
Integrative transcriptome data mining for identification of core lncRNAs in breast cancer
- Published
- Accepted
- Received
- Academic Editor
- Motoki Takaku
- Subject Areas
- Bioinformatics, Oncology
- Keywords
- lncRNA, Breast cancer, Biomarkers, Bioinformatics, WGCNA
- Copyright
- © 2019 Zhang et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2019. Integrative transcriptome data mining for identification of core lncRNAs in breast cancer. PeerJ 7:e7821 https://doi.org/10.7717/peerj.7821
Abstract
Background
Cumulative evidence suggests that long non-coding RNAs (lncRNAs) play an important role in tumorigenesis. This study aims to identify lncRNAs that can serve as new biomarkers for breast cancer diagnosis or screening.
Methods
First, the linear fitting method was used to identify differentially expressed genes from the breast cancer RNA expression profiles in The Cancer Genome Atlas (TCGA). Next, the diagnostic value of all differentially expressed lncRNAs was evaluated using a receiver operating characteristic (ROC) curve. Then, the top ten lncRNAs with the highest diagnostic value were selected as core genes for clinical characteristics and prognosis analysis. Furthermore, core lncRNA-mRNA co-expression networks based on weighted gene co-expression network analysis (WGCNA) were constructed, and functional enrichment analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID). The differential expression level and diagnostic value of core lncRNAs were further evaluated by using independent data set from Gene Expression Omnibus (GEO). Finally, the expression status and prognostic value of core lncRNAs in various tumors were analyzed based on Gene Expression Profiling Interactive Analysis (GEPIA).
Results
Seven core lncRNAs (LINC00478, PGM5-AS1, AL035610.1, MIR143HG, RP11-175K6.1, AC005550.4, and MIR497HG) have good single-factor diagnostic value for breast cancer. AC093850.2 has a prognostic value for breast cancer. AC005550.4 and MIR497HG can better distinguish breast cancer patients in early-stage from the advanced-stage. Low expression of MAGI2-AS3, LINC00478, AL035610.1, MIR143HG, and MIR145 may be associated with lymph node metastasis in breast cancer.
Conclusion
Our study provides candidate biomarkers for the diagnosis and prognosis of breast cancer, as well as a bioinformatics basis for the further elucidation of the molecular pathological mechanism of breast cancer.
Introduction
Long non-coding RNAs (lncRNAs) are a type of non-coding RNA molecules with a length of more than 200 nucleotides (McFadden & Hargrove, 2016). Studies have shown that lncRNAs are involved in a variety of biological processes (e.g., genetic imprinting, cell differentiation, apoptosis, cell proliferation, and immune response) (Zhu et al., 2018). The value of lncRNAs in the diagnosis and prognosis of tumors has now been widely recognized (Yang et al., 2016).
Breast cancer is the most common malignant tumor in women (Bertoli, Cava & Castiglioni, 2015). In recent years, the clinical management of breast cancer has improved, but the cancer is still a serious threat to women’s health due to its poor prognosis and high mortality rate (Pelosi et al., 2014). With the rapid development of tumor genomics, the discovery of highly specific biomarkers has become an intrinsic requirement for clinical diagnosis and treatment of breast cancer. Numerous transcripts of lncRNAs provide a wealth of materials for our search for quality biomarkers.
The Cancer Genome Atlas (TCGA), as the most authoritative cancer genome database, provides abundant raw data for large-scale tumor researches (Gao et al., 2019). A series of highly sensitive and specific tumor biomarkers based on TCGA data mining have been discovered and applied with the assistance of various assistive technologies (Gao et al., 2018). WGCNA, featuring soft threshold settings and a weighted network, can efficiently identify highly synergistically altered gene sets (Langfelder & Horvath, 2007; Sutherland et al., 2016). The construction of key target co-expression networks based on WGCNA provides methodological support for exploring the molecular mechanisms of diseases.
In this study, we used the limma package for identifying differentially expressed lncRNAs in breast cancer. The ROC curve was used to evaluate and select the top ten differentially expressed lncRNAs with the highest diagnostic value as core genes. We then comprehensively analyzed the clinical features and prognostic value of these core genes in breast cancer. The WGCNA co-expression networks and their functional enrichment analysis assisted us in exploring the role of core genes and their key targets in breast cancer. GEO’s independent data set was used to evaluate the expression level and diagnostic value of core lncRNAs. Based on GEPIA, we explored the expression status and prognostic value of core genes in a variety of tumors. Through this research, we hope to identify high-quality biomarkers for breast cancer from the perspective of lncRNAs, and provide a bioinformatics basis for further elucidation of the molecular pathogenesis of breast cancer.
Materials & Methods
Downloading breast cancer RNA expression profile data from TCGA
We downloaded high-throughput data of breast cancer RNA-Seq from TCGA (Wu et al., 2016. The access time for the TCGA data is February 1, 2019, and the species is human. These RNA-Seq data were derived from 837 breast cancer tumor samples and 105 adjacent non-cancerous breast tissue samples. This study strictly adheres to the TCGA publication guidelines.
Identification of differentially expressed lncRNAs in breast cancer
The breast cancer RNA-Seq data we downloaded covers 12,727 lncRNAs described by NCBI (http://www.ncbi.nlm.nih.gov) or Ensembl (http://asia.ensembl.org/index.html). We used the limma package (in R language) to identify differentially expressed lncRNAs (P < 0.05 and —log2FC—>1) (Ritchie et al., 2015). The limma package is an excellent analytical statistical tool for differential expression analysis of gene expression chip data. ROC curves were used to assess the diagnostic effect of all differentially expressed lncRNAs in breast cancer (Patel et al., 2019). ROC curves were plotted based on the expression value of each core lncRNA and the type of samples (either breast cancer tissue or non-cancerous breast tissue). According to the expression value of each core lncRNA, we classified the 837 breast cancer tumor samples and 105 adjacent non-cancerous breast tissue samples (downloaded from TCGA) into the breast cancer group and non-breast cancer group. The results of the grouping according to the classification were compared with the actual species of the sample (breast cancer tissue or non-breast cancer tissue) to obtain a false positive rate and a true positive rate. These rates were respectively represented on the abscissa (X axis) and the ordinate (Y axis) to plot the ROC curves. The area under the curve (AUC) of the ROC curve was calculated, and the statistical standard of AUC > 0.7 and P < 0.05 was used (the higher the AUC, the better the diagnostic effect). The top 10 lncRNAs of most diagnostic values were further analyzed as core lncRNAs.
Clinical characteristics analysis and prognostic value evaluation of core genes
In order to get a more accurate result, a Student’s t-test was also used to evaluate the differential expression level of core lncRNAs in breast cancer tissues and non-cancer breast tissues. A Student’s t-test can highlight a difference between two groups. (Ryu et al., 2018). Similarly, differences in the expression of core genes between different clinical groups were evaluated. K-M analysis (Gökbuget et al., 2018) was used to assess the prognostic value of core lncRNAs. The sample data for the K-M survival analysis was derived from the corresponding clinical data (including survival time and survival status) of the 837 breast cancer tumor samples originally downloaded from TCGA. According to the median value of each core lncRNA expression value in R language, the patients were divided into the high expression group and low expression group. Genes of two groups of patients with significantly different overall survival (OS) rate (P < 0.05) were selected as prognostic factor. The p-value of logrank <0.05 was statistically significant. In order to select better prognostic biomarkers, we also added univariate cox analysis (Zhao et al., 2018b) to these core lncRNAs.
WGCNA-based construction of core lncRNA-mRNA co-expression network and its functional enrichment analysis
The core lncRNA-mRNA co-expression networks based on WGCNA were constructed to explore the molecular mechanism of core genes in breast cancer (Chen et al., 2015). The threshold is 0.02. We used Cytoscape to visualize the co-expression networks. The DAVID database (david.ncifcrf.gov) was used for GO analysis of co-expression networks. The widely used GO analysis can perform functional enrichment analysis of gene collections from three aspects: biology pathway (BP), cell component (CC), and molecular function (MF) (Chai et al., 2016). P < 0.05 was considered statistically significant.
