
Molecular evolution of virulence genes and non-virulence genes in clinical, natural and artificial environmental Legionella pneumophila isolates
- Published
- Accepted
- Received
- Academic Editor
- Mario Alberto Flores-Valdez
- Subject Areas
- Ecosystem Science, Environmental Sciences, Evolutionary Studies, Genetics, Microbiology
- Keywords
- Legionella pneumophila, Non-virulence genes, Virulence genes, Molecular phylogeny, Genetic diversity, Molecular evolution
- Copyright
- © 2017 Zhan and Zhu
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2017. Molecular evolution of virulence genes and non-virulence genes in clinical, natural and artificial environmental Legionella pneumophila isolates. PeerJ 5:e4114 https://doi.org/10.7717/peerj.4114
Abstract
Background
L. pneumophila is the main causative agent of Legionnaires’ disease. Free-living amoeba in natural aquatic environments is the reservoir and shelter for L. pneumophila. From natural water sources, L. pneumophila can colonize artificial environments such as cooling towers and hot-water systems, and then spread in aerosols, infecting the susceptible person. Therefore, molecular phylogeny and genetic variability of L. pneumophila from different sources (natural water, artificial water, and human lung tissue) might be distinct because of the selection pressure in different environments. Several studies researched genetic differences between L. pneumophila clinical isolates and environmental isolates at the nucleotide sequence level. These reports mainly focused on the analysis of virulence genes, and rarely distinguished artificial and natural isolates.
Methods
We have used 139 L. pneumophila isolates to study their genetic variability and molecular phylogeny. These isolates include 51 artificial isolates, 59 natural isolates, and 29 clinical isolates. The nucleotide sequences of two representative non-virulence (NV) genes (trpA, cca) and three representative virulence genes (icmK, lspE, lssD) were obtained using PCR and DNA sequencing and were analyzed.
Results
Levels of genetic variability including haplotypes, haplotype diversity, nucleotide diversity, nucleotide difference and the total number of mutations in the virulence loci were higher in the natural isolates. In contrast, levels of genetic variability including polymorphic sites, theta from polymorphic sites and the total number of mutations in the NV loci were higher in clinical isolates. A phylogenetic analysis of each individual gene tree showed three to six main groups, but not comprising the same L. pneumophila isolates. We detected recombination events in every virulence loci of natural isolates, but only detected them in the cca locus of clinical isolates. Neutrality tests showed that variations in the virulence genes of clinical and environmental isolates were under neutral evolution. TrpA and cca loci of clinical isolates showed significantly negative values of Tajima’s D, Fu and Li’s D* and F*, suggesting the presence of negative selection in NV genes of clinical isolates.
Discussion
Our findingsreinforced the point that the natural environments were the primary training place for L. pneumophila virulence, and intragenic recombination was an important strategy in the adaptive evolution of virulence gene. Our study also suggested the selection pressure had unevenly affected these genes and contributed to the different evolutionary patterns existed between NV genes and virulence genes. This work provides clues for future work on population-level and genetics-level questions about ecology and molecular evolution of L. pneumophila, as well as genetic differences of NV genes and virulence genes between this host-range pathogen with different lifestyles.
Introduction
Legionella pneumophila (L. pneumophila) is the main causative agent of Legionnaires’ disease (Fields, Benson & Besser, 2002; Gomez-Valero et al., 2014). It is a gram-negative bacterium in natural water environments such as pools, rivers and lakes, as well as in various artificial water systems worldwide (Gomez-Valero, Rusniok & Buchrieser, 2009). Free-living amoeba in natural aquatic environments is the reservoir and shelter for L. pneumophila. From natural environments, it can colonize artificial water environments such as cooling towers and hot-water systems, and then spread in aerosols, infecting the susceptible person (Escoll et al., 2013; Lau & Ashbolt, 2009). It is clear the intra-amoebal lifestyle of L. pneumophila in natural environments is important to L. pneumophila genome. It provides the primary evolutionary pressure to this microorganism and shapes the genomic structure of this bacterium. In this process, amoeba may act as a gene smelter, allowing diverse microorganisms to evolve by gene acquisition and loss, and then makes L. pneumophila better adapt to the intra-amoebal lifestyle or evolve into new pathogenic forms (Gomez-Valero et al., 2011; Khodr et al., 2016; Richards et al., 2013; Zhan, Hu & Zhu, 2016). Within L. pneumophila lifestyles, artificial environments are an intermediate post for this microorganism from the natural environments to human. Since person-to-person transmission of L. pneumophila has rarely been reported, the infection of human lung tissue may be an evolutive end for L. pneumophila (Correia et al., 2016; Costa et al., 2014). Therefore, the three environments (natural water, artificial water, and human lung tissue) where L. pneumophila inhabits, play different roles in its life and may influence the shape of L. pneumophila genome. Several reports proved that L. pneumophila clinical isolates displayed less genetic diversity than environmental isolates (Coscolla & Gonzalez-Candelas, 2009; Costa et al., 2012; Costa et al., 2014). These results could illustrate that at the evolutive end, there is no more selective pressure to shape the L. pneumophila genome. Another explanation is that isolates of L. pneumophila recovered from clinical cases are a limited and specific subset of all genotypes existing in nature, and they may be an especially adapted group of clones (Coscolla & Gonzalez-Candelas, 2009). However, these reports focused on the analysis of virulence-related genes, and rarely distinguished artificial and natural isolates (Coscolla & Gonzalez-Candelas, 2009; Costa et al., 2012; Costa et al., 2014).
