
Bunyaviruses are common in male and female Ixodes scapularis ticks in central Pennsylvania
- Published
- Accepted
- Received
- Academic Editor
- Dina Fonseca
- Subject Areas
- Ecology, Entomology, Genomics, Microbiology, Virology
- Keywords
- Tick, Virus, Metagenomics, Vector-borne pathogen
- Copyright
- © 2016 Sakamoto et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2016. Bunyaviruses are common in male and female Ixodes scapularis ticks in central Pennsylvania. PeerJ 4:e2324 https://doi.org/10.7717/peerj.2324
Abstract
The blacklegged tick Ixodes scapularis is widely distributed in the United States and transmits multiple pathogens to humans, wildlife and domestic animals. Recently, several novel viruses in the family Bunyaviridae (South Bay virus (SBV) and Blacklegged tick phlebovirus (BTPV)) were identified infecting female I. scapularis ticks collected in New York State. We used metagenomic sequencing to investigate the distribution of viruses infecting male and female I. scapularis ticks collected in Centre County, Pennsylvania. We identified both SBV and BTPV in both male and female ticks from all collection locations. The role of male I. scapularis in pathogen epidemiology has been overlooked because they rarely bite and are not considered important pathogen vectors. However, males may act as reservoirs for pathogens that can then be transmitted to females during mating. Our data highlight the importance of examining all potential avenues of pathogen maintenance and transmission throughout the vector-pathogen life cycle in order to understand the epidemiology of tick-borne pathogens.
Introduction
The blacklegged tick Ixodes scapularis is widely distributed in the United States (Sakamoto, Goddard & Rasgon, 2014) and transmits multiple zoonotic pathogens including Borrelia burgdorferi (the agent of Lyme disease (LD)), Anaplasma phagocytophilum (the agent of human anaplasmosis), Babesia microti (the agent of human babesiosis), Deer Tick Virus/Powassan virus (two closely related tick-borne flaviviruses that cause encephalitis), and potentially nematodes (Zhang, Norris & Rasgon, 2011; Namrata et al., 2014; Henning et al., 2016; Diuk-Wasser, Vannier & Krause, 2016). Epidemiologically, I. scapularis nymphs are the most important stage of pathogen transmission to humans because they are more difficult to detect and remove prior to the transmission event (Diuk-Wasser, Vannier & Krause, 2016). Adult I. scapularis females are also important in transmission, both directly and by producing new offspring that can subsequently maintain the transmission cycle. Conversely, male I. scapularis are not as well studied in relation to I. scapularis pathogen epidemiology because they rarely bite and are not considered important pathogen vectors (De Meeûs, Lorimier & Renaud, 2004). However, while biting is uncommon, it does occur and represents an underexplored avenue of pathogen transmission to humans. In addition, some lab studies have shown that male ticks can sexually transfer pathogens to females, suggesting that males could potentially act as reservoirs (Plowright, Perry & Greig, 1974; Hayes, Burgdorfer & Aeschlimann, 1980; Chunikhin et al., 1983; Gonzalez et al., 1992; Alekseev et al., 1999) and may contribute to the epidemiology of pathogens in unexpected ways.
The use of massively parallel sequencing technology has been shown to be very effective in discovery of novel (even unculturable) microbes. Multiple metagenomics studies have recently been published describing known and novel viral sequences from a diverse array of arthropods (e.g. Ng et al., 2011; Tokarz et al., 2014; Xia et al., 2015; Temmam et al., 2015). In one study of mosquito viromes, nearly 50% of approximately 500,000 viral sequences were unidentified (Ng et al., 2011). The rich data set generated from viral sequences purified from mosquitoes’ revealed novel viruses related to those that infect animals, plants, insects, and bacteria (Ng et al., 2011). Virome studies have been conducted in multiple tick species (Tokarz et al., 2014; Xia et al., 2015; Temmam et al., 2015). Tokarz et al. (2014) recently used virome sequencing to detect and identify multiple novel viruses in female Amblyomma americanum, Dermacentor variabilis, and I. scapularis ticks from New York. In addition to the previously identified Powassan virus, they identified several novel bunyaviruses in the genera Nairovirus (South Bay virus (SBV)) and Phlebovirus (blacklegged tick phlebovirus (BTPV)). These viruses were highly divergent from previously identified tick-borne bunyaviruses (Swei et al., 2013; Tokarz et al., 2014).