Evaluation of core lncRNAs using GEO data sets
We downloaded the data of GEO’s independent data set GSE125677. GSE125677 contained lncRNAs sequencing data derived from three paired breast cancer and their adjacent normal tissues, and the identified 10 core lncRNAs were included. Independent sample t-test was conducted to analyze differential expression levels of core lncRNAs between breast cancer tumor and adjacent breast tissues (Al-Hashel et al., 2018). A test result with —t—>2 and p < 0.05 was considered statistically significant. ROC curves were used to assess the diagnostic effect of core lncRNAs in breast cancer. Patel et al. (2019). AUC is the area under the curve of the ROC curve. The higher the AUC, the better the diagnostic effect is considered to be. ROC curves were plotted based on the expression value of each core lncRNA and the type of samples, namely either breast cancer tissue or non-cancerous breast tissue. An lncRNA with AUC >0.7 and P < 0.05 is considered to have a good diagnostic value for breast cancer.
GEPIA-based analysis of expression patterns and prognostic value of core lncRNAs in a variety of tumors
GEPIA was developed by Zefang Tang, Chenwei Li and Boxi Kang of Zhang’s Lab in Peking University (Tang et al., 2017). The RNA-Seq datasets GEPIA used is based on the UCSC Xena project (http://xena.ucsc.edu), which computed all expression raw data with a standard pipeline. Therefore, with GEPIA we could efficiently obtain information about the core lncRNAs from TCGA database. We used GEPIA to analyze the differential expression of core lncRNAs in 31 tumor tissues and adjacent non-cancerous tissues. A difference or result with P < 0.01 and —log2FC—>1 was considered statistically significant. Meanwhile, we assessed the single-factor prognostic value of core lncRNAs in these 30 tumors other than breast cancer. P < 0.05 was considered statistically significant.
Results
Differentially expressed lncRNAs selected from breast cancer RNA expression data in TCGA
We identified 276 differentially expressed lncRNAs in breast cancer (Fig. 1), among which 164 were up-regulated and 112 were down-regulated. The statistical criteria were P value < 0.05 and —log2FC—> 1. The top 22 in the results of ROC analysis of differentially expressed lncRNAs was shown in Table S1 (P < 0.05 and AUC >0.95). The following top 10 lncRNAs with highest diagnostic value were used as core genes for subsequent analysis: AC093850.2, MAGI2-AS3, LINC00478, PGM5-AS1, AL035610.1, MIR143HG, RP11-175K6.1, AC005550.4, MIR497HG, and MIR145 (see Table 1 for details).
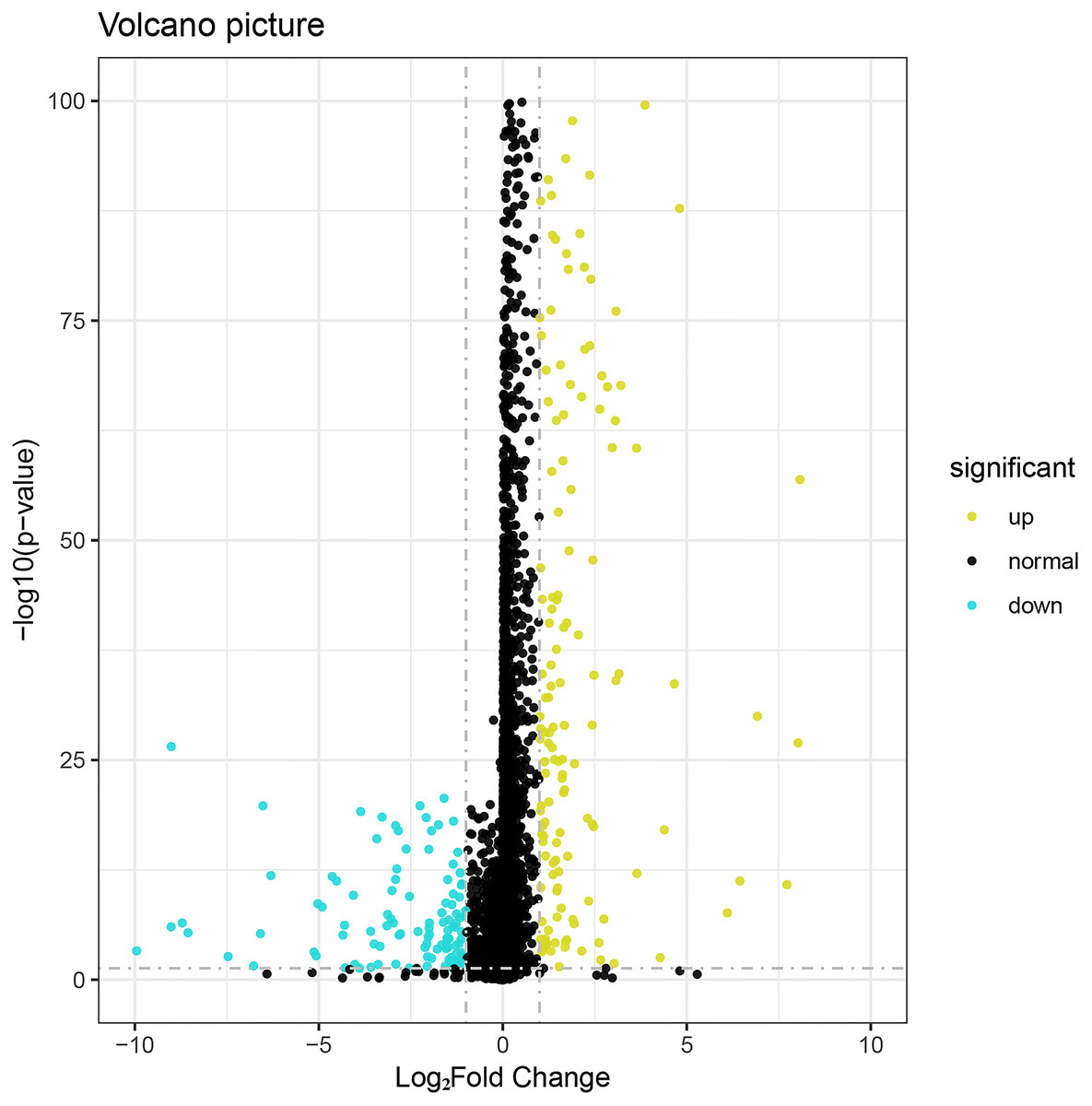
Figure 1: Volcano plot of the aberrantly expressed lncRNAs between breast cancer and para-tumorous breast tissues.
Deep yellow: high expression; Blue: low expression; Black dots: the lncRNAs with expression of |log2FC| < 1. P > 0.05.{kind=link}
| LncRNA | Ensemble | Regulation | FC | AUC | CI | P-value |
|---|---|---|---|---|---|---|
| AC093850.2 | ENSG00000230838 | Up | 0.010908018 | 0.9872 | 0.980,0.993 | <0.001 |
| MAGI2-AS3 | ENSG00000234456 | Down | 6.91764815 | 0.9853 | 0.976,0.994 | <0.001 |
| LINC00478 | ENSG00000215386 | Down | 2.33209709 | 0.9843 | 0.977,0.991 | <0.001 |
| PGM5-AS1 | ENSG00000224958 | Down | 2.012299062 | 0.9805 | 0.971,0.989 | <0.001 |
| AL035610.1 | ENSG00000232079 | Down | 2.180782938 | 0.9803 | 0.970,0.990 | <0.001 |
| MIR143HG | ENSG00000249669 | Down | 5.21641063 | 0.9798 | 0.961,0.998 | <0.001 |
| RP11-175K6.1 | ENSG00000245812 | Down | 4.107274497 | 0.9755 | 0.964,0.986 | <0.001 |
| AC005550.4 | ENSG00000229108 | Down | 5.250828547 | 0.9727 | 0.962,0.983 | <0.001 |
| MIR497HG | ENSG00000267532 | Down | 5.76572037 | 0.9724 | 0.957,0.987 | <0.001 |
| MIR145 | ENSG00000269936 | Down | 79420.71504 | 0.9693 | 0.947,0.990 | <0.001 |
Notes:
- FC
-
fold change
- AUC
-
area under the curve
- CI
-
confidence interva p < 0.05
An analysis of clinical characteristics of core lncRNAs in breast cancer
The level of AC093850.2 in breast cancer was significantly higher than that in adjacent breast tissues. In contrast, nine other lncRNAs were down-regulated in breast cancer tissues; and the low expression of MIR145 was most significant (—log2FC—>16) (Fig. 2). These ten core lncRNAs had high diagnostic value for distinguishing between breast cancer tissues and non-cancer tissues, with the smallest AUC = 0.969 (Fig. 3). K-M survival analysis showed that AC093850.2 was significantly correlated with breast cancer survival (P = 0.0075, Fig. 4). Figure 4 shows the K-M curves and contains the p-value of logrank. In addition, univariate cox analysis also showed that AC093850.2 was an independent prognostic indicator for breast cancer (P = 0.000482235, Table S2). Several core lncRNAs were closely related to some clinical parameters of breast cancer. The main results are shown in Table 2 and Fig. 5 (P < 0.05, —t— ≥ 1.987). The expression levels of AC005550.4 and MIR497HG can distinguish the breast cancer patients in early-stage from the advanced-stage. Compared with the clinical group of breast cancer without lymph nodal involvement, these five core lncRNAs (MAGI2-AS3, LINC00478, AL035610.1, MIR143HG, and MIR145) had lower expression level in the group with lymph nodal involvement.