Our goal was to determine the genetic diversity and population structure of L. pneumophila isolates from different sources at none-virulence (NV) gene and virulence gene levels, respectively, and to identify the molecular mechanisms operating in the evolution of these genes. We have studied the genetic and population diversity by comparing nucleotide sequences from five representative gene loci. These loci included two NV loci which were common among a set of bacterial genomes (tryptophan synthase α subunit-encoding gene, trpA and tRNA nucleotidyltransferase gene, cca), and three virulence loci (icmK, lspE, and lssD). These nucleotide sequences were from 51 artificial isolates, 59 natural isolates and 29 clinical or disease-related isolates. The trpA gene controls the sequence of reactions from chorismic acid to tryptophan, and the cca gene catalyzes the accurate synthesis of the -C-C-A terminus of tRNA (Dounce, Morrison & Monty, 1955; Gebran et al., 1994). They all play fundamental roles in the survival of bacteria. The virulence genes belong to different protein secretion systems, including the Dot/Icm type IVB protein secretion system (icmK), the Lsp type II secretion system (lspE), and the type I Lss secretion system(lssD). They represent key virulence and play crucial roles in ecology and pathogenesis of L. pneumophila (De Buck, Anne & Lammertyn, 2007; Fuche et al., 2015; Korotkov, Sandkvist & Hol, 2012).
Our results showed the levels of genetic variability in the virulence loci were higher in natural isolates. In contrast, levels of genetic variability in the NV loci were higher in clinical isolates. Molecular phylogeny of these genes showed three to six main groups, but none of them contained the same origin of L. pneumophila isolates. Intragenic recombination of cca, lssD, lspE and icmK genes was also detected in this study. It was favored as an important evolutionary mechanism for natural and clinical isolates by influencing the population genetic structure of L. pneumophila.
Materials and Methods
L. pneumophila isolates
One hundred and thirty-nine strains of L. pneumophila were enrolled in this study. These isolates included 51 artificial isolates, 59 natural isolates, and 29 clinical isolates. The source natures, geographic locations, collection dates and sequence types (STs) based on the Sequence-Based Typing (SBT) scheme (Gaia et al., 2005; Ratzow et al., 2007) of these isolates are shown in Table S1. Briefly, the 29 clinical strains were isolated between 1947 and 2012, from the non-China regions, including different cities of USA, Germany, UK, France, etc. Their details were obtained from the NCBI database (https://www.ncbi.nlm.nih.gov). The environmental isolates were from 14 different sites in two cities (Guangzhou and Jiangmen) of Guangdong Province, China between October 2003 and September 2007. All the environmental isolates were selected for sequencing partial trpA, cca , lssD, lspE, and icmK genes. We selected the most variable regions of these genes through a sequence alignment with the known sequences in the NCBI database, in order to achieve maximum genetic variability. The variability of the gene regions was measured by calculating the single-nucleotide polymorphisms of the sequence using Dnasp v5 (Librado & Rozas, 2009; Rozas, 2009) (Table S2).
Genomic DNA extraction, PCR, and DNA sequencing
Genomic DNA extraction of the artificial and natural strains was performed as shown in our previous report (Zhan, Hu & Zhu, 2016). PCR was employed to amplify fragments of DNA. The corresponding oligonucleotide primers are shown in Table S2. The PCR was performed using an EasyPfu PCR SuperMix (Transgene Biotech, Beijing, China) according to the manufacturer’s instructions and carried out using the GeneAmp PCR system (MJ Research PTC-200) with the following thermal conditions: 95 °C for 3 min followed by 35 cycles of 95 °C for 20 s, 60 °C for 20 s and 72 °C for 30 s (lspE, lssD, and icmK loci) or 70 s (cca and trpA loci), and a final extension at 72 °C for 5 min. For confirmation purposes, each PCR reaction was performed with a positive control (L. pneumophila strain ATCC33152 genomic DNA as the PCR template) and a negative control (sterile water as the PCR template). PCR products were purified using an EasyPure Quick Gel Extraction (Transgene Biotech, Beijing, China) and then transferred to Guangzhou IGE Biotechnology Ltd for sequencing.
Sequence analysis
The quality of DNA sequencing was manually checked by Chromas (http://technelysium.com.au). The corresponding gene sequences of 29 clinical isolates were obtained from the NCBI database. Multiple sequence alignments were performed using ClustalX 2.1 (Chenna et al., 2003; Thompson et al., 1997). Genetic variability analyses were performed using DnaSP v5 (Librado & Rozas, 2009; Rozas, 2009). Phylogenetic analyses were conducted by a MEGA7 package (Kumar, Stecher & Tamura, 2016). Neighbor-Joining (NJ) phylogenetic trees were obtained for each locus separately with the MEGA7 based on the Kimura 2-parameter model (Kimura, 1980; Saitou & Nei, 1987). The tree was drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. NJ tree nodes were evaluated by bootstrapping with 1,000 replicates. The ratios of synonymous and non-synonymous substitutions were calculated according to the Nei-Gojobori method with Jukes-Cantor correction as implemented in the MEGA7 (Kumar, Stecher & Tamura, 2016).