In this study, we used metagenomic sequencing to examine the occurrence and distribution of viruses from 18 pools of I. scapularis ticks collected in 2014 (nine male, nine female) from multiple populations in and surrounding the State College area of Centre County, Pennsylvania. We identified both SBV and BTPV as the major viruses present in these populations. SBV was identified in all pools and was always predominant, while BPTV was more variable and present at lower levels. These data show that tick-associated bunyaviruses are common in both male and female I. scapularis ticks in central Pennsylvania.
Materials and Methods
Field collection
Adult male and female I. scapularis were collected from Centre County, Pennsylvania in the fall of 2014 using a drag cloth (91.44 × 114.3 cm, Fig. 1). Male and females were separated and ticks stored alive in a 5 ml scintillation vial until returned to the laboratory for visual identification. After identification, live ticks were washed in 70% ethanol for 15 s, then 10% bleach for 1 min, then washed three times in autoclaved, nuclease-free water, and dried on autoclaved filter paper and placed at −80 °C until extraction.
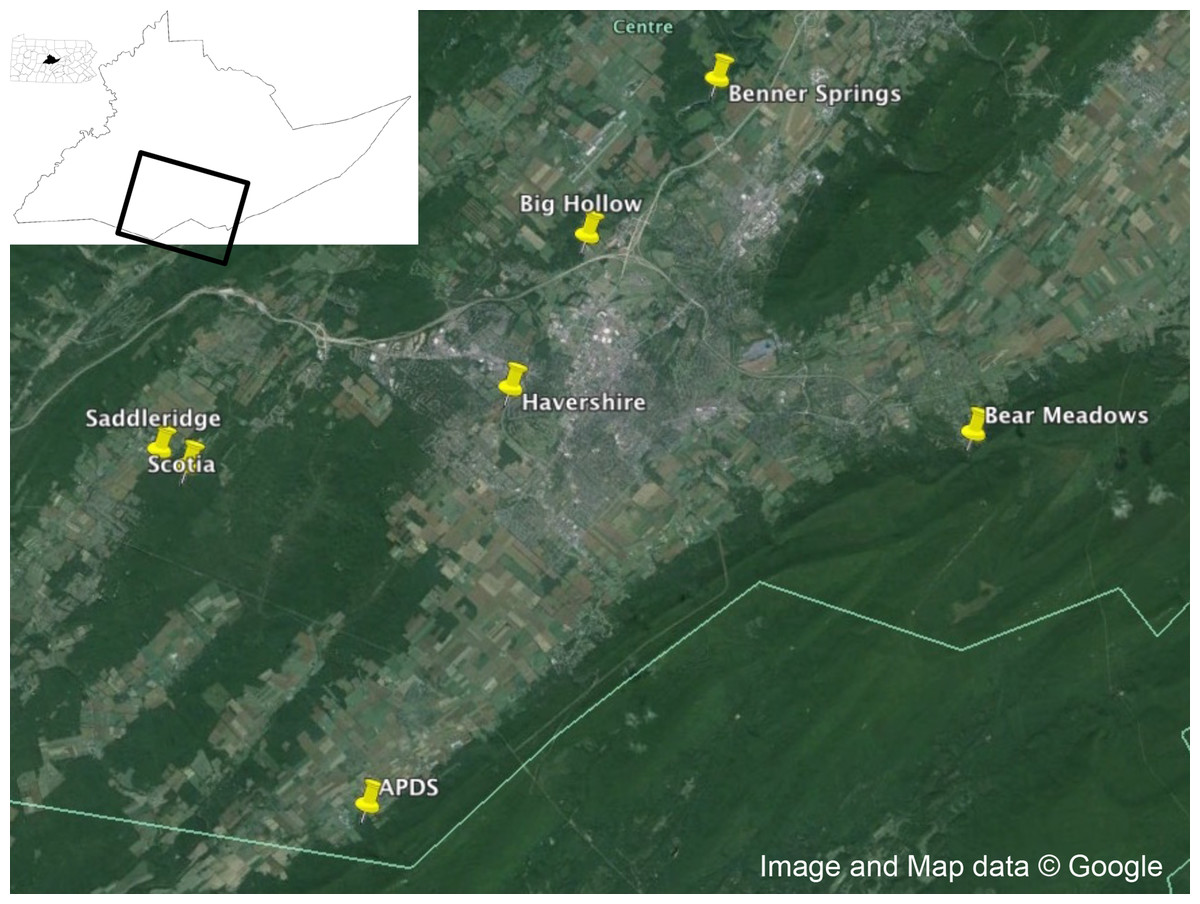
Figure 1: Collection locations.