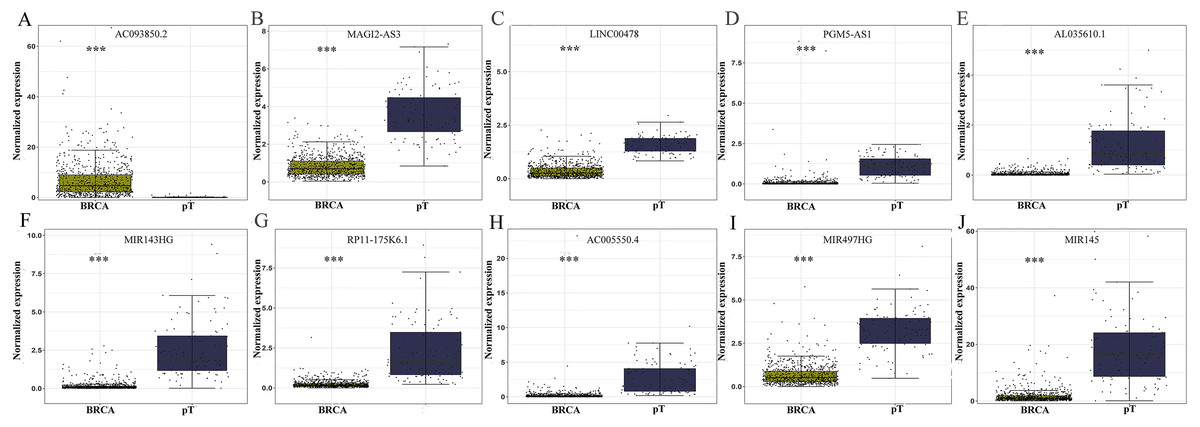
Figure 2: Differential expression analysis of the core genes between breast cancer and para-tumorous breast tissues.
Deep yellow column: breast cancer tissues (BRCA). Blue column: breast para-tumorous tissue (pT). X axis: tissue types. Y axis: Normalized expression. (A) AC093850.2; (B) MAGI2-AS3; (C) LINC00478; (D) PGM5-AS1; (E) AL035610.1; (F) MIR143HG; (G) RP11-175K6.1; (H) AC005550.4; (I) MIR497HG; (J) MIR145. ∗∗∗P < 0.001.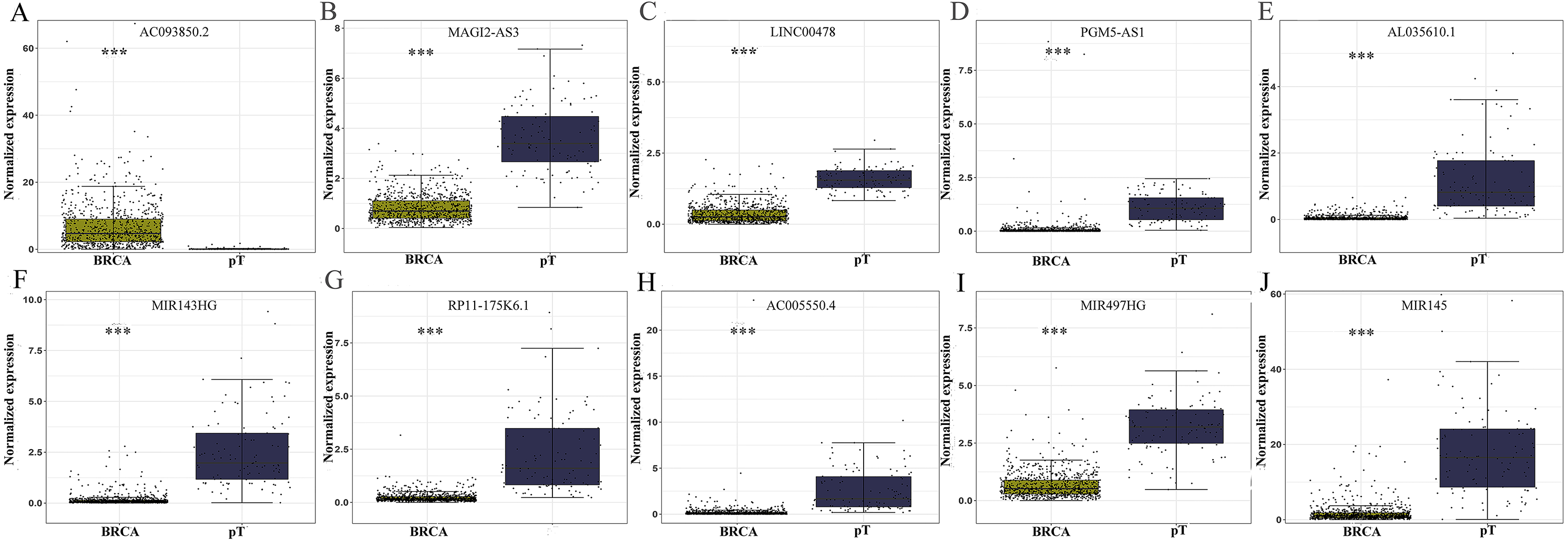{kind=link}
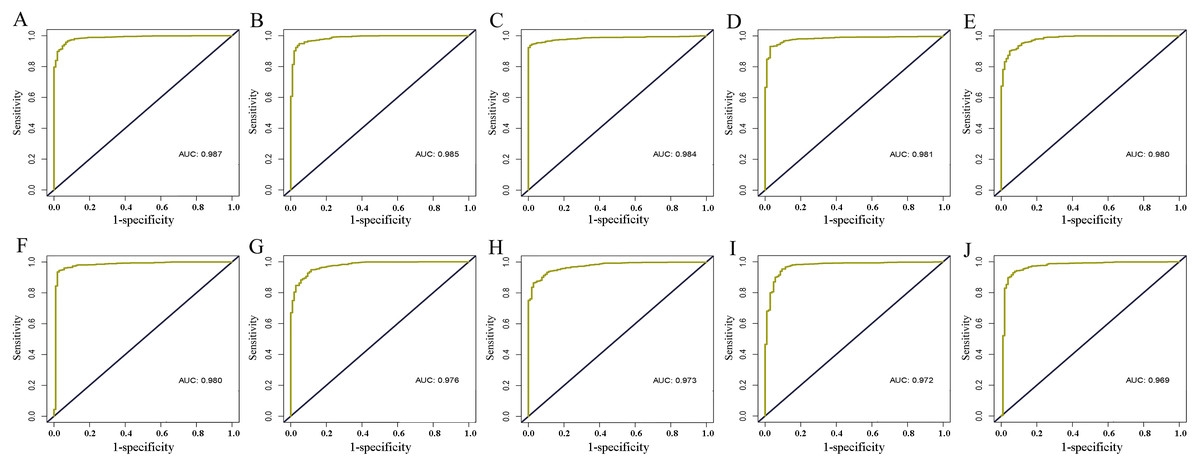
Figure 3: ROC curves of the core lncRNAs in breast cancer.