Molecular evolution analysis
The aligned sequences of the five loci were screened using RDP4 to detect intragenic recombination (Martin et al., 2015; Martin et al., 2017). Six methods implemented in the program RDP4 were utilized. These methods were RDP (Martin & Rybicki, 2000), GENECONV (Padidam, Sawyer & Fauquet, 1999), BootScan (Martin et al., 2005), MaxChi (Smith, 1992), Chimaera (Posada, 2002), and SiScan (Gibbs, Armstrong & Gibbs, 2000). Potential recombination event was considered as that identified by at least two methods according to Coscolla’s report (Coscolla & Gonzalez-Candelas, 2009). Common settings for all methods were to consider sequences as linear, statistical significance was set at the P < 0.05 level, with Bonferroni correction for multiple comparisons and requiring phylogenetic evidence and polishing of breakpoints.
Tajima’s D, Fu and Li’s D* and F* were calculated for testing the mutation neutrality hypothesis as previously described by Coscolla and colleagues (2007). These statistics were calculated with the program Dnasp v5 (Rozas, 2009) using a statistical significance level P < 0.05 and applying the false discovery rate to correct for multiple comparisons, and 1000 replicates in a coalescent simulation. Non-neutrality evolution was considered as that identified by at least two of the methods.
Population structure analysis
Hierarchical analysis of molecular variance (AMOVA) for clinical and environmental sequences using the alignment of the five loci was performed using Arlequin Ver3.5.2 (Excoffier & Lischer, 2010). This analysis provides estimates of variance components and F-statistics analogues speculating the correlation of haplotype diversity at different levels of the hierarchical subdivision. We defined the hierarchical subdivision of these isolates at three levels. At the upper level, the three groups considered were clinical, artificial and natural isolates. As populations within groups, the intermediate level, we reckoned the strains isolated from the same geographic location as subpopulations. Therefore, natural and artificial isolates were both split into two subgroups based on the cities where they were isolated (Guangzhou and Jiangmen subgroups), and clinical strains were also divided into two subgroups based on the continents where they were isolated (eg. America or non-America, including Europe and Australia). The third level corresponded to the different haplotypes which were found within the six subgroups considered in the previous level. The statistical significance of fixation indices was tested using a non-parametric permutation approach (Excoffier, Smouse & Quattro, 1992).
Nucleotide sequence accession numbers
The 550 sequences from L. pneumophila environmental isolates determined in this study were deposited in the GenBank Nucleotide Sequence Database with accession numbers KY708328 –KY708437 (cca), KY708438 –KY708547 (trpA), KY708768 –KY708877 (lssD), KY708658 –KY708767 (lspE) and KY708548 –KY708657 (icmK).
Results
Sequence analysis and genetic variability of L. pneumophila isolates from different sources
