
Patterns of hepatitis B virus S gene escape mutants and reverse transcriptase mutations among genotype D isolates in Jordan
- Published
- Accepted
- Received
- Academic Editor
- Bettina Böttcher
- Subject Areas
- Virology, Epidemiology, Gastroenterology and Hepatology, Infectious Diseases
- Keywords
- Hepatitis B, HBV, Mutation, Phylogeny, Epidemiology
- Copyright
- © 2019 Ababneh et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2019. Patterns of hepatitis B virus S gene escape mutants and reverse transcriptase mutations among genotype D isolates in Jordan. PeerJ 7:e6583 https://doi.org/10.7717/peerj.6583
Abstract
Background
Hepatitis B virus (HBV) is an important infectious cause of morbidity and mortality in Jordan. HBV genotype D is the most prevalent in the country. Virus escape mutants in the HBV S region is an important public health problem halting preventive efforts. The aim of the current study was to investigate patterns of HBV escape and resistance mutations and to assess domestic transmission of the virus.
Methods
Patients infected with HBV were recruited at Jordan University Hospital (n = 56) and were diagnosed during (1984–2012). A total of 37 partial HBV S sequences were generated using Sanger’s method. Mutation analysis was done using the HIV grade HBV drug resistance interpretation online tool and Geno2pheno (HBV) online tools. Domestic transmission of HBV was assessed using maximum likelihood phylogenetic inference with similar GenBank sequences.
Results
Genotyping revealed an exclusive presence of sub-genotype D1. Typical HBV escape mutants were identified in seven patients. These mutations included: L109R, Q129R, M133L, S143L and D144E with overall prevalence of 18.9% (95% CI [9.5–34.2]). Reverse transcriptase (RT) sequence analysis showed mutations in three patients with overall prevalence of 8.1% (95% CI [2.8–21.3]). RT mutations included: V173L, S202I, L180M, M204V and T184A. Transmission cluster analysis revealed a relatively high proportion of infections taking place as a result of domestic spread (29.7%).
Conclusions
Based on our findings, RT mutation analysis appears to be of high value before the initiation of therapy in patients with chronic HBV infection in Jordan. Phylogenetic analyses revealed a considerable proportion of local spread in the country which should be considered in the preventive infection control efforts.
Introduction
The infection by hepatitis B virus (HBV) is considered a major cause of hepatic-related morbidity and mortality globally with an estimated 257 million people living with chronic infection and approximately 887,000 HBV-related deaths by the end of 2015 (World Health Organization, 2017).
The distinguished features of HBV among other human viruses can be summarized as follows: HBV genome is a partially double-stranded circular DNA of extremely small size (about 3,300 bases) with overlapping open reading frames. In addition, HBV replication cycle is peculiar in respect of an intermediate step involving an error-prone reverse transcriptase (RT), and is manifested in a faster evolution compared to other DNA viruses (Fares & Holmes, 2002; Zhou & Holmes, 2007). The high evolutionary rate of HBV have resulted in its diversification in the human population with emergence of several genotypes (A–J), based on inter-genotype sequence variation of more than 8% (Okamoto et al., 1988; Schaefer, 2007; Stuyver et al., 2000; Tatematsu et al., 2009; Yu et al., 2010). The HBV genotypes are further split into subgenotypes designated by numbers based on intra-genotype sequence variation of 4–8% (Norder et al., 2004). Moreover, recombination events have expanded the genetic diversity of HBV with evidence of substantial influence on shaping the evolutionary history of the virus (Simmonds & Midgley, 2005).
The unique features of HBV genome, particularly for the wide spread of overlapping open reading frames, have implications on treatment and prevention of the virus spread (Liang, 2009). This is particularly evident as follows: mutations arising in the RT region as a result of selective pressure by antiviral drugs can end up in the emergence of virus escape mutants in the adjacent S region with subsequent lack of response to HBV vaccine (Caligiuri et al., 2016; Croagh, Desmond & Bell, 2015). Among the most commonly described mutations in the RT region are mapped at the codons I169, L180, A181, T184, S202, M204, N236 and M250 (Locarnini & Yuen, 2010). These mutations can arise due to inadequate HBV therapy particularly with drugs that have low genetic barrier for resistance (lamivudine; 3TC) with some mutations having enhanced replication and competence effect on the virus (Bock et al., 2002; Locarnini & Yuen, 2010; Luber, 2005).