Deep yellow: sensitive curve. Blue: identify line. X axis: 1-specificity. Y axis: sensitivity. AUC: area Under Curve. The higher the AUC, the better the diagnostic effect. (A) AC093850.2; (B) MAGI2-AS3; (C) LINC00478; (D) PGM5-AS1; (E) AL035610.1; (F) MIR143HG; (G) RP11-175K6.1; (H) AC005550.4; (I) MIR497HG; (J) MIR145.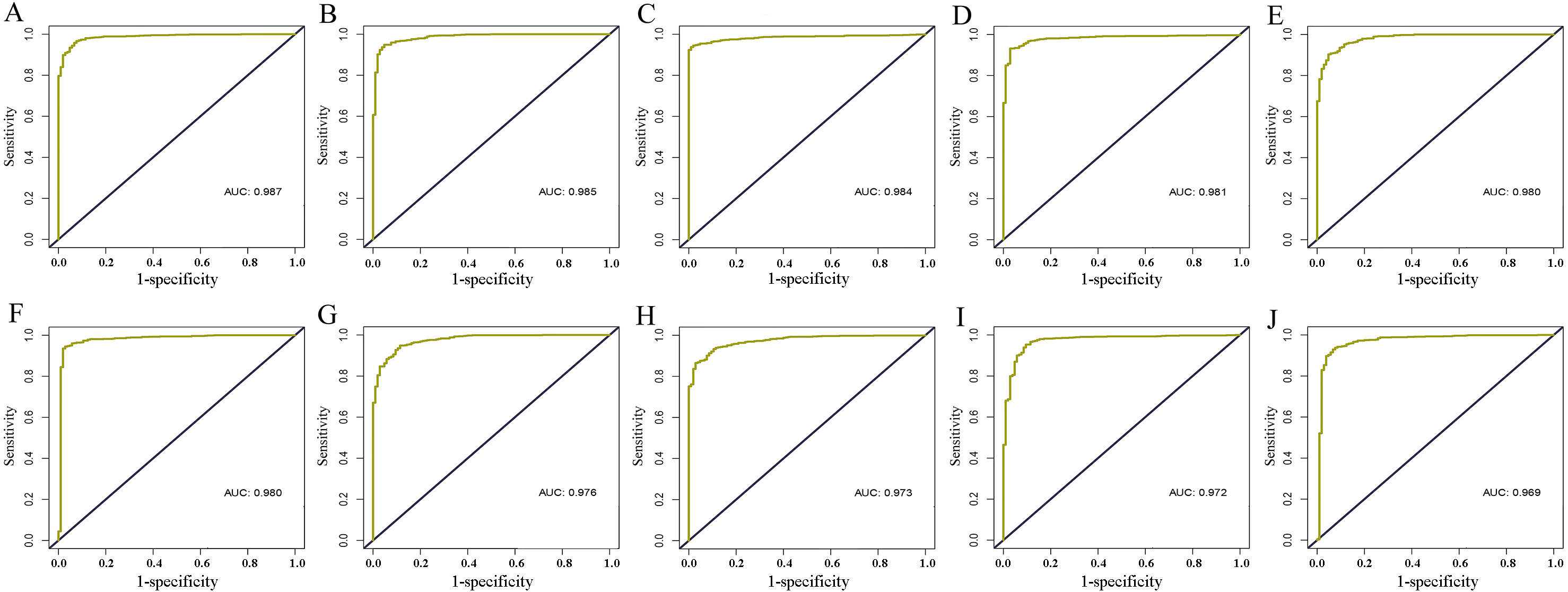{kind=link}
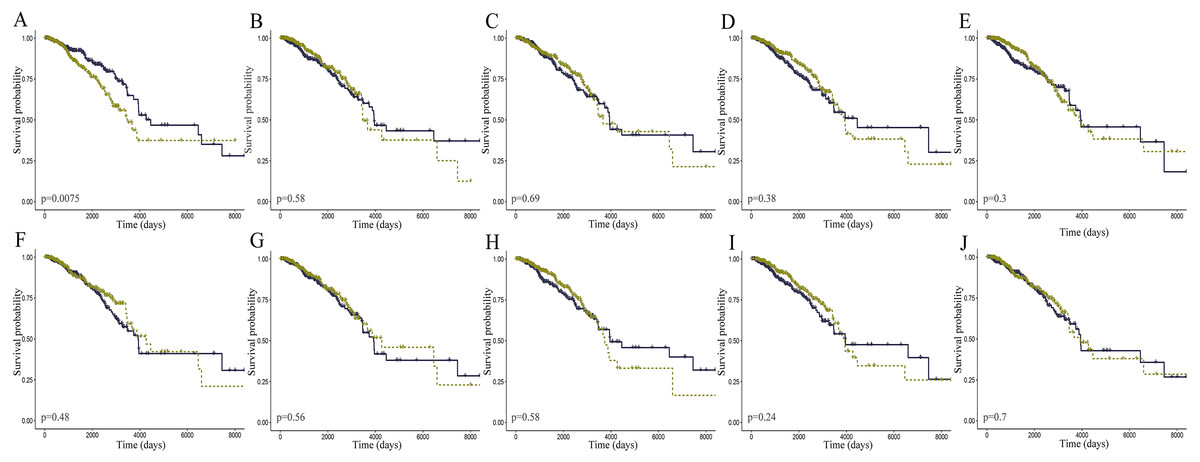
Figure 4: K-M curves of the core lncRNAs in breast cancer.
The prognostic significance of the core lncRNAs in patients with breast cancer, according to the Kaplan–Meier analysis (in R) of data from TCGA. The deep yellow lines represent patients with high gene expression, and blue lines represent patients with a low gene expression. (A) AC093850.2; (B) MAGI2-AS3; (C) LINC00478; (D) PGM5-AS1; (E) AL035610.1; (F) MIR143HG; (G) RP11-175K6.1; (H) AC005550.4; (I) MIR497HG; (J) MIR145.{kind=link}
| lncRNA factor | Gender (female/male) | T (T1/2 vs. T3/4) | N (no/yes) | M (no/yes) | Pathological stage (I/II vs. III/IV) | Targeted molecular therapy (no/yes) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| t | P | t | P | t | P | t | P | t | P | t | P | |
| AC093850.2 | −1.675 | 0.132 | 1.322 | 0.188 | −1.217 | 0.224 | −0.764 | 0.459 | −1.376 | 0.167 | −2.681 | 0.009 |
| MAGI2-AS3 | 0.418 | 0.686 | 0.908 | 0.365 | −2.824 | 0.005 | −0.32 | 0.754 | −0.706 | 0.481 | −3.801 | 0.00032 |
| LINC00478 | 5.277 | 0.0003 | −1.559 | 0.122 | −3.199 | 0.0014 | −0.463 | 0.651 | −1.669 | 0.0964 | 0.497 | 0.622 |
| PGM5-AS1 | 1.245 | 0.235 | 1.651 | 0.099 | −0.544 | 0.586 | 0.719 | 0.48 | 1.4899 | 0.137 | −2.295 | 0.023 |
| AL035610.1 | 4.638 | 0.0007 | 0.1461 | 0.884 | −1.987 | 0.048 | 0.338 | 0.741 | 0.386 | 0.701 | −1.735 | 0.088 |
| MIR143HG | 2.471 | 0.03 | −0.52 | 0.604 | −2.882 | 0.004 | −0.452 | 0.659 | −0.504 | 0.616 | −4.538 | 8.44E−06 |
| RP11-175K6.1 | 0.209 | 0.839 | 1.195 | 0.234 | −1.559 | 0.12 | 0.347 | 0.734 | −0.312 | 0.756 | −0.8 | 0.428 |
| AC005550.4 | 5.514 | 5.03E−08 | 2.194 | 0.029 | 0.978 | 0.329 | 1.738 | 0.102 | 1.191 | 0.234 | −0.727 | 0.47 |
| MIR497HG | 1.479 | 0.175 | −2.257 | 0.026 | −0.62 | 0.535 | 0.087 | 0.932 | −1.091 | 0.277 | −1.347 | 0.183 |
| MIR145 | 0.455 | 0.66 | −0.037 | 0.971 | −2.545 | 0.012 | −0.437 | 0.67 | −0.562 | 0.575 | −3.161 | 0.002 |
Notes:
- T
-
tumor stage
- N
-
lymph node
- M
-
metastasis
P < 0.05 and — t —> 2 are statistically significant.