Collection locations in Centre County, Pennsylvania for Ixodes scapularis examined in this study. Map data: Google.{kind=link}
Homogenization of ticks
Ticks were homogenized individually in 100 μl sterile 1X PBS using sterilized micro-pestles. The homogenate was centrifuged at 1,500 g for 30 min at 4 °C to pellet the tick debris (Ng et al., 2011; Thurber et al., 2009). After centrifugation, 50 μl of the supernatant was pooled with the supernatant from ∼20 other ticks of the same sex and collection site (Table 1). Pooled supernatants were filtered through a 0.45 μm filter.
| Population | N in pool | Sex | SBV L segment reads | SBV S segment reads | BLTV1 L segment reads | BLTV1 S segment reads | Total SBV reads | Total BLTV1 reads |
|---|---|---|---|---|---|---|---|---|
| APDS | 16 | Female | 10,572 | 53,870 | 15,090 | 22 | 64,442 | 15,112 |
| APDS | 14 | Male | 86,173 | 112,175 | 334 | 57 | 198,348 | 391 |
| Bear Meadows | 20 | Female | 20,100 | 42,848 | 5 | 0 | 62,948 | 5 |
| Bear Meadows | 20 | Male | 50,477 | 60,168 | 20 | 0 | 110,645 | 20 |
| Benner Springs | 20 | Female | 64,356 | 125,388 | 2,031 | 6 | 189,744 | 2,037 |
| Benner Springs | 20 | Male | 319,538 | 412,178 | 31 | 22 | 731,716 | 53 |
| Big Hollow | 20 | Female | 10,231 | 50,032 | 5 | 0 | 60,263 | 5 |
| Big Hollow | 15 | Male | 132,159 | 203,425 | 15,276 | 2,368 | 335,584 | 17,644 |
| Havershire | 18 | Female | 10,684 | 25,660 | 136 | 0 | 36,344 | 136 |
| Havershire | 15 | Male | 45,211 | 57,494 | 135 | 0 | 102,705 | 135 |
| Saddleridge | 20 | Female | 30,505 | 38,028 | 749 | 0 | 68,533 | 749 |
| Saddleridge | 20 | Female | 9,084 | 15,741 | 760 | 0 | 24,825 | 760 |
| Saddleridge | 20 | Male | 47,635 | 74,986 | 9 | 152 | 122,621 | 161 |
| Saddleridge | 20 | Male | 31,767 | 46,160 | 15 | 2 | 77,927 | 17 |
| Scotia | 20 | Female | 144,187 | 314,304 | 6,066 | 408 | 458,491 | 6,474 |
| Scotia | 20 | Female | 146,145 | 288,615 | 9,425 | 857 | 434,760 | 10,282 |
| Scotia | 20 | Male | 849,797 | 1,083,252 | 28,885 | 9,576 | 1,933,049 | 38,461 |
| Scotia | 20 | Male | 148,980 | 244,248 | 13,998 | 1,779 | 393,228 | 15,777 |
RNase/DNase treatment and viral total nucleic acid extraction
Prior to virion nucleic acid extraction, filtrates were treated with nucleases to remove exogenous nucleic acids (Ng et al., 2011). Each pool was incubated with 14 units Turbo DNase I (Life Technologies/Ambion), 25 units Benzonase (Millipore/Novagen), and 20 units RNase I (Thermo Scientific/Fermentas) for 1.5 h at 37 °C and stopped with DNase stop solution according to the manufacturer’s protocol. Total nucleic acid was extracted immediately after nuclease treatment using the MagMAX viral RNA Isolation purification kit (Life Technologies, Inc.) following the manufacturer’s protocol. Samples were stored at −80 °C until sequenced.
Next generation sequencing and bioinformatics analysis
Illumina compatible libraries were generated from enriched viral particle preparations using the Nextera XT library prep kit (Illumina, San Diego, CA, USA). Sequencing libraries were normalized using the library quantification kit for Illumina platforms (Kapa Biosystems, Wilmington, MA, USA) prior to sequencing so that the same amount of input material was sequenced for each barcoded library. Next generation sequencing was performed on the MiSeq platform (2 × 250 bp paired-end sequencing). Resulting sequence reads were trimmed, de-duplicated and de novo assembled using a customized NGS pipeline at the Blood Systems Research Institute as described previously (Deng et al., 2015). The assembled contigs and unassembled singlets were compared with a viral proteome database using BLASTx using E-value cutoff 0.01.