In general, we obtained the gene sequences from 29 clinical, 59 natural and 51 artificial environmental isolates. Genetic diversity estimates in all the clinical, natural and artificial isolates are presented in Table 1. The highest nucleotide diversity (π) in the L .pneumophila isolates was found in the lssD locus, varied from 0.04213 to 0.05399, while the lowest nucleotide diversity was found in trpA locus, varied from 0.01041 to 0.01246. Both the most haplotype (h) and highest haplotype diversity (Hd) of the five gene loci was found in natural isolates. For the trpA locus, the nucleotide diversity, number of polymorphic nucleotide sites (S), population mutation ration (θ), average number of pairwise nucleotide differences (k), and the total number of mutations (η) were all higher in clinical isolates. In contrast, another NV locus, cca did not show higher nucleotide diversity and nucleotide differences in clinical isolates, but the number of polymorphic nucleotide sites, population mutation ratio and the total number of mutations were higher in clinical isolates. The highest nucleotide diversity and the average number of pairwise nucleotide differences of cca locus were found in artificial isolates. Different results were found in the three virulence loci: the haplotype, haplotype diversity, nucleotide diversity and the average number of pairwise nucleotide differences were higher in natural isolates.
| Gene type | locus | Strain type | Sequence, (n) | Sequence length | h | Hd | SD of Hd | π | SD of π | S | θ | SD of θ | k | η | dN/dS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NV genes | cca | C | 29 | 1082 | 10 | 0.692 | 0.092 | 0.01749 | 0.00542 | 130 | 0.03059 | 0.00268 | 18.921 | 136 | 0.1034 |
| N | 59 | 1082 | 16 | 0.892 | 0.019 | 0.01935 | 0.00072 | 70 | 0.01392 | 0.00166 | 20.936 | 71 | 0.1085 | ||
| A | 51 | 1082 | 6 | 0.660 | 0.062 | 0.02212 | 0.00439 | 121 | 0.02486 | 0.00226 | 23.929 | 124 | 0.0864 | ||
| trpA | C | 29 | 748 | 9 | 0.690 | 0.091 | 0.01216 | 0.00486 | 76 | 0.02587 | 0.00297 | 9.094 | 82 | 0.0633 | |
| N | 59 | 748 | 14 | 0.852 | 0.026 | 0.01041 | 0.00066 | 44 | 0.01266 | 0.00191 | 7.786 | 47 | 0.1001 | ||
| A | 51 | 748 | 7 | 0.590 | 0.075 | 0.01214 | 0.00374 | 58 | 0.01723 | 0.00226 | 9.078 | 62 | 0.0801 | ||
| Virulence genes | lssD | C | 29 | 330 | 7 | 0.640 | 0.092 | 0.04213 | 0.01229 | 63 | 0.04861 | 0.00612 | 13.901 | 68 | 0.0502 |
| N | 59 | 330 | 11 | 0.793 | 0.039 | 0.05399 | 0.00932 | 72 | 0.04696 | 0.00553 | 17.817 | 79 | 0.0353 | ||
| A | 51 | 330 | 4 | 0.404 | 0.082 | 0.04241 | 0.01122 | 71 | 0.04782 | 0.00568 | 13.994 | 77 | 0.0398 | ||
| lspE | C | 29 | 331 | 7 | 0.608 | 0.100 | 0.02834 | 0.00571 | 37 | 0.02846 | 0.00468 | 9.379 | 39 | 0.0165 | |
| N | 59 | 331 | 13 | 0.839 | 0.031 | 0.03748 | 0.00223 | 49 | 0.03186 | 0.00455 | 12.407 | 53 | 0.0378 | ||
| A | 51 | 331 | 8 | 0.696 | 0.064 | 0.02937 | 0.00608 | 54 | 0.03626 | 0.00493 | 9.721 | 61 | 0.0341 | ||
| icmK | C | 29 | 385 | 9 | 0.690 | 0.091 | 0.03363 | 0.00647 | 65 | 0.04299 | 0.00533 | 12.948 | 71 | 0.0913 | |
| N | 59 | 385 | 50 | 0.989 | 0.008 | 0.04775 | 0.00217 | 77 | 0.04305 | 0.00491 | 18.382 | 104 | 0.2543 | ||
| A | 51 | 385 | 37 | 0.978 | 0.011 | 0.03995 | 0.00655 | 76 | 0.04387 | 0.00503 | 15.380 | 91 | 0.1603 |
Notes:
- h
-
Haplotypes
- Hd
-
Haplotype diversity
- π
-
Nucleotide diversity
- S
-
Polymorphic sites
- θ
-
Theta (per site) from S, population mutation ration
- k
-
Nucleotide differences
- η
-
Total number of mutations
The rates of non-synonymous substitutions per non-synonymous site (dN) were extremely low and different between these genes, ranging from 0.00243 (for lspE in clinical isolates) to 0.0295 (for icmK in natural isolates), despite the relatively similar values of polymorphic sites (37 vs. 77). Synonymous substitutions (dS) ranged from 0.0338 for trpA in natural isolates to 0.3091 for lssD in natural isolates (Table S3). An obviously different dN/dS ratio was observed, ranging from 0.0165 (for lssD in clinical isolates) to 0.2543 (for icmK in natural isolates) (Table 1). We found different dN/dS ratios among clinical, natural, and artificial isolates. dN/dS ratios of the cca, trpA, lspE, and icmK locus were higher in natural isolates than in artificial and clinical isolates (Table 1).