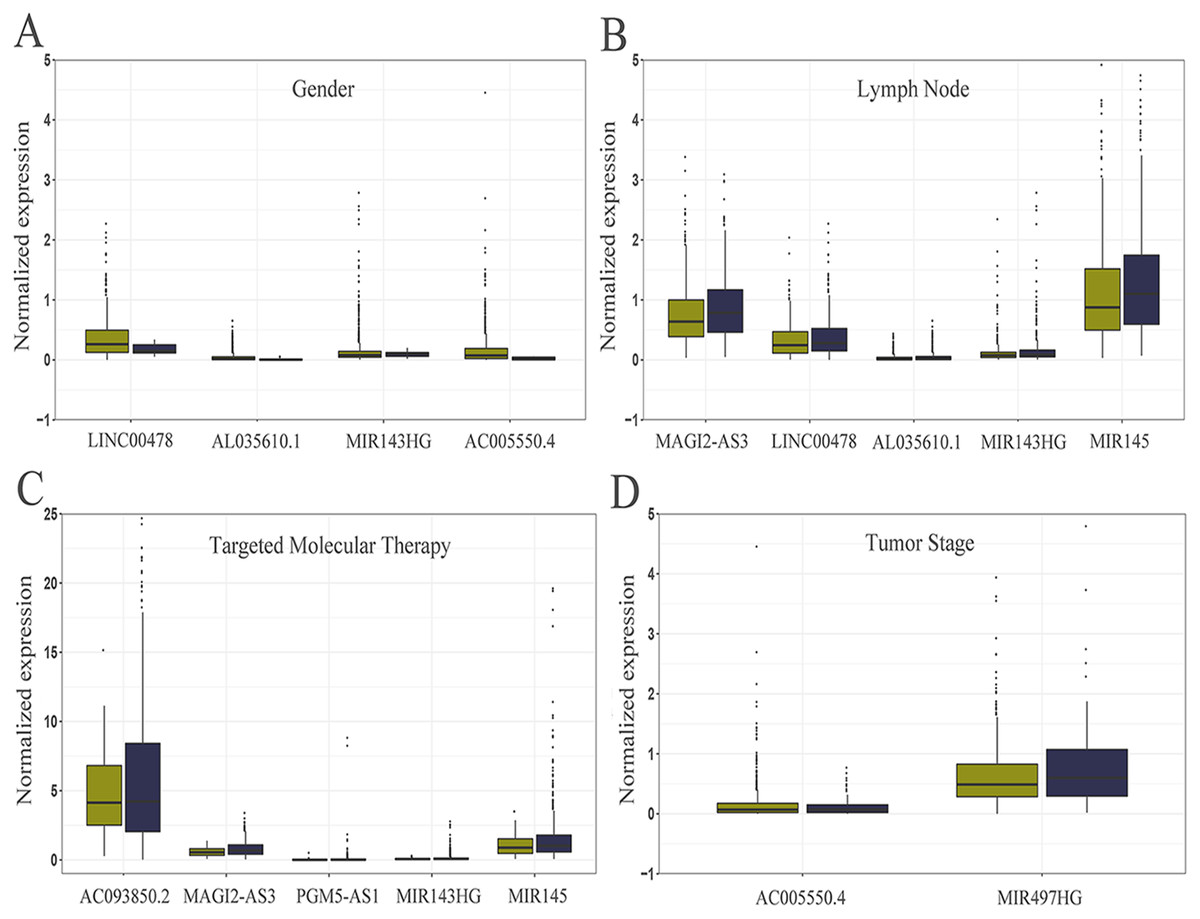
Figure 5: Relationship between the expression of core lncRNAs and different clinical parameters in breast cancer from TCGA.
The statistical significance of differences was calculated by the Student’s t-test. P < 0.05, |t| > 1.987. (A) Gender: deep yellow, female; blue, Male. (B) Lymph Node: deep yellow, no; blue, yes. (C) Targeted Molecular Therapy: deep yellow, no; blue, yes. (D) Tumor Stage: deep yellow, T1-T2; blue, T3-T4.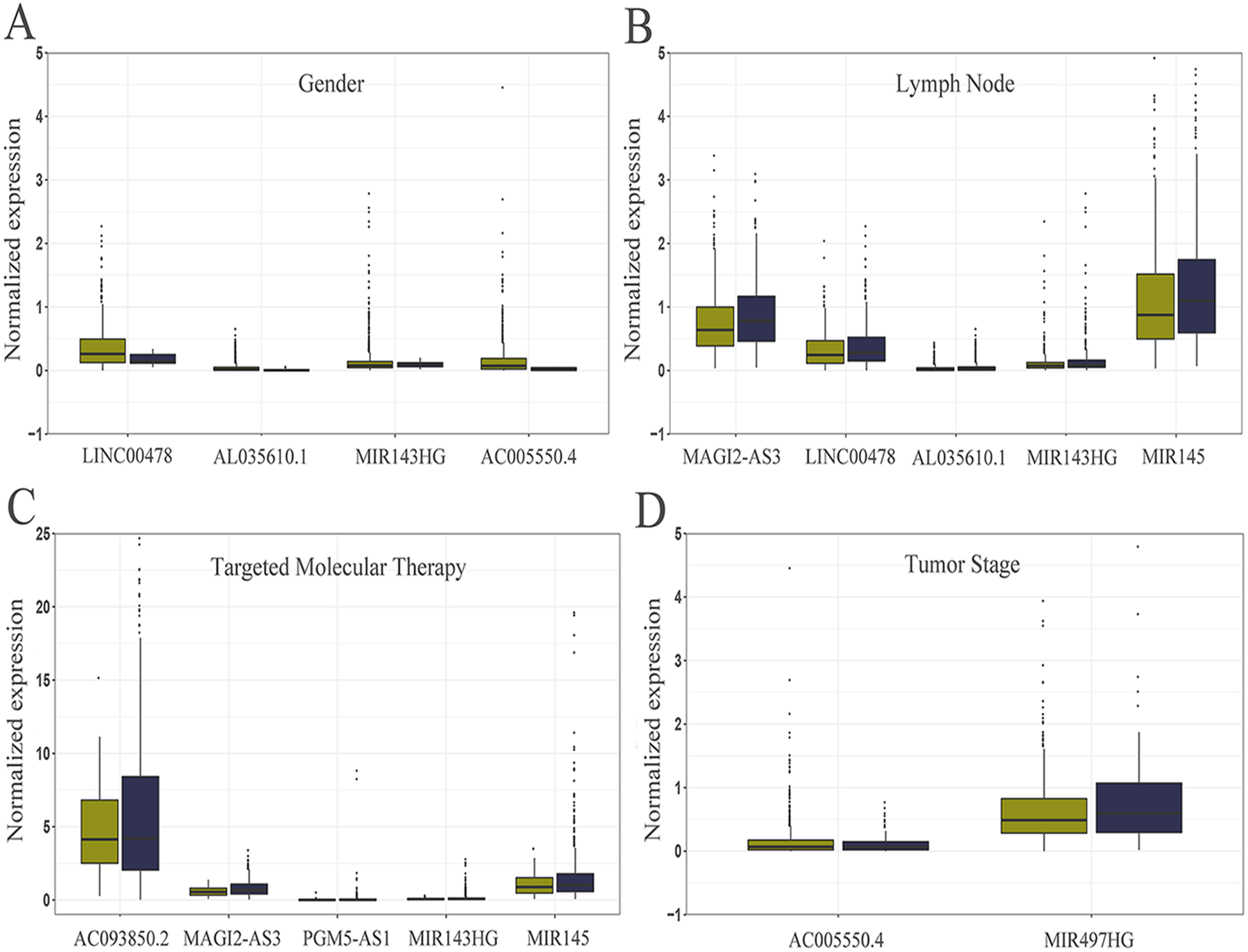{kind=link}
The roles of core lncRNAs in breast cancer based on WGCNA network
The results show that six core genes (AC005550.4, AL035610.1, PGM5-AS1, RP11-175K6.1, MAGI2-AS3, and MIR497HG), especially the first three, in the lncRNA-mRNA WGCNA networks were closely related. In addition, AC093850.2 and MIR143HG had a large number of co-expressed mRNAs (Fig. 6). The GO analysis showed that AC005550.4 and AL035610.1 were significantly enriched in biology pathways such as glucose metabolism, lipid metabolism, and alcohol metabolism. Meanwhile, co-expressed mRNAs of RP11-175K6.1 were significantly enriched in biology pathways including protein kinase B signaling pathway, regulation of angiogenesis, regulation of circadian rhythms, and the reaction of steroid hormones. The co-expressed mRNA of AC093850.2 was closely related to biology pathways such as collagen metabolism and glycosaminoglycan binding. It is worth noting that both MIR143HG and LINC00478 were closely related to biology pathways such as taste stimulation and bitterness perception. The main results are shown in Table S3 (P < 0.05, count >4).