Validation of SBV S segment assembly in individual field-collected ticks
We used the purified virion RNA extracted from pools to generate first-strand cDNA using the ProtoScript® II First Strand cDNA Synthesis Kit (NEB # E6560) following the manufacturer’s guidelines. Confirmation primers (F: AAC-AAG-AGG-TCT-CCG-TTC-CA; R: CTC-GGA-CTT-TTG-GGT-GTG-TG) specific to the SBV S segment assembly were designed using Primer3 (http://frodo.wi.mit.edu) and used to confirm the structure of the viral genome. PCR products cloned, purified, and sequenced in both directions on an ABI 3130/Genetic Analyzer. Sequences were aligned to the viral genome assembly.
Phylogenetic analysis
We used Maximum Likelihood, implemented in MEGA v. 5.2.2 (Tamura et al., 2011), to phylogenetically compare the full-length aligned L and S segment nucleotide sequences of the bunyaviruses found in this study to complete full-length segments from GenBank. Sequence alignment was performed using ClustalW in MEGA. Tree robustness was assessed through 1,000 bootstrap replications. GenBank numbers included in the phylogenetic analysis are listed in Fig. 3.
Results
Metagenomic sequencing of viral cDNA from wild-caught ticks indicated that viral communities in Pennsylvania I. scapularis were very non-diverse. Of reads of viral origin (35% of total reads; remainder mapped to the tick host), approximately 98% belonged to members of the family Bunyaviridae. An additional ∼1% of reads did not map to any known virus families. The remaining ∼1% of viral sequences mapped to viral families other than Bunyaviridae—whether these represent viral infections at low levels or minor contamination during library construction and/or sequencing remains to be determined (Fig. 2). It is clear, however, that if contamination occurred in this study the frequency was very low. Raw sequence data was deposited in the NIH Sequence Read Archive under accession number SRP075634.

Figure 2: Viral families.
Viral families identified in central Pennsylvanian I. scapularis. Note break in Y-axis scale.{kind=link}
Within the bunyavirus data, viral sequences belonged to both the genus Nairovirus and the genus Phlebovirus (Nairovirus: 98%, Phlebovirus: 2%). The nairovirus SBV was found in all pools, regardless of population or sex. The phlebovirus BTPV was found in all pools but the abundance was highly variable, with very low read counts in several populations (Table 1). We were able to assemble the full-length L and S segments of both SBV and BTPV. There were no significant differences between viral sequences isolated from males vs. females. The obtained L segments matched 98 and 99% to SBV and BTPV1 