It is now confirmed by several reports (Lin et al., 2005; Luongo et al., 2015; Ye, Shang & Li, 2015), that transmission of HBV can occur despite successful vaccination as evidenced by the persistence of hepatitis B surface antibody (anti-HBs) in the patients. In addition, mutant viruses may evade serologic detection by the currently used enzyme immunoassays (Sheldon & Soriano, 2008; Teo & Locarnini, 2010). This can end up in the occurrence of occult hepatitis B infection (Purdy, 2007). On the other hand, a few studies indicated that the public health concerns of escape mutants might not be as previously feared (Leong, Lin & Nguyen, 2016). The prototypic and most stable vaccine-escape mutant is G145R (Hsu et al., 2004; Purdy, 2007). Among other commonly detected vaccine-escape mutants are: P120Q, Q129H, F134Y/L, S143L and D144A/E (Coppola et al., 2015).
In Jordan, the sero-prevalence of chronic HBV was estimated to range between 1.4% and 3.5% indicating a low-intermediate endemicity of the virus (Batayneh & Bdour, 2002; Hamoudi, Ghazzawi & Hamoudi, 2016; Hayajneh, Masaadeh & Hayajneh, 2010). Few studies investigated the risk factors for HBV acquisition in the country and the results pointed to horizontal familial transmission, unhygienic dental care, long-term hemodialysis and living abroad for at least 1 year as the risk factors most frequently associated with the spread of the virus (Al Hijazat & Ajlouni, 2008; Hayajneh, Masaadeh & Hayajneh, 2010). Similar to countries in the Middle East and North Africa (MENA) region, genotype D appeared to be predominating HBV genotypes in Jordan despite the low number of studies investigating this question (Hamoudi, Ghazzawi & Hamoudi, 2016; Lin & Kao, 2015; Masaadeh, Hayajneh & Alqudah, 2008).
Phylogenetic inference can be used to investigate the proportion of domestic spread of viruses and to test hypotheses related to variables that are associated with higher likelihood of transmission (Lin et al., 2005; Pybus & Rambaut, 2009; Sallam et al., 2017).
The aim of the current study was to investigate the patterns of HBV S gene escape mutants and the antiviral drug resistance mutations among HBV isolates in Jordan. In addition, we aimed to investigate possible risk factors and proportion of local transmission of the virus in the country.
Methods
Study population
A total of 76 individuals with chronic HBV infection that were diagnosed between 1984 and 2012 were included in the study. Of those, 56 were followed-up completely till the study closure and provided serum samples. Diagnosis of chronic infection was defined as the presence of hepatitis B surface antigen (HBsAg) for more than six months. Serum samples were collected between 2010 and 2012. Demographic and clinical data were collected and included information on age, gender, year of diagnosis, possible risk factor(s), antiviral drug treatment status and the viral load. The year of study closure was 2012.
Ethical permission
The study was approved by the Jordan University Hospital ethical review board (IRB/13/2010) in accordance with the declaration of Helsinki. An informed consent was obtained from all study subjects.