Figure 1: Neighbor-Joining tree of L. pneumophila isolates from DNA sequences of cca locus.
Strain types, source natures and geographic locations of these isolates were shown in Table S1. Bootstrap support values (1,000 replicates) for nodes higher than 50% are indicated next to the corresponding node. Three main groups of the clades could be found.{kind=link}
Figure 2: Neighbor-Joining tree of L. pneumophila isolates from DNA sequences of trpA locus.
Strain types, source natures and geographic locations of these isolates were shown in Table S1. Bootstrap support values (1,000 replicates) for nodes higher than 50% are indicated next to the corresponding node. Six main groups of the clades could be found.{kind=link}
Figure 3: Neighbor-Joining tree of L. pneumophila isolates from DNA sequences of lssD locus.
Strain types, source natures and geographic locations of these isolates were shown in Table S1. Bootstrap support values (1,000 replicates) for nodes higher than 50% are indicated next to the corresponding node. Five main groups of the clades could be found.{kind=link}
Figure 4: Neighbor-Joining tree of L. pneumophila isolates from DNA sequences of lspE locus.
Strain types, source natures and geographic locations of these isolates were shown in Table S1. Bootstrap support values (1,000 replicates) for nodes higher than 50% are indicated next to the corresponding node. Five main groups of the clades could be found.{kind=link}
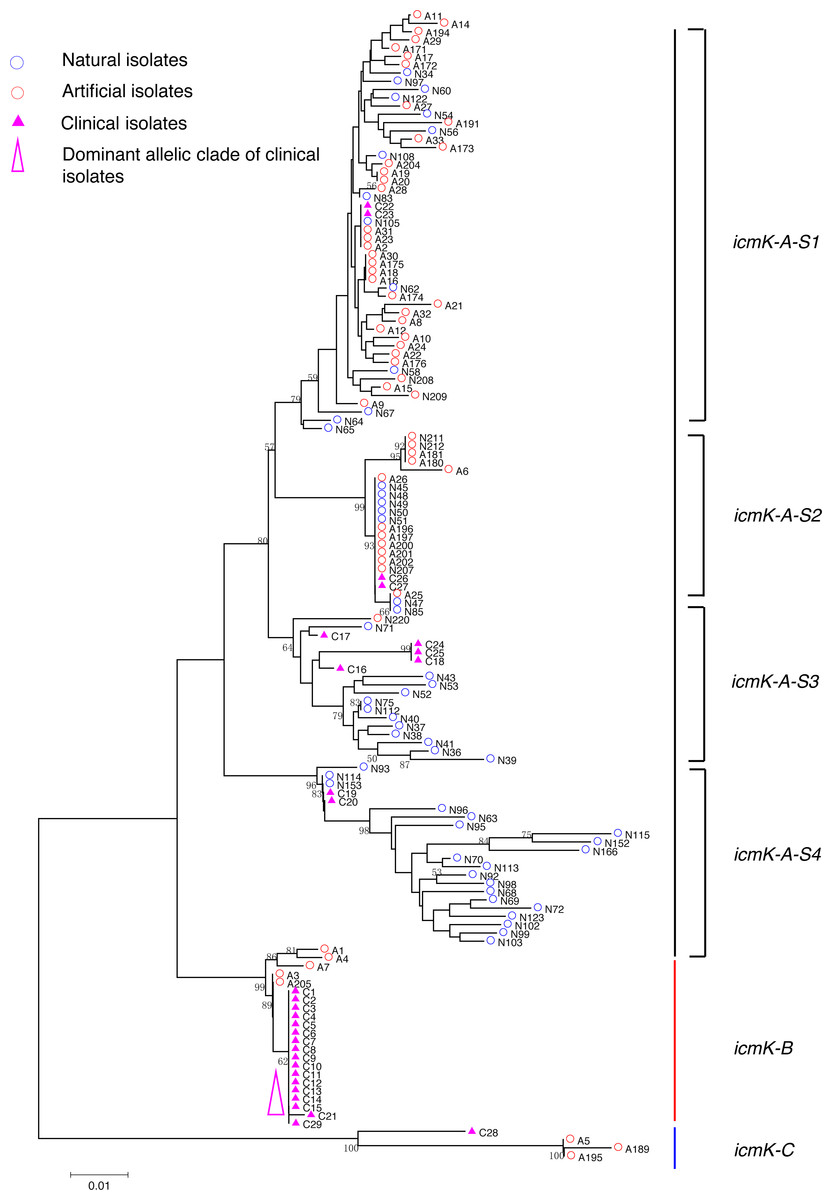
Figure 5: Neighbor-Joining tree of L. pneumophila isolates from DNA sequences of icmK locus.
Strain types, source natures and geographic locations of these isolates were shown in Table S1. Bootstrap support values (1,000 replicates) for nodes higher than 50% are indicated next to the corresponding node. Three main groups of the clades could be found.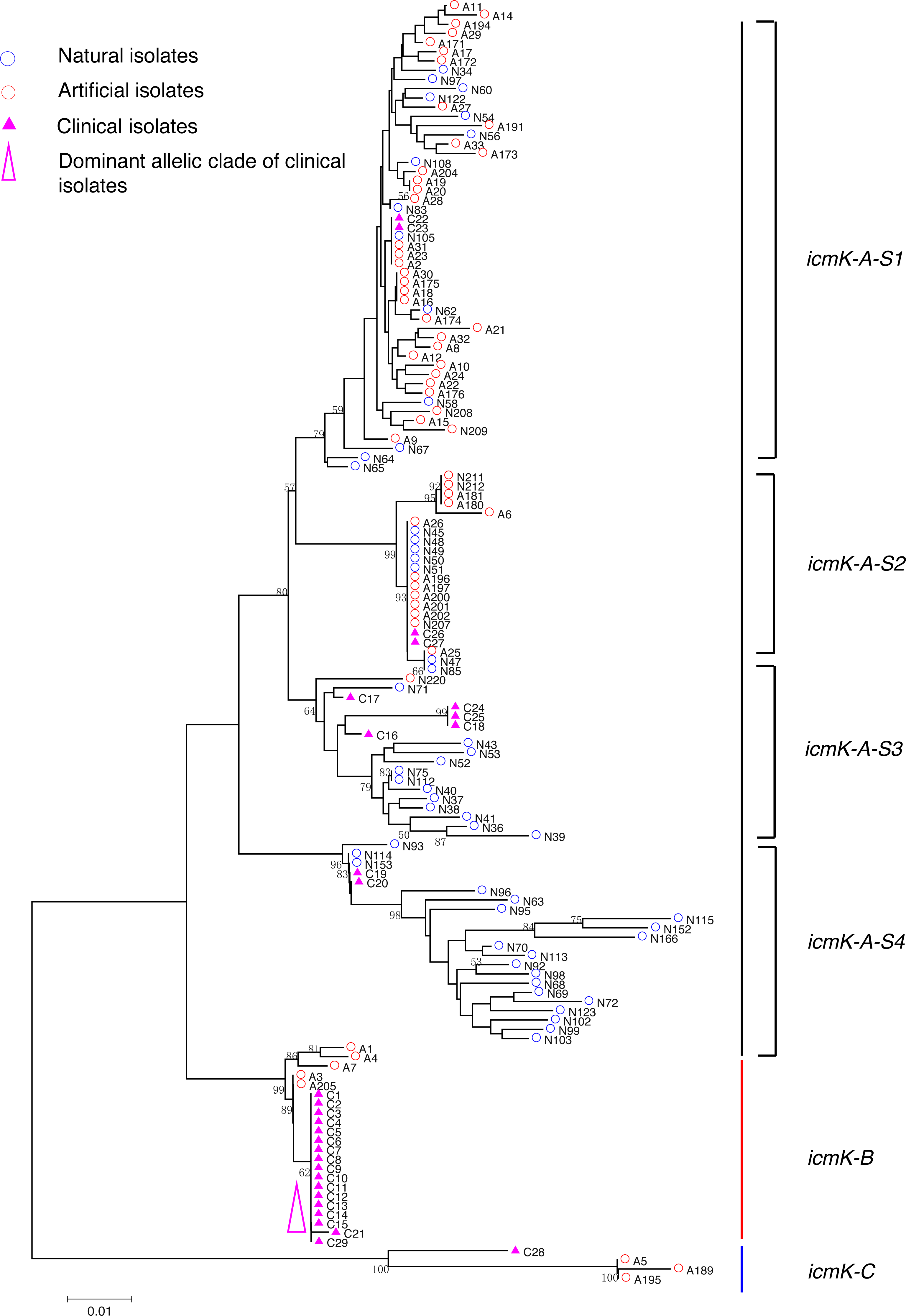{kind=link}
Phylogenetic analysis of the five gene loci
Phylogenetic trees were derived separately for each locus to test the phylogenetic relationships between these isolates. The NJ trees showed three to six main groups (Figs. 1 to 5). The number of haplotypes in these loci ranged from 15 (lssD locus) to 89 (icmK locus). The NV loci displayed a similar number of haplotypes (23 for cca and 22 for trpA), while the virulence loci displayed large variable range of haplotypes (16 for lssD, 19 for lspE and 89 for icmK). More than half of the clinical isolates (16/29 to 18/29, 55.17% to 62.07%) presented a single dominant allelic profile in the five loci (Figs. 1 to 5). Similarly, more than half (27/51 to 38/51, 52.94% to 74.51%) of the artificial isolates presented a distinct dominant allelic profile of the four loci (Figs. 1 to 4). Some of the allelic profiles were unique to clinical, artificial or natural isolates. Six allelic profiles of cca were unique to clinical isolates, comprising 26.09% of all profiles. Two to six allelic profiles of trpA, lssD, lspE and icmK loci were unique to clinical isolates, constituting 6.74% to 25% of the select profiles. A significantly different proportion of unique alleles of the five loci between natural and artificial strains was found (P = 0.0008, paired t-test). 