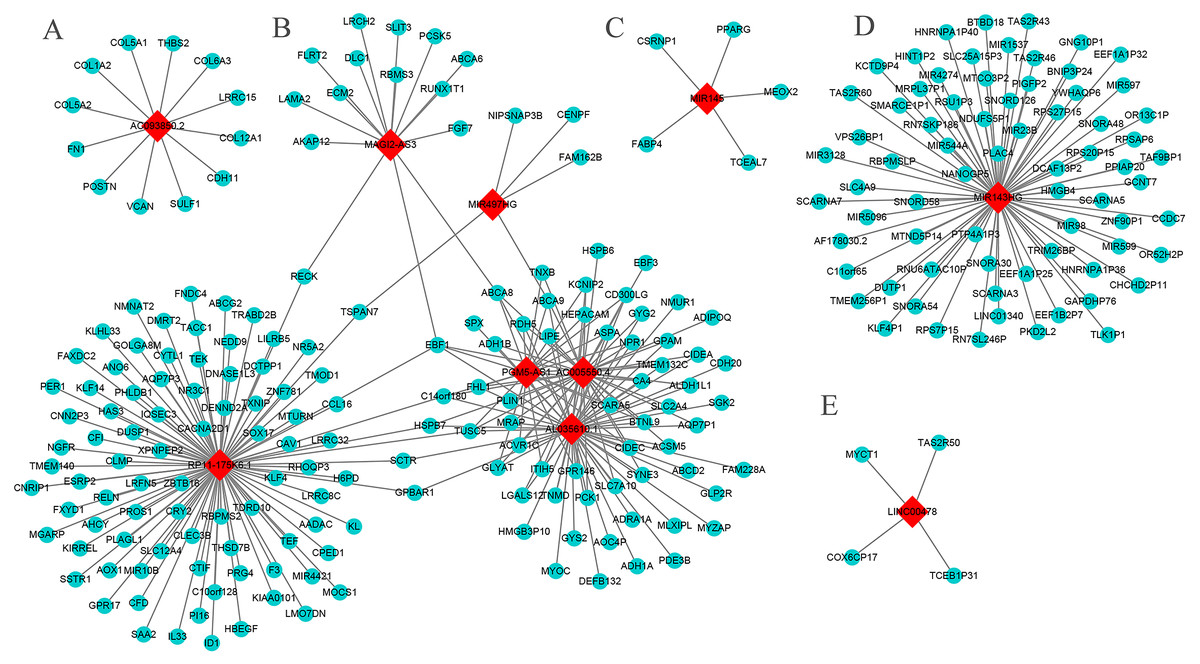
Figure 6: The core lncRNA-mRNA co-expression network based on WGCNA.
Red diamonds: core lncRNAs. Blue balls: core lncRNAs co-expressed mRNAs. The threshold is 0.02. (A) AC093850.2; (B) MAGI2-AS3; PGM5-AS1; AL035610.1; RP11-175K6.1; AC005550.4; MIR497HG; (C) MIR145; (D) MIR143HG; (E) LINC00478.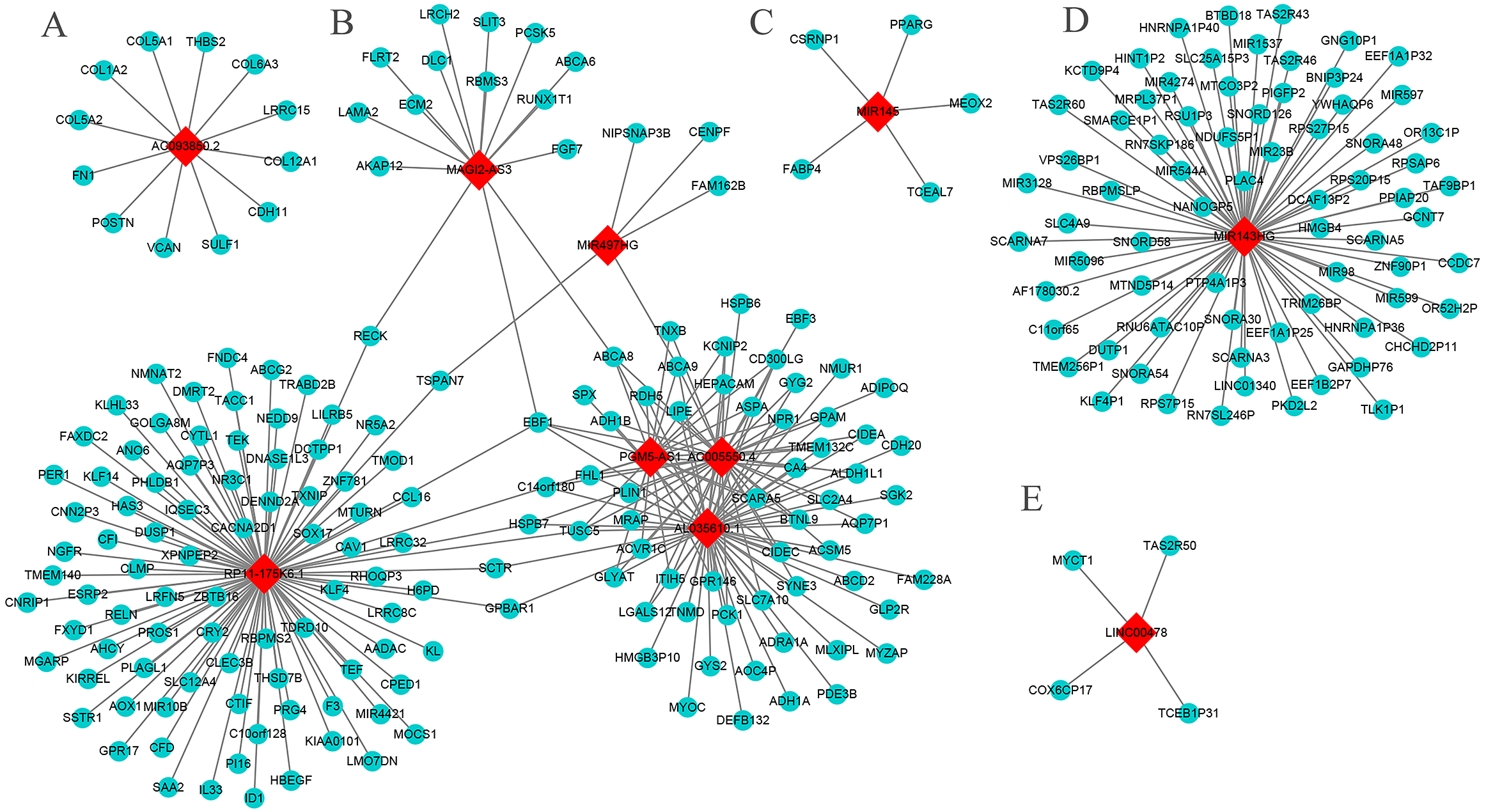{kind=link}
In addition, we also analyzed the most significant GO terms for all mRNAs in the co-expression network. The cell component was mainly enriched in the membrane area. Significantly enriched biology pathways include: angiogenesis, regulation of vascular development, positive regulation of cell migration, lipid metabolism, and peptide activity. Molecular function was mainly enriched in serine hydrolase activity, heparin binding, glycosaminoglycan binding, and sulfur compound binding. The main results are shown in Table S4.