Viral DNA extraction, amplification and sequencing
HBV DNA was extracted from serum using a QIAamp DNA blood mini kit (QIAGEN, Hilden, Germany). Spin columns were loaded with 200 μL of serum. Following treatment of samples according to the manufacturer’s protocol, HBV DNA was eluted in 40 μL of HyPure water (Thermo Fisher Scientific, Waltham, MA, USA). The S region was amplified using primers P1 (nucleotide positions: 1240–1260, 5′-GCGCTGCAGAAGGTTTGTGGCTCCTCTG-3′) and P2 (nucleotide positions: 1928–1948, 5′-GAGTAACTCCACAGTAGCTCC-3′) for the polymerase chain reaction (PCR). The PCR details were as follows: 95 °C for 2 min to activate the enzyme, followed by 35 cycles (denaturation at 95 °C for 30 s, primer annealing at 56 °C for 30 s and elongation 72 °C for 35 s) and finally 10 min at 72 °C. The amplification reaction was performed using a total volume of 25 μL with three μL of DNA template from the extraction in addition to 22 μL of the reaction mix (DNase/RNase free H2O 14.4 μL + GoTaq Reaction Buffer 5.0 μL + primer 1 (P1, 100 ng/μL) 1.0 μL + primer 2 (P2, 100 ng/μL) 1.0 μL + 10 μM dNTPs 0.5 μL + GoTaq DNA Polymerase (5U/μL) 0.1 μL using a locally optimized protocol modified from the manufacturer’s instructions) (Promega, Madison, WI, USA). A volume of 1.0 μL of the final product was taken for electrophoresis using 1% agarose gel for evaluation of suitability of DNA for sequencing. The expected amplicon size was 700 bp. Prior to sequencing, all PCR products were purified. To ensure a DNA concentration ideal for sequencing, the purified products were titrated by Nanodrop. Sequencing was performed with primers P1 and P2 using a BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) according to manufacturer’s instructions through Macrogen commercial sequencing facility (http://foreign.macrogen.co.kr/eng/). Sequence material was assembled using BioEdit software.
Virus genotyping and mutation analysis
Genotyping was done using HBV database genotyping tool available online (https://hbvdb.ibcp.fr/HBVdb/). Genotype assignments were confirmed using GenBank BLAST tool and the HBVseq tool from the HIV Drug Resistance Database (Shafer, 2006). Escape mutant analysis and drug resistance analysis were conducted using HIV-GRADE HBV drug resistance interpretation online tool and Geno2pheno (HBV) online tool (Neumann-Fraune et al., 2014; Obermeier et al., 2012).
Phylogenetic analysis of possible transmission links
Phylogenetic inference of possible links among the Jordanian sequences was performed using the maximum likelihood (ML) approach as implemented in PhyML 3.0 (Guindon et al., 2010). A search for similar HBV GenBank sequences was done using the BLAST tool, with retention of the best 10 target sequences. The redundant sequences were removed to exclude potential multiple intra-patient sequences, using the Skipredundant tool from EMBOSS package with 0.98 as the similarity cut-off (Rice, Longden & Bleasby, 2000). The final dataset comprising the Jordanian sequences with similar reference sequences was subjected to five runs of ML analysis using the GTR + G + I nucleotide substitution model with an estimated proportion of invariable sites (p = 0.695). Statistical support of the nodes in phylogenetic trees was estimated using the approximate Likelihood Ratio Test Shimodaira-Hasegawa like (aLRT-SH) with 0.85 as the significance level (Anisimova et al., 2011). The ML tree with the highest likelihood was retained for subsequent analysis.
Statistical analysis
The 95% confidence interval of the prevalence (Wilson score interval, binomial distribution) was calculated using EpiTools epidemiological calculator available online (http://epitools.ausvet.com.au).
Sequence accession numbers
A total of 37 sequences analyzed in this study were deposited in GenBank. These sequences were assigned with the following accession numbers: MK033359–MK033395.
Results
Characteristics of the study population
Out of 76 subjects who were initially enrolled in the study, 56 study subjects who were diagnosed with HBV infection at Jordan University Hospital during 2010–2012, had serum samples and were included for subsequent analysis. The characteristics of the study subjects are illustrated in (Table 1). A total of 75% of the study subjects were males. The median age at the time of diagnosis was 36 years (mean: 39 years, range: 11–77 years). The median time between diagnosis and sampling was 24 months (range: 1–312 months, with three study subjects lacking information).
Determination of HBV genotype
The HBV RT sequences that were utilized in our study were generated using the Sanger population sequencing method. We were able to successfully obtain a total of 37 sequences (66% success rate) with open reading frames each having a final length of 573 bp (RT domain codons: 25–217; HBs antigen (SHB protein) codons: 17–208). All Jordanian HBV sequences were of genotype D using the various genotyping analysis tools. Further analysis based on Geno2pheno (HBV) 2.0 and GenBank blast tools revealed that all Jordanian sequences were of sub-genotype D1. Of the 37 HBV sequences characterized in our study, 10 sequences (27%) were retrieved from individuals who were treatment-naïve, 10 sequences (27%) were retrieved from individuals with chronic HBV infection that underwent treatment.
