syn conformer is predominant for compounds 4a–b (thus, the stereochemistry Ec=c sp EC=N), while for compounds 4c–h, the anti conformer is favored (Table 5). It is noteworthy that the proportion of the anti conformer decreases in favor of the syn conformer when transitioning from compounds 4c–d to 4g–h and from 4g–h to 4e–f.
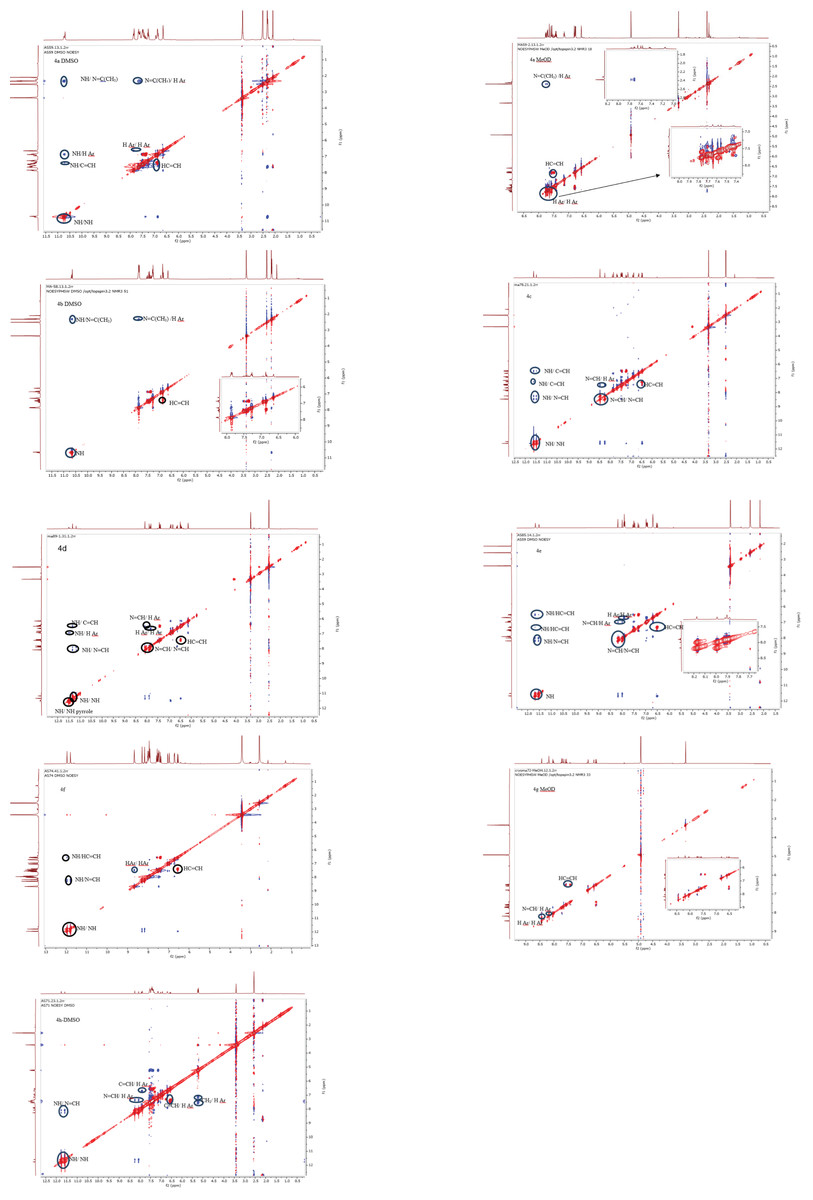
Figure 5: NOESY spectra in DMSO-d6 of compounds 4a–f, 4h.