Evaluation of the expression and diagnostic value of all core lncRNAs in GEO independent datasets
Information of all core lncRNAs can be obtained from GSE125677. With the exception of the high expression of AC093850.2 in breast cancer tissues, the other nine lncRNAs were lowly expressed in cancer tissues. Moreover, the ROC curves of these core lncRNAs indicated they had a good diagnostic value for distinguishing breast cancer patients from the healthy population. This supported our findings of the analysis based on the TCGA database (see Table 3 and Fig. 7).
| Variable | pT | BC | T-test | ROC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Mean | SD | n | Mean | SD | t | P | AUC | SE | 95% CI | P | |
| AC093850.2 | 6 | 0.34478499 | 0.94650167 | 6 | −0.344785 | 0.8436006 | 1.6316 | 0.1225 | 0.852 | 0.0997 | 0.656–1.000 | 0.0004236 |
| MAGI2-AS3 | 6 | −0.4193923 | 0.6252397 | 6 | 0.4193923 | 1.0432272 | −2.069 | 0.0589 | 0.79 | 0.1184 | 0.558–1.000 | 0.01428 |
| LINC00478 | 6 | −0.4452022 | 0.9184215 | 6 | 0.4452022 | 0.7652063 | −2.2345 | 0.04056 | 0.852 | 0.0907 | 0.674–1.000 | 0.0001052 |
| PGM5-AS1 | 6 | −0.4562647 | 0.638028 | 6 | 0.45626467 | 0.99976049 | −2.3082 | 0.03728 | 0.827 | 0.113 | 0.606–1.000 | 0.003783 |
| AL035610.1 | 6 | −0.78516717 | 0.49833236 | 6 | 0.7851672 | 0.4894569 | −6.7445 | <0.0001 | 1 | 0 | 1.000–1.000 | <0.0001 |
| MIR143HG | 6 | −0.4520927 | 0.6770228 | 6 | 0.4520927 | 0.9781449 | −2.2802 | 0.03849 | 0.753 | 0.0918 | 0.519–0.987 | 0.034 |
| RP11-175K6.1 | 6 | −0.6691243 | 0.6003143 | 6 | 0.6691243 | 0.7122052 | −4.3102 | <0.0001 | 0.914 | 0.0682 | 0.780–1.000 | <0.0001 |
| AC005550.4 | 6 | −0.559967 | 0.9184722 | 6 | 0.55996702 | 0.57086945 | −3.1068 | 0.008093 | 0.827 | 0.1189 | 0.594–1.000 | 0.00593 |
| MIR497HG | 6 | −0.8101302 | 0.5172673 | 6 | 0.8101302 | 0.3615728 | −7.702 | <0.0001 | 1 | 0 | 1.000–1.000 | <0.0001 |
| MIR145 | 6 | −0.3768513 | 0.7582684 | 6 | 0.3768513 | 0.9901974 | −1.813 | 0.08992 | 0.728 | 0.1245 | 0.484–0.973 | 0.06665 |
Notes:
- pT
-
para-tumor tissue
- BC
-
breast cancer
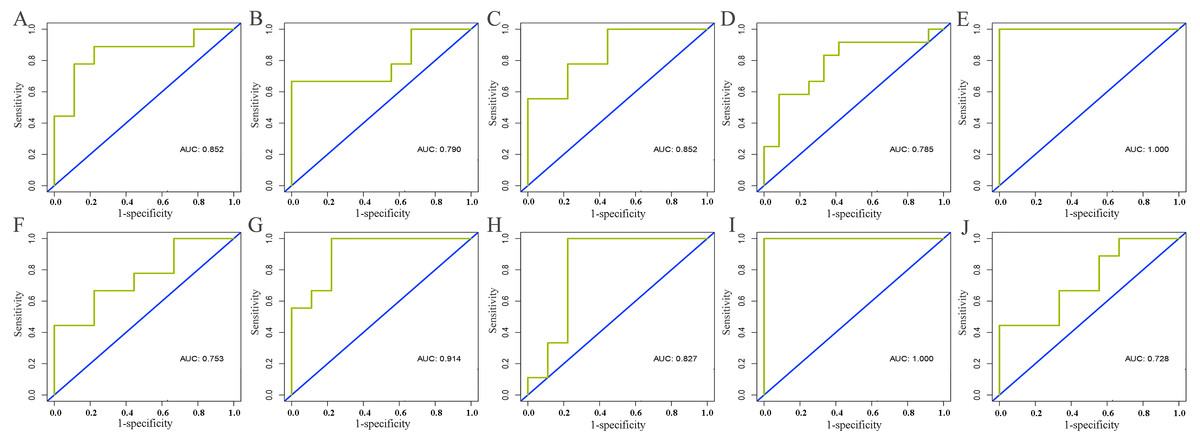
Figure 7: Validation of diagnostic value of core lncRNAs by ROC curves based on GEO dataset (GSE125677).
Deep yellow: sensitive curve. Blue: identify line. X axis: 1-specificity. Y axis: sensitivity. AUC: area under curve. The higher the AUC, the better the diagnostic effect. (A) AC093850.2; (B) MAGI2-AS3; (C) LINC00478; (D) PGM5-AS1; (E) AL035610.1; (F) MIR143HG; (G) RP11-175K6.1; (H) AC005550.4; (I) MIR497HG; (J) MIR145. P < 0.05.{kind=link}
Expression pattern and prognostic value of core lncRNAs in various tumors based on GEPIA
According to GEPIA, seven core lncRNAs (AC093850.2, MAGI2-AS3, PGM5-AS1, AL035610.1, MIR143HG, RP11-175K6.1, and MIR497HG) were differentially expressed in various tumors. Meanwhile, these seven core lncRNAs had a good single-factor prognostic value for a variety of tumors other than breast cancer. Details are shown in Table 4.
| core lncRNAs | In which diseases the core gene is up-regulation (P < 0.01) | In which diseases the core gene is down-regulation (P < 0.01) | In which diseases the core gene have prognostic value (P < 0.05) |
|---|---|---|---|
| AC093850.2 | BRCA/LUAD/LUSC/PAAD | GBM/KICH/LUSC/MESO/STAD/ | |
| MAGI2-AS3 | PAAD/SKCM/THYM | BRCA/ACC/BLCA/CESC/CHOL/COAD/ESCA/ KICH/KIRC/KIRP/LIHC/LUAD/ LUSC/OV/PRAD/READ/TGCT/THCA/ UCEC/UCS | BLCA/LGG/LUSC/STAD/UVM |
| PGM5-AS1 | BLCA/PCPG/UCEC | LGG/MESO | |
| AL035610.1 | KIRC/KIRP/OV | BRCA/ESCA/HNSC/KICH/SARC | KIRC/UCEC |
| MIR143HG | BRCA/BLCA/CESC/COAD/ESCA/KICH/ LUAD/LUSC/OV/PRAD/READ/ SKCM/TGCT/THCA/UCEC/UCS | BLCA/CESC/COAD/KIRP/STAD | |
| RP11-175K6.1 | DLBC/GBM | BRCA/BLCA/CESC/SKCM/THCA/UCEC/ | KIRP/LAML/MESO/PAAD/STAD/ UCS/UVM |
| MIR497HG | DLBC/KIRC/LGG/THYM | BRCA/ACC/BLCA/CESC/COAD/ESCA/KICH/ LUAD/LUSC/OV/PCPG/READ/ SKCM/TGCT/THCA/UCEC/UCS | BLCA/GBM/KICH/LUAD/SKCM/UVM |
Notes:
Full names of these diseases can be found in Table S5.
Discussion
With the rapid development of molecular genomics, highly specific diagnostic and prognostic biomarkers have provided molecular evidence for improvements in conventional diagnostic and therapy protocols. More and more lncRNAs are recognized as oncogenes or tumor suppressors. As a complement to the coding genes and miRNAs, lncRNAs have brought new hope for the diagnosis and prognosis of breast cancer. In this study, from the perspective of lncRNAs, we selected biomarkers that are closely related to the diagnosis, clinical features, and prognosis of breast cancer.
lncRNAs have an advantage in the application of diagnosis (Richard & Eichhorn, 2018), and the detection of specific lncRNAs has been applied to the early diagnosis of cancers (Zhang & Tang, 2018). In this study, the 10 differentially expressed core lncRNAs (AC093850.2, MAGI2-AS3, LINC00478, PGM5-AS1, AL035610.1, MIR143HG, RP11-175K6.1, AC005550.4, MIR497HG, and MIR145) selected have high diagnostic value for breast cancer. This suggests that they were closely linked to the risk of breast cancer. After further evaluation using the independent data set (GSE125677) in GEO, we gained results (see Table 3) showing that the differential expression levels and diagnostic value of the following seven core lncRNAs all reached statistical standards: LINC00478, PGM5-AS1, AL035610.1, MIR143HG, RP11-175K6.1, AC005550.4, and MIR497HG. However, a shortcoming of this study should be acknowledged, that is, the sample size of the data set used for evaluation is small. It is necessary to actively search for data sets of larger sample size in the follow-up studies in order to further explore and improve related biological experiments.