RT mutation analysis
Using the Geno2pheno HBV drug resistance tool and HIV grade HBV drug resistance interpretation tool, three patients (8.1% (95% CI [2.8–21.3])), were found to harbor at least a single RT mutation conferring possible or confirmed resistance to RT inhibitors. The details of these mutations are in (Table 2). The first patient was a male with history of treatment with lamivudine who had mutations at established drug resistance positions: L180M, T184A, M204V which confer resistance to lamivudine, entecavir and telbivudine. The second patient was a female with unknown history of treatment who had mutations at established drug resistance positions: L180M, M204V which confer resistance to lamivudine, telbivudine and partial resistance to entecavir. The third patient was a male with history of treatment with lamivudine who had mutations at established drug resistance positions: V173L, L180M, M204V which confer resistance to lamivudine, telbivudine and partial resistance to entecavir.
| Sample ID | Age | Gender | Year of Dx1 | Sub-genotype | Rx status2 | Mutations RT3 | Escape mutations |
|---|---|---|---|---|---|---|---|
| 22 | 38 | Male | 2009 | D1 | Naïve | N53S, F122I, H124D, Y135S, H216L, L217F | – |
| 39 | 35 | Male | 2009 | D1 | Unknown | Y135S, H216L, L217F | – |
| 84 | 35 | Male | Unknown | D1 | Unknown | F122I, H124Y, Y135S, H216L, L217F | – |
| 97 | 36 | Male | 1995 | D1 | Naïve | R110G, Y135S, H216L | – |
| 23 | 68 | Male | 2009 | D1 | Unknown | F122I, H124Y, Y135S, H216L | – |
| 66 | 38 | Male | 2004 | D1 | Unknown | H124Y, Y135S, I163V, H216I, L217F | – |
| 85 | 33 | Female | 2004 | D1 | Naïve | S119P, H124Y, Y135S, H216L | – |
| 99 | 33 | Male | 2007 | D1 | Experienced (Lamivudine) | F122I, H124Y, Y135S, L217F | – |
| 4 | 30 | Male | 2006 | D1 | Naïve | L91I, H124Y, N131D, Y135S, H216L, L217F | – |
| 24 | 22 | Female | 2009 | D1 | Experienced (Lamivudine) | Y135S, H216I, L217F | – |
| 45 | 38 | Female | 1998 | D1 | Experienced (Lamivudine) | H124Y, Y135S, Q215H, H216L | – |
| 101 | 30 | Male | 2010 | D1 | Unknown | F122I, H124Y, Y135S, H216L, L217F | – |
| 2 | 60 | Male | 2009 | D1 | Experienced (Lamivudine) | Y135S, H216L, L217F | R122K |
| 46 | 35 | Female | 2000 | D1 | Experienced (Lamivudine) | H124Y, Y135S, Q215H, H216F, L217W | – |
| 57 | 39 | Female | 2010 | D1 | Unknown | A38E, Y54H, M129L, Y135S, V173L, H216L | – |
| 87 | 30 | Male | 2011 | D1 | Unknown | K32M, Y135S, S213T, H216I | – |
| 106 | 47 | Male | 2011 | D1 | Unknown | F122I, H124Y, Y135S, H216L | – |
| 6 | 13 | Male | 2009 | D1 | Experienced (Lamivudine) | H124Y, Y135S, Q149K, H216L | – |
| 49 | 32 | Female | 2008 | D1 | Unknown | S85A, Y135S, N139D, L199V, H216I, L217W | L109R |
| 76 | 51 | Female | 2009 | D1 | Naïve | L91I, H124D, Y135S, K212N, H216P | S143L |
| 81 | 38 | Male | 1998 | D1 | Experienced (Lamivudine) | S78T, F122I, H124Y, Y135S, H216I | – |