30.43% (7/23), 40.91% (9/22), 40% (6/15), 42.11% (8/19), and 50.56% (45/89) allelic profiles of the cca, trpA, lssD, lspE and icmK loci were unique to natural isolates, respectively. By contrast, only 4.35% (1/23), 13.64% (3/22), 6.67% (1/15), 15.79% (3/19), and 35.96% (32/89) profiles of the cca, trpA, lssD, lspE and icmK loci were unique to artificial isolates. Most of the clinical isolates distributed in group A cluster of cca locus (86.21%, 25/29) and mostly in the subgroup 2 (cca-A-S2, 68.97%, 20/29); group B cluster of trpA locus (68.97%, 20/29) and mostly in the subgroup 2 (trpA-B-S2, 55.17%, 16/29); group B cluster of lssD locus (58.62%, 17/29), group A cluster of lspE locus (79.31%, 23/29) and mostly in the subgroup 2 (lspE-A-S2, 62.07%, 18/29); group B cluster of icmK locus (58.62%, 17/29). Artificial environmental isolates mainly distributed in group B cluster of cca locus (64.71%, 33/51), group A cluster of trpA locus (70.59%, 36/51), group A clusters of lssD locus (76.47%, 39/51), group A clusters of lspE (82.35%, 42/51) and group A cluster of icmK (84.31%, 43/51) (Figs. 1 to 5). The natural isolates in the NJ trees of cca, trpA, lssD, and lspE were more dispersed, while in the NJ tree of icmK, there mainly distributed in the group A, subgroup 3 and 4 clusters (Fig. 5).
Relationships between clinical, natural and artificial isolates
We performed a hierarchical analysis of molecular variance (AMOVA) for the 139 isolates based on five loci considered above. The largest proportion of the genetic variation in each locus was found within populations, as this level accounted for 76.39% to 90.88% of the total variation (Table 2). The proportion of the total genetic variation explained by differences between clinical, artificial and natural isolates was small, ranging from −5.04% for lssD to 18.87% for cca, and it was not significant (Table 2). In contrast, geographical difference contributed to a part of genetic variation, especially for the virulence loci (accounting for 10.38% to 21.78% variation), and was all significant.
| Gene locus | Source of variation | d.f. | Sum of squares | Variance components | Percentage of variation | F-statistics |
|---|---|---|---|---|---|---|
| cca | Among groups | 2 | 259.278 | 2.49795 Va | 18.67 | FCT = 0.18668 |
| Among populations within groups | 3 | 42.796 | 0.25207 Vb | 1.88 | FSC = 0.02316 | |
| Within populations | 133 | 1413.883 | 10.63070 Vc | 79.45 | FST = 0.20552* | |
| Total | 1715.957 | 13.38072 | ||||
| trpA | Among groups | 2 | 66.418 | 0.47727 Va | 9.73 | FCT = 0.05807 |
| Among populations within groups | 3 | 23.622 | 0.25700 Vb | 5.24 | FSC = 0.14977* | |
| Within populations | 133 | 554.392 | 4.16836 Vc | 85.02 | FST = 0.09735* | |
| Total | 644.432 | 4.90263 | ||||
| lssD | Among groups | 2 | 48.770 | −0.41909 Va | −5.04 | FCT = − 0.05043 |
| Among populations within groups | 3 | 73.572 | 1.17713 Vb | 14.17 | FSC = 0.13486* | |
| Within populations | 133 | 1004.356 | 7.55155 Vc | 90.88 | FST = 0.09123* | |
| Total | 1126.698 | 8.30959 | ||||
| lspE | Among groups | 2 | 85.132 | −0.08745 Va | −1.39 | FCT = − 0.01387 |
| Among populations within groups | 3 | 74.467 | 1.37339 Vb | 21.78 | FSC = 0.21481* | |
| Within populations | 133 | 667.660 | 5.02000 Vc | 79.61 | FST = 0.20393* | |
| Total | 827.259 | 6.30594 | ||||
| icmK | Among groups | 2 | 198.660 | 1.34135 Va | 13.23 | FCT = 0.13230 |
| Among populations within groups | 3 | 68.765 | 1.05258 Vb | 10.38 | FSC = 0.11964* | |
| Within populations | 133 | 1030.086 | 7.74501 Vc | 76.39 | FST = 0.23611* | |
| Total | 1297.511 | 10.13893 |
Notes:
Evolution and recombination analysis
Tajima’s D, Fu and Li’s D* and F* statistics were calculated for testing the mutation neutrality hypothesis. The results showed most of the genes were in accord with the neutral hypothesis, except the NV genes of clinical isolates (Fig. 6, Table 3).