Further, the analysis of the clinical characteristics of the core lncRNAs showed that AC005550.4 and MIR497HG could better distinguish between early and late stages of breast cancer: the expression level of AC005550.4 in the early stage of breast cancer, was higher than the late stage; and the expression level of MIR497HG in early breast cancer was lower than that in advanced stage. It is worth noting that, compared with those in the clinical group of breast cancer without lymph node metastasis, the five core lncRNAs (MAGI2-AS3, LINC00478, AL035610.1, MIR143HG, and MIR145) were expressed at lower levels due to lymph node metastasis in breast cancer. Lymphatic metastasis is generally recognized as an important indicator for prognosis (Yamashita et al., 1997), so that results support that the low expression of MAGI2-AS3, LINC00478, AL035610.1, MIR143HG, and MIR145 is associated with metastasis and progression of breast cancer. It has been reported that the down-regulation of MIR145 is closely correlated with the invasion behavior of malignant breast tumors (Radojicic et al., 2011).
There are many reports on MIR143HG, LINC00478, and MIR145. It has been reported that miR-143 (host gene: MIR143HG) reduces cell proliferation and migration, and the host gene MIR143HG can be regulated by miR-143 (Du et al., 2016). Zhao et al. (2018a) used miRNA microarray and RT-qPCR data to confirm that the expression of miR-143/145 and its host gene MIR143HG were down-regulated in tumor tissues of HBV-related liver cancer patients. LINC00478 was lowly expressed in cancer tissues of oral and oropharyngeal squamous cell carcinoma, which was verified by three data sets GSE42743, GSE9844, and GSE6791 (Feng et al., 2017). One of the introns of LINC00478, Let-7c, can exert a tumor suppressor effect by targeting oncogenes (Kumar & Purohit, 2014). It has also been reported that LINC00478 binds to androgen to down-regulate the expression of the miR-99a/let7c/125b-2 cluster produced by introns of LINC00478, then consequently prostate cancer growth factors IGF1R (targeted directly by miR-99a/let7c/125b-2 cluster) derepresses, triggering a series of downstream signaling cascades, which may contribute to prostate cancer progression (Sun et al., 2014). In our study, MIR145 was significantly down-regulated in breast cancer tissues (—log2FC—>16). MIR145 with Ensembl ID ENSG00000269936 (see Table 1) is miRNA-145′s host gene, and miRNA-145 could be used as a candidate biomarker for breast cancer diagnosis (Xiong et al., 2017). It has been reported that miR-SNP rs353291 (located in miRNA-145) is associated with the white population’s susceptibility to breast cancer (Chacon-Cortes et al., 2015). Studies on breast cancer cell lines have shown that miRNA-145 has a regulatory effect on genes that regulate apoptosis (Wang et al., 2009). A study found that the promoter region of MIR145 is hypermethylated in breast cancer clinical samples and breast cancer cell lines, and miRNA-145 is clearly down-regulated, which can inhibit breast cancer cells migration and invasion by directly targeting angiopoietin 2 gene (ANGPT2) (Liu et al., 2017). Our functional enrichment analysis of MIR145 co-expressed mRNAs also showed it has a significant correlation with biology pathways including negative regulation of blood vessel endothelial cell migration.
In addition, we also discovered a small number of reports on AC093850.2, MAGI2-AS3, and PGM5-AS1. A report has shown that MAGI2-AS3 inhibited the proliferation of breast cancer (Liu et al., 2017). MAGI2-AS3 has been reported to be a hub gene of the triple negative breast cancer lncRNA-mRNA co-expression network, and triple-negative breast cancer patients with elevated levels of MAGI2-AS3 have better recurrence-free survival rate (HR = 0.51) (Du et al., 2016). There are also reports that MAGI2-AS3 was down-regulated in cancer tissues of patients with bladder cancer, and it had low expression level and consequently poor prognosis (Wang et al., 2018a). As an immune-related lncRNA, PGM5-AS1 has prognostic value for regenerative glioma. The expression of PGM5-AS1 in the high-risk group was higher than that in the low-risk group (Wang et al., 2018b). Moreover, patients with high expression of PGM5-AS1 had lower OS (Zhu et al., 2017). At present we have not seen reports of four lncRNAs such as AL035610.1, RP11-175K6.1, AC005550.4, and MIR497HG. According to our functional enrichment analysis, AC093850.2 co-expressed mRNAs were closely related to biology pathways such as collagen metabolic process and glycosaminoglycan binding. Very similar to AC005550.4 co-expressed mRNAs, AL035610.1 co-expressed mRNAs are mainly involved in biological pathways including the metabolism of carbohydrates, lipids, alcohol, coenzyme metabolism, and coping peptides. RP11-175K6.1, closely related to the regulation of circadian rhythm, participated in the protein kinase B signaling pathway, regulated angiogenesis, and controlled the response of cells to steroid stimulation. These results provide guidance for further studies of the action mechanism of these lncRNAs in breast cancer.
It is worth noting that the six lncRNAs including MAGI2-AS3, PGM5-AS1, AL035610.1, RP11-175K6.1, AC005550.4, and MIR497HG were closely related in the WGCNA network, and we preliminarily deduced that their functions are similar, and this provides a good molecular function module for exploration of the disease mechanisms of breast cancer. These new lncRNAs (AL035610.1, RP11-175K6.1, AC005550.4, and MIR497HG) and the less frequently reported lncRNAs (AC093850.2, MAGI2-AS3, and PGM5-AS1) provide us with new ideas for understanding breast cancer. However, their mechanism of action remains to be explored, and their clinical potential in breast cancer needs further validation.
The K-M survival analysis and univariate cox analysis in this study showed that the core lncRNA AC093850.2, whose expression was significantly up-regulated in breast cancer (—log2FC—>6.5), has a certain prognostic value for breast cancer patients. Guo-Wei Huang (Huang et al., 2018) reported a result validated in a GEO dataset (GSE53624) that AC093850.2 was closely associated with OS and DFS in patients with esophageal squamous cell carcinoma. They found that with elevated AC093850.2 levels, the patient’s overall survival became shorter, and the relapse happened earlier. Moreover, their results indicate that knockdown of AC093850.2 had an inhibitory effect on both cell proliferation and cell migration. Other studies have shown that AC093850.2 is associated with survival in patients with invasive breast carcinoma (Zhao et al., 2018c).
To further understand the roles of core lncRNAs in tumors, we analyzed the expression levels of core lncRNAs in 31 tumor tissues and adjacent non-cancerous tissues based on GEPIA. According to the results of GEPIA, the expression pattern (up- or down-regulation) of AC093850.2, PGM5-AS1, and MIR143HG in other cancers was consistent with that in breast cancer. MAGI2-AS3, MIR143HG, and MIR497HG had significant differences in the expression in a variety of cancers. Based on these results, we deduced that they might have primarily been involved in the underlying pathological processes of cancer development. In particular, the expression of MIR143HG was significantly down-regulated in 16 cancers including breast cancer, suggesting that MIR143HG may be an important tumor suppressor gene. At the same time, we evaluated the single-factor prognostic value of core lncRNA in various tumors based on GEPIA and found 7 core lncRNAs (AC093850.2, MAGI2-AS3, PGM5-AS1, AL035610.1, MIR143HG, RP11-175K6.1, MIR497HG) has a good prognostic value for a variety of tumors other than breast cancer. This greatly expands our understanding of these core lncRNAs and provides a bioinformatics basis for further in-depth researches.
Conclusion
In summary, our results indicate that seven core lncRNAs (LINC00478, PGM5-AS1, AL035610.1, MIR143HG, RP11-175K6.1, AC005550.4, and MIR497HG) have good single-factor diagnostic value for breast cancer, while AC093850.2 has a certain prognostic value for breast cancer. AC005550.4 and MIR497HG could better distinguish the breast cancer patients in early-stage from the advanced-stage. Low expression of MAGI2-AS3, LINC00478, AL035610.1, MIR143HG, and MIR145 may be associated with lymph node metastasis in breast cancer. These findings not only provide potential biomarkers for the future diagnosis and prognosis of breast cancer, but also provide a new perspective for elucidating the molecular mechanisms underlying.
Supplemental Information
Analysis results of 22 lncRNAs with the highest diagnostic value for breast cancer (AUC > 0.95)
FC: fold change. P > 0.05.