| 83 | 51 | Male | 2000 | D1 | Unknown | A38E, R110G, F122I, H124Y, Y135S, Q149K, S176T, S185I, H197P, C198R, S202I, V208I, A211G, K212G, S213Q, V214Y, Q215L, H216L, L217F | – |
| 5 | 11 | Male | 2009 | D1 | Experienced (Lamivudine) | Y135S, H216L, L217F | – |
| 34 | 26 | Male | 2008 | D1 | Unknown | F122I, H124Y, Y135S, H216L | – |
| 52 | 25 | Male | 2009 | D1 | Naïve | H124Y, Y135S, H216P | – |
| 70 | 44 | Male | 2011 | D1 | Naïve | Y54G, N76D, Y135S, Y141F, H216I, L217F | M133L |
| 89 | 24 | Male | 2009 | D1 | Unknown | Y54H, Y135S, H216L | D144E |
| 105 | 48 | Female | 2011 | D1 | Naïve | H124Y, T128P, Y135S, L145M, H216P | – |
| 8 | 60 | Male | 1984 | D1 | Experienced (Lamivudine) | A38E, Y54H, H124Y, M129L, Y135S, V173L, L180M, M204V, H216L | – |
| 35 | 29 | Male | 2009 | D1 | Unknown | S109P, Y135S, H216L, L217F | – |
| 56 | 36 | Male | 2010 | D1 | Unknown | T37A, F122L, N123D, Y135S, H216L | – |
| 77 | 65 | Male | 2011 | D1 | Naïve | H124Y, Y135S, G152K, K154N, I187L, H216P, L217F | – |
| 103 | 23 | Female | 2011 | D1 | Naïve | N53K, F122I, H124Y, Y135S, H216L | – |
| 63 | 77 | Male | 2004 | D1 | Unknown | H124Y, Y135S, Q215H, H216T, L217F | R122K |
| 38 | 34 | Female | 2007 | D1 | Unknown | F122I, H124Y, Y135S, H216I, L217W | – |
| 58 | 65 | Female | 2006 | D1 | Unknown | V27A, A38E, S117N, N118D, H124D, Y135S, S137T, L180M, M204V, H216L | R122K, Q129R |
| 104 | 37 | Male | 2007 | D1 | Experienced (Lamivudine) | A38E, H124Y, Y135S, L180M, T184A, M204V, H216I | – |
HBsAg escape mutations
The overall prevalence of typical HBsAg escape mutants was 18.9% (95% CI [9.5–34.2]). The R122K mutation which affects HBV detection was found in three patients. The L109R mutation that is a vaccine-escape mutant was found in a single patient. The S143L mutation which is both a vaccine-escape mutant and affects HBV detection was found in a single patient. The M133L and D144E mutations that are vaccine-escape mutants, affecting HBV detection and immunoglobulin therapy were found each in a single patient. Finally, a single patient had Q129R mutation that is a vaccine-escape mutant and affects HBV detection besides the R122K mutation.
Possible domestic transmission of HBV in Jordan
To estimate the proportion of HBV sequences that are possibly linked in transmission clusters indicating domestic transmission, ML analysis was conducted which revealed that the proportion of phylogenetic clustering was 29.7% (11 of the 37 Jordanian sequences; Fig. 1). The clustering sequences were distributed among a dyad (two sequences) and a network of 10 sequences (with nine Jordanian sequences and a single Israeli sequence). The non-clustering Jordanian sequences were present in supported monophyletic clades together with sequences that were collected in Iran, Turkey, Syria and Egypt (Fig. 1).