Figure 6: Tajima’s D, Fu and Li’s D* and F* test for the five gene loci of L. pneumophila from different sources.
C indicates clinical isolates, N indicates natural isolates, and A indicates artificial isolates.{kind=link}
| Gene type | Locus | Strain type | Tajima’s D | Fu and Li’s D* test | Fu and Li’s F* test | |
|---|---|---|---|---|---|---|
| NV genes | cca | C | −1.75425, 0.10>P>0.05 | −2.57998, P < 0.05 | −2.72586, P < 0.05 | Purifying selection |
| N | 1.27280, P > 0.10 | 0.61870, P > 0.10 | 1.03831, P > 0.10 | Neutral | ||
| A | −0.47005, P > 0.10 | 2.15319, P < 0.02 | 1.39413, P > 0.10 | Neutral | ||
| trpA | C | −2.17325, P < 0.01 | −3.38643, P < 0.02 | −3.52727, P < 0.02 | Purifying selection | |
| N | −0.77493, P > 0.10 | −0.13862, P > 0.10 | −0.44983, P > 0.10 | Neutral | ||
| A | −1.18806, P > 0.10 | 2.03417, P < 0.02 | 1.00976, P > 0.10 | Neutral | ||
| Virulence genes | lssD | C | −0.77832, P > 0.10 | 0.42930, P > 0.10 | 0.03804, P > 0.10 | Neutral |
| N | 0.16541, P > 0.10 | 2.01583, P < 0.02 | 1.56550, 0.05<P<0.10 | Neutral | ||
| A | −0.64072, P > 0.10 | 2.07816, P < 0.02 | 1.27560, P > 0.10 | Neutral | ||
| lspE | C | −0.20029, P > 0.10 | 0.82007, P > 0.10 | 0.57579, P > 0.10 | Neutral | |
| N | 0.29705, P > 0.10 | 1.33893, P > 0.10 | 1.31199, P > 0.10 | Neutral | ||
| A | −0.98456, P > 0.10 | 1.29642, P > 0.10 | 0.54402, P > 0.10 | Neutral | ||
| icmK | C | −1.07929, P > 0.10 | −1.12272, P > 0.10 | −1.30880, P > 0.10 | Neutral | |
| N | −0.62353, P > 0.10 | 0.17035, P > 0.10 | −0.16288, P > 0.10 | Neutral | ||
| A | −0.84707, P > 0.10 | 1.29035, P > 0.10 | 0.58803, P > 0.10 | Neutral |
The intragenic recombination events of each gene locus in different types of isolates were detected using RDP4 individually. For clinical isolates, RDP reported recombination events happened in the cca locus of C17, C21, C22, and C23, in the lssD locus of C17, and in the lspE locus of C16. No recombination event was identified on these loci of artificial environmental isolates. However, recombination events were detected only in virulence loci of natural isolates including the lssD locus of N71 and N220, the lspE locus of N52 and N53 and the icmK locus of N115 and N152. Details are shown in Table 4. These results might indicate that horizontal exchange of genetic material of virulence genes in clinical and natural isolates was more widespread than that in artificial isolates, and it was more prevalent in natural isolates.