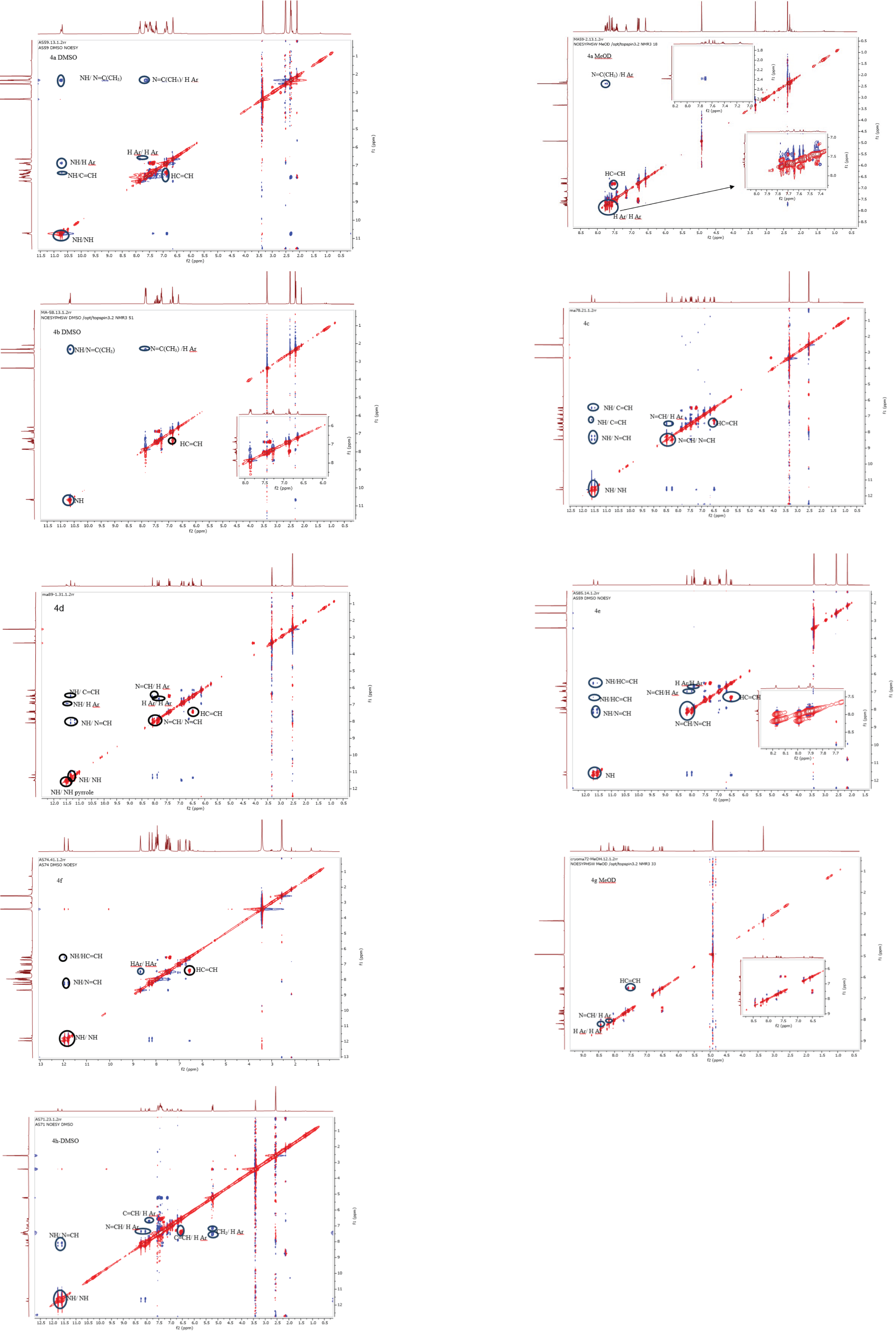{kind=link}
| Compounds | Stereoisomerism |
|---|---|
| 4a | Ec=c sp EC=N |
| 4b | Ec=c sp EC=N |
| 4c | Ec=c ap EC=N |
| 4d | Ec=c ap EC=N |
| 4e | Ec=c ap EC=N |
| 4f | Ec=c ap EC=N |
| 4g | Ec=c ap EC=N |
| 4h | Ec=c ap EC=N |
Comparing the ratios of different conformers between compounds 4c–h and 4a–b reveals a reversal of the anti conformer in favor of the syn conformer when transitioning from compounds 4c–h to 4a–b. This inversion of conformers could be explained by the bulkier nature of the azoethylidine fragment (N=C(CH3)) compared to the azomethine fragment (N=CH).
Conclusion
This study successfully synthesized eight new derivatives of (E)3-furan-2-yl acrylohydrazide. The obtained compounds were comprehensively characterized through spectroscopic analyses, including 1D and 2D NMR, as well as high-resolution mass spectrometry.
The analysis of 1H NMR data unveiled significant conformational diversity among the synthesized compounds. Compounds 4a–b demonstrated an Ec=c sp EC=N configuration, while all other compounds exhibited an Ec=c ap EC=N configuration. Notably, compounds 4g displayed an Ec=c ap configuration and likely EC=N, although confirmation of the latter was challenging.
Ultimately, this research contributes to the continum of prior investigations on N-acylhydrazones, emphasizing the stability of (E)-3-(furan-2-yl) acrylohydrazides. This understanding of their conformation properties opens promising avenues for future applications in diverse fields, such as the design of new materials.
In our research, we have developed N-acylhydrazones featuring diverse passivation groups. The incorporation of these groups has the potential to significantly influence their utility in solar energy applications, and also could exhibit promising bioactive properties. Our preliminary studies with our synthesis compounds have demonstrated their efficacy in inhibiting the growth of S. aureus, motivating further investigation into their potential as treatments for methicillin-resistant S. aureus infections.