respectively, while the obtained S segments both matched 98% to SBV and BPTV1 (Tokarz et al., 2015). Results were confirmed by phylogenetic analysis (Fig. 3). Similar to previous studies (Tokarz et al., 2014) we were unable to identify any contigs with homology to the bunyavirus M segment. PCR using specific primers to the SBV S segment resulted in amplification of an approximately 600 bp fragment that mapped 100% to the predicted assembly. SBV and BTPV1 L and S segment sequences were deposited in GenBank under accession numbers KX184198–KX184201.
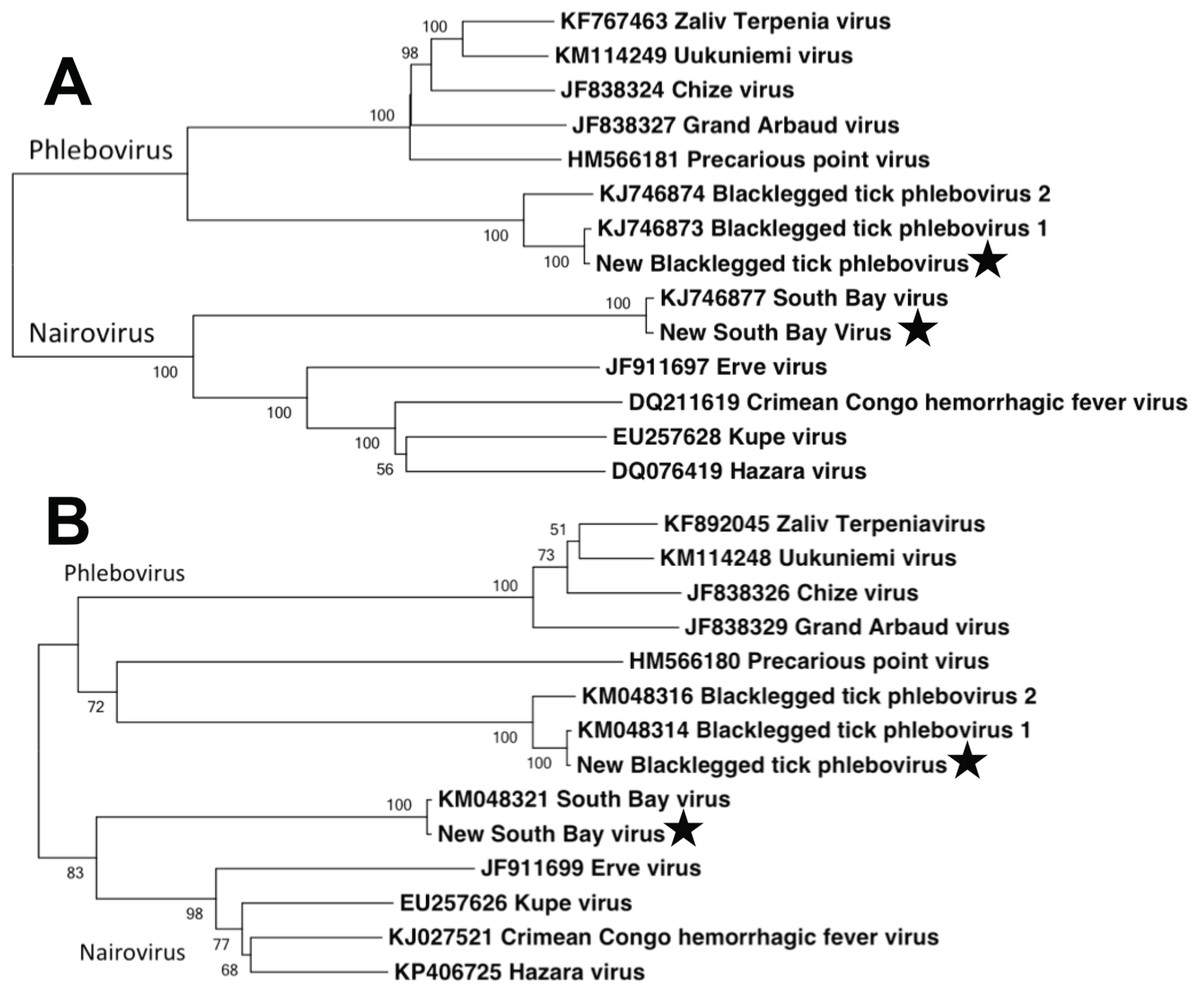
Figure 3: Phylogenetic analysis.
Maximum likelihood phylogenetic tree of full-length nairovirus and phlebovirus L segment (A) and S segment (B) nucleotide sequences. GenBank numbers are listed in taxon names. Numbers at tree nodes represent bootstrap support values (1,000 replications). Stars represent sequences obtained in this study.{kind=link}
Discussion
In terms of disease case numbers, ticks are the most important arthropod pathogen vectors in the United States (Diuk-Wasser, Vannier & Krause, 2016). Most attention has focused on bacterial pathogens such as Borrelia, but ticks are important vectors of viral pathogens as well (Swei et al., 2013; Tokarz et al., 2014; Xia et al., 2015; Temmam et al., 2015; Diuk-Wasser, Vannier & Krause, 2016). Tokarz et al. (2014) used next generation sequencing to identify several novel viruses in three species of ticks. In I. scapularis, they identified the novel bunyaviruses SBV and BTPV 1 & 2 (two very closely related phleboviruses; Fig. 3). Our results extend the findings of Tokarz et al. (2014), and show that these novel bunyaviruses are present and widespread in I. scapularis ticks in central Pennsylvania. We found that these viruses are not only present in females, but are widely distributed at high abundance in male ticks as well.
The role of male Ixodes scapularis in pathogen epidemiology has been overlooked, because males often do not take a blood meal. However, males may act as reservoirs for pathogens that can then be transmitted to females during mating (Plowright, Perry & Greig, 1974; Hayes, Burgdorfer & Aeschlimann, 1980; Chunikhin et al., 1983; Gonzalez et al., 1992; Alekseev et al., 1999). These acquired pathogens could then conceivably be transmitted to the vertebrate host during blood feeding or transmitted transovarially to offspring. Our data highlights the importance of examining all potential avenues of pathogen maintenance and transmission throughout the vector-pathogen life cycle in order to understand the epidemiology of these novel tick-borne viruses, and conceptually other pathogens.