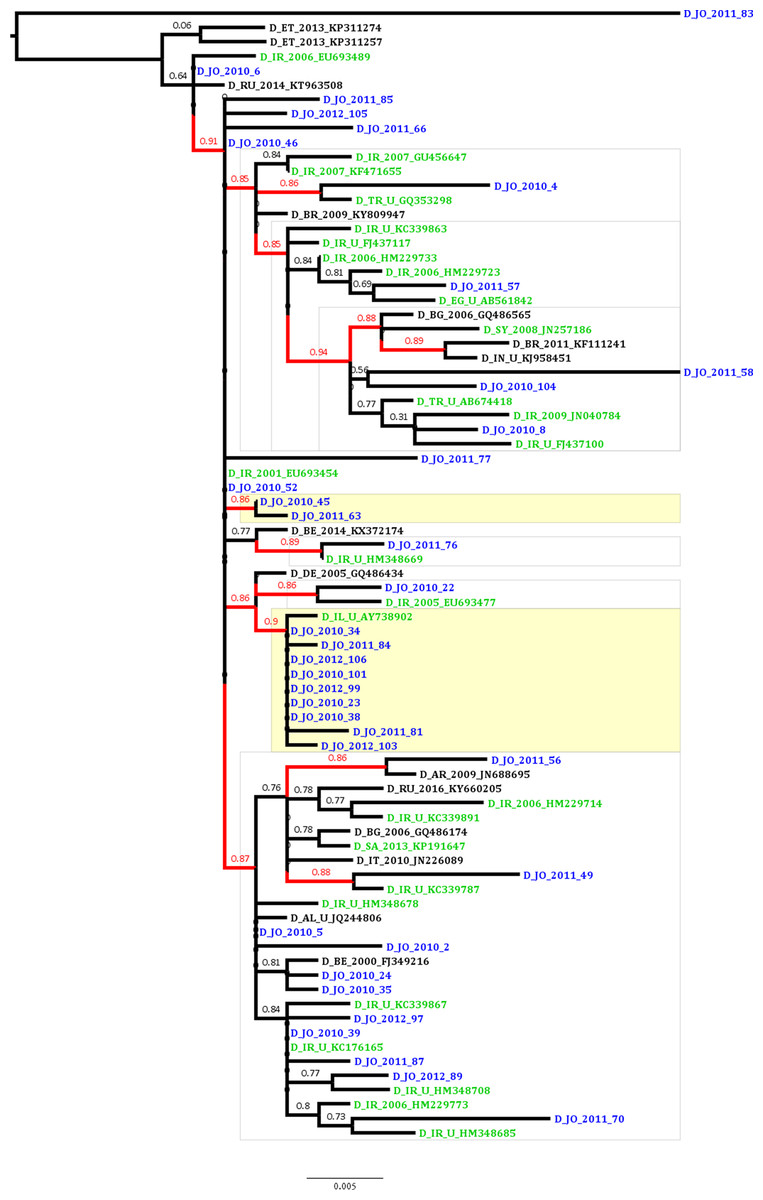
Figure 1: Maximum likelihood tree of the Jordanian HBV sequences with similar GenBank sequences.
The Jordanian sequence names are highlighted in blue color. The statistically supported branches (with approximate Likelihood Ratio Test Shimodaira-Hasegawa like values ≥ 0.85) are highlighted in red color. The GenBank sequences collected in the Middle East and North Africa (MENA) region are highlighted in green color.{kind=link}
Discussion
The epidemiological and antiviral drug resistance data of HBV in Jordan are limited. The results of our study are strengthened by the good coverage of sampling from different regions in the country (Fig. 2). Genotyping results showed that subgenotype D1 was found exclusively, which is consistent with previous studies from Jordan and the MENA region where this sub-genotype predominates (Abdelnabi et al., 2014; Al Baqlani et al., 2014; Asaad et al., 2015; Ciccozzi et al., 2014; El-Mowafy et al., 2017; Hamoudi, Ghazzawi & Hamoudi, 2016; Ziaee et al., 2016). The finding that all HBV isolates in our study were exclusively of genotype D is possibly related to origins of infections in the MENA in which genotype D predominates. This is supported by the finding of intermingling of sequences from Iran, Turkey, Syria and Saudi Arabia (Fig. 1).