| Gene type | Locus | Strain typeg | Recombination events | Recombinant isolates | Major parenta | Minor parentb | Detection methods implemented in RDP softwarec | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RDP | GENECONV | Bootscan | Maxchi | Chimaera | SiSscan | |||||||
| NV genes | cca | C | 1 | C17 | C2 | C23d | Ne | N | N | Yf | Y | Y |
| 2 | C21, C22, C23 | C27d | C24 | N | Y | N | Y | Y | Y | |||
| Virulence genes | lssD | C | 1 | C17 | C28 | C16 | Y | Y | Y | Y | Y | Y |
| N | 1 | N71, N220 | N67 | N93d | N | N | Y | Y | Y | Y | ||
| lspE | C | 1 | C16 | C25 | C29 | N | N | N | Y | Y | Y | |
| N | 1 | N52, N53 | N112 | N67d | N | N | N | Y | Y | Y | ||
| icmK | N | 1 | N115, N152 | N43d | N113 | N | N | N | Y | Y | Y | |
Notes:
Discussion
Although whole-genome sequencing (WGS) is a more informative technology to study genetic differences between different L. pneumophila isolates (Borges et al., 2016; Qin et al., 2016), investigations on special genes still provide clues in understanding L. pneumophila pathogenic mechanisms and epidemiological characteristics (Costa et al., 2012; Costa et al., 2014; Costa et al., 2010). Many studies have compared the genetic differences of L. pneumophila isolates from different sources, but these studies paid less attention to NV genes (Costa et al., 2014; Ko et al., 2003). Therefore, we have compared genetic variability of clinical, artificial and natural isolates of L. pneumophila at the nucleotide level in five coding loci, including two NV genes and three key virulence genes. These loci were researched as representative NV genes and virulence genes in this study. In addition to the two NV genes we studied, three NV genes including rpoB, DNA topoisomerase I gene and DNA polymerase III subunits gamma gene were also included in the study as our initial plan. However, alignment of the sequences from the NCBI database showed a very small variability (<10% sequence variation, Table S4) of these genes within L. pneumophila strains. We supposed that they might not provide sufficient resolution in determining the genetic difference between isolates from different source. Thus, they were not included in the following study. Globally representative clinical isolates were included in this study and compared with the environmental isolates due to the lack of L. pneumophila clinical isolates in China. For the NV genes, most of the genetic diversity parameters (π for cca and S, θ, η; for both cca and trpA) that were not directly dependent on sample size, were higher in the clinical isolates. In contrast, genetic diversity parameters of virulence loci (π, S, k of the three loci; and θ of lssD, icmK loci) were higher in the natural isolates. These results suggested that different evolutionary patterns existed between virulence genes and NV genes. It is well believed that environmental protozoa inhabiting the natural water sources, provided the primary evolutionary pressure for Legionella to obtain and maintain virulence factors (Moliner, Fournier & Raoult, 2010; Richards et al., 2013) and lead to a relatively higher genetic diversity in virulence genes. This was supported by our study. (Table 1). Similar ratios of dN/dS were found in the same loci of L. pneumophila isolates from different sources. Low ratios of dN/dS in both NV loci and virulence loci indicated these loci might be under purifying selection (Sobrinho Jr & De Brito, 2012). In this case, genetic variation occurs when it does not confer a significant disadvantage on surviving variant, and dN/dS ratios reflect general restrictions on gene and protein variability (Costa et al., 2014).
NJ trees showed that some alleles (including NV and virulence loci) were restricted to the clinical isolates although most of them were shared with both the clinical and environmental isolates. More than half of the clinical isolates and artificial isolates presented a single dominant allele of cca, trpA, lssD, lspE (Figs. 1 to 4). This result supported the hypothesis proposed by Coscolla that clinical isolates were a small specific subset of all genotypes existing in nature, perhaps representing an especially adapted group of clones (Coscolla & Gonzalez-Candelas, 2009). We could also conclude that artificial isolates were also a non-random subset of all genotypes existing in nature, and only those more adaptive could inhabit an artificial environment. Some alleles were natural, artificial or clinical isolates tropic, indicating that isolates with these alleles were better-adapted in the selected environment. The relatively small proportion of unique allelic profiles in cca, trpA, lssD and lspE loci of artificial isolates further illustrated the intermediate post role of the artificial environment. Four L. pneu mophila isolates (A5, A189, A195, and C28) were more likely to experience a much more complex evolutionary history of cca, trpA, and icmK genes, because they were situated on their own distinct clade, separated from other isolates (Figs. 1, 2 and 5), envisioning the existence of several evolutionary reticulated events acting on these isolates (Costa et al., 2012). We found more allelic profiles the icmK locus (139 isolates possessed 89 allelic profiles, Fig. 5). This result indicated that icmK might be a suitable candidate locus for improving discrimination level in sequence-based epidemiological typing scheme of L. pneumophila. AMOVA results showed small differences among clinical, natural and artificial isolates, while a relatively larger genetic variation was found in the cca and icmK loci although the FCT values were not significant. Significant values of FST were found in all cases, supporting that some genetic differentiation (in specific genes) might exist between clinical and environmental isolates (Table 2).
Recombination events tend to happen in the genes with genetic plasticity, such as virulence genes (Coscolla & Gonzalez-Candelas, 2007; Gomez-Valero et al., 2011; Sánchez-Busó et al., 2014). These events are important for increasing L. pneumophila genetic pool by allowing the selection of new allelic patterns with increasing fitness or in a more neutral perspective (Zawierta et al., 2007). We observed recombination events in the three virulence loci and they mostly (6/8, 75%) happened in natural environmental isolates. We also found recombination events in the cca locus, but they all happened in clinical isolates. Neutrality tests showed that variations in the virulence genes of clinical and environmental isolates were under neutral evolution, indicating that the given virulence genes were probably implicated in conserved virulence mechanisms. This result was partly in accordance with Costa’s report that lspE locus maintained neutral evolution (Costa et al., 2012). Nevertheless, trpA and cca of clinical isolates showed significantly negative values of Tajima’s D, Fu and Li’s D* and F*. This could be interpreted as the presence of negative selection, an increased population size or subpopulation structure (Hughes, 2008; Jukes, 2000; Nei, Suzuki & Nozawa, 2010). These results also suggested that different evolutionary patterns existed between NV genes and virulence genes of L. pneumophila strains from different sources.
Conclusions
In sum, we have characterized the genetic variability of two NV genes and three virulence genes in clinical, natural and artificial isolates of L. pneumophila. The results unveiled different genetic variability between NV genes and virulence genes in L. pneumophila from the clinical, artificial and natural sources. Recombination events played an important role in the molecular evolution of the NV genes of clinical isolates and the virulence genes of natural isolates, and might lead to the change of population structure of this bacterium. This work provides clues for future work on population-level and genetics-level questions about ecology and molecular evolution of L. pneumophila, as well as the genetic differences of NV genes and virulence genes between this host-range pathogen with different lifestyles.