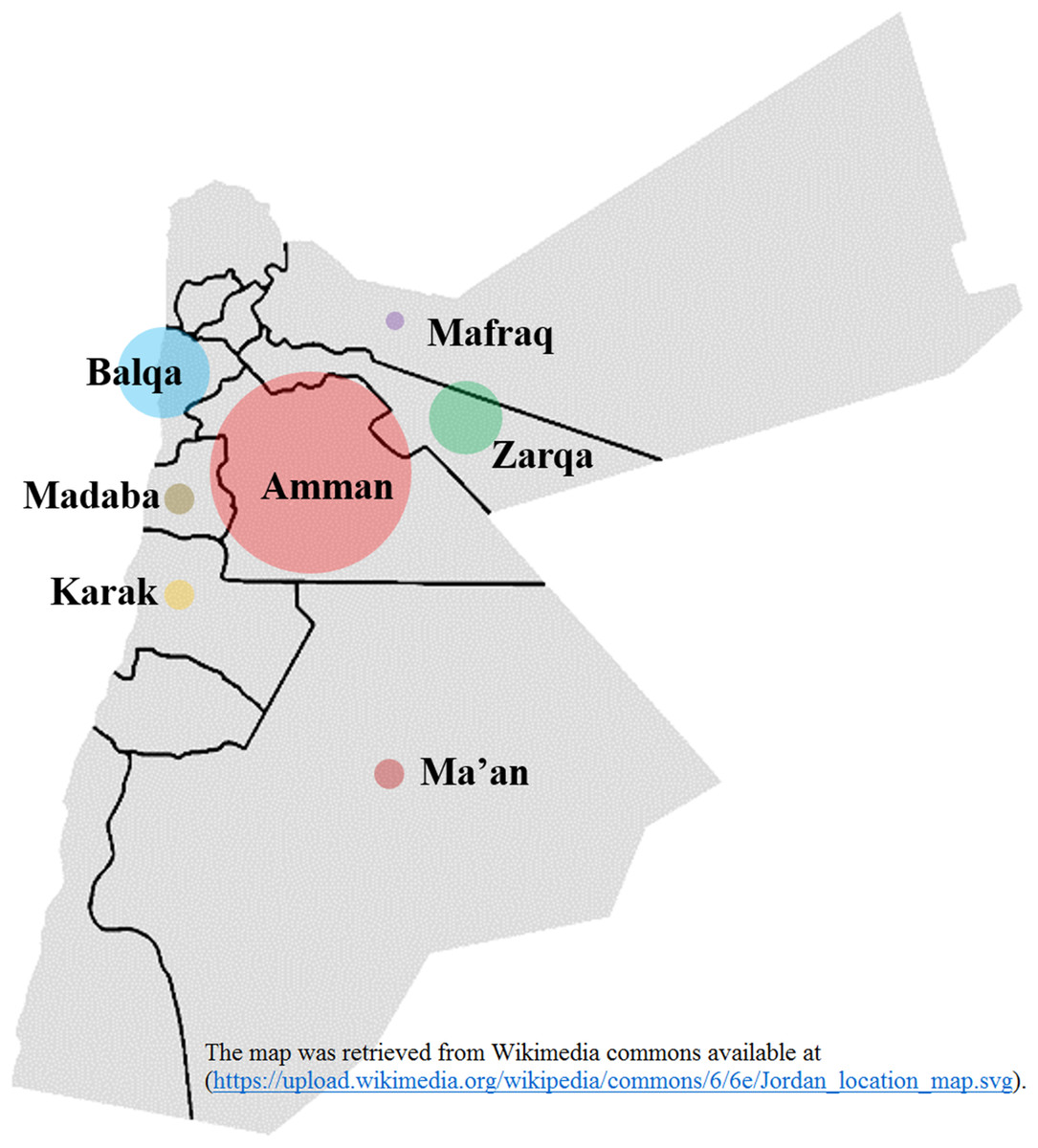
Figure 2: Hepatitis B virus sample distribution from different Jordanian governorates.
The diameter of each circle is proportional to the number of samples; Amman: 25, Balqa: 11, Zarqa: 9, Madaba: 3, Ma’an: 3, Karak: 3, Mafraq: 1. The map was retrieved from Wikimedia commons available at: (https://upload.wikimedia.org/wikipedia/commons/6/6e/Jordan_location_map.svg).{kind=link}
{kind=link}
In the current study, we investigated the patterns of HBsAg escape mutants and HBV RT mutations for the first time in Jordan to the best of our knowledge. The prevalence of HBsAg escape mutants in Jordan was 18.9%, with the R122K mutation which affects HBV detection being the most prevalent type. This is comparable to a study done in Iran, which showed that the frequency of 14% major hydrophilic region mutations (MHR), with the most frequent ones of P120T/S and R122K/T (Moradi et al., 2012). In Turkey, the most frequent mutations observed were T143M and K122R, whereas in Egypt 14.8% presented with mutations in the MHR, and eight different mutations were discovered: R122K, S143L, L109P, S114P, S117N, P127S, P127T and Y134F (Zeid, Ramadan & Shemis, 2016). Previous studies have shown the significance of R122 as a serologic determinant for HBV subtype, with subsequent effect on the antigenicity and immunogenicity of the virus (Ashton-Rickardt & Murray, 1989; Hou et al., 2001; Wu et al., 2012).
The mutations in the HBV RT have been shown to affect the management of HBV infection (Caligiuri et al., 2016; Terrault et al., 2016). For instance, the patients in our study sample who developed HBV RT mutations showed complete or partial resistance to treatment by lamivudine, telbivudine and entecavir. In the three patients with lamivudine resistance mutations, partial or complete resistance to entecavir was detected which is likely related to selection of HBV entecavir mutants in those patients. This finding is in line with recent findings by Geipel et al. (2015).
The most common resistance mutations in Jordan were 180M and 204V, which were always present as co-mutations. Other identified mutations were 173L and 184A mutations, and they were co-mutated with 180M, 204V. M204I/V mutations are frequently accompanied by compensatory mutations in other domains such as 59 rtV173L, rtL180M, rtT184S/G, 58 rtI169T, rtS202I, rtL80V/I and rtQ215S which enhanced the replication efficiency of rt204I/V mutants without significantly affecting lamivudine resistance, by compensating the decrease in efficiency due to resistance-associated changes (Bartholomeusz & Locarnini, 2006a, 2006b; Caligiuri et al., 2016; Zoulim & Locarnini, 2009). For example, dual rtL180M and M204V/I mutants were frequently found in patients in Italy, with three patients having triple mutation of rtV173L, rtL180M and M204V (Quiros-Roldan et al., 2008). Upon comparing the mutant strains with wild HBV strains, patients with rtM204V/I more frequently presented with severe acute hepatitis B and lower serum HBV DNA values (Coppola et al., 2013).
A recent meta-analysis has shown that the incidence of spontaneous primary and secondary mutations among untreated chronic hepatitis B patients was 4.9% for primary mutations of rtM204V/I, while the natural incidence of secondary rtL180M mutations was 2.7% (Zhang et al., 2015). Previous literature suggests that lamivudine is the main cause of YMDD (tyrosine-methionine-aspartateaspartate) mutations (M204I/V) within the catalytic sites (C domain) in HBV P-ORF (Caligiuri et al., 2016; Ji et al., 2011). In Turkey, Japan and China, the a high rates of spontaneous YMDD mutation may increase the unresponsiveness to lamivudine, leading to prolonged therapy time and extra cost (Zhang et al., 2015), for which we need to perform further studies to investigate the probability of development of antiviral resistant mutants worldwide, and to study the effects of the infection with mutant viruses on the clinical course of the disease.
Upon conducting ML analysis to investigate the possibility of domestic transmission of HBV in Jordan, the proportion of phylogenetic clustering was 29.7%. Horizontal transmission is the main mode of transmission of HBV in Jordan (André, 2000). Clustering of chronic HBV infections within the family is common in areas of where HBV is endemic, and horizontal transmission during early childhood is a major route of HBV transmission (Dumpis et al., 2001; Lin et al., 2005; Zampino et al., 2002). The high proportion of clustering among the study subjects highlights the need for intervention measures by public health control strategies to stop the chains of forward transmission of the virus.
Several caveats of our study should be addressed including the low sampling coverage and the short length of RT sequences that were used to conduct the transmission cluster analysis. The study was closed in 2012, which highlights the need for a follow up study to assess the recent trends of genotype and risk factors for HBV infection in the country. Another caveat of our study is the lack of clinical and serological data from the study subjects.
Conclusions
To conclude, we investigated the genotype distribution, risk factors, RT and HBsAg vaccine escape mutations and the transmission of HBV in Jordan over a large area of the country. Based on the findings of our study, we recommend RT mutation analysis before the initiation of therapy in patients with chronic HBV infection in Jordan. Lamivudine monotherapy is strongly discouraged due to high risk of resistance development. In addition, the relatively high proportion of phylogenetic clustering highlights the need for public health control measures to prevent forward transmission of the virus in the country and regionally.