
Immune-neural regulatory mechanism of osteoporosis induced by androgen deficiency
- Published
- Accepted
- Received
- Academic Editor
- Vladimir Uversky
- Subject Areas
- Biochemistry, Andrology, Immunology, Orthopedics
- Keywords
- Andrology, Osteoporosis, Pathway, Macrophage polarization
- Copyright
- © 2026 Zhou et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2026. Immune-neural regulatory mechanism of osteoporosis induced by androgen deficiency. PeerJ 14:e20737 https://doi.org/10.7717/peerj.20737
Abstract
This article explores the pathophysiology of osteoporosis associated with androgen deficiency, a metabolic disorder characterized by inadequate levels of androgens or disruptions in androgen signaling. A focus is placed on the collaborative regulation of bone health by the immune and nervous systems. Notably, androgens are identified as critical modulators of immune responses, significantly influencing the development and progression of osteoporosis. The review highlights that androgens enhance erythropoietin (EPO) signaling, which exhibits dose-dependent effects on bone metabolism—physiological EPO levels promote osteoblast differentiation, while excessive EPO can induce osteoclastogenesis through the Janus kinase 2/Signal transducer and activator of transcription 5 (JAK2/STAT5) pathway. Importantly, the expression of EPO receptors (EPOR) is not confined to osteoblasts and osteoclasts but is also found on numerous immune cells, which suggests the potential for androgens to affect the EPO/EPOR signaling system and its implications for bone homeostasis. Additionally, androgens contribute to the management of osteoporosis by inhibiting osteoclastogenesis mediated by immune cells, promoting M2 macrophage polarization, decreasing Th17 cell differentiation, and lowering the Th1/Th2 ratio, ultimately leading to reduced Receptor Activator of Nuclear Factor Kappa-B Ligand (RANKL) expression. Furthermore, the central nervous system plays a pivotal role by mitigating the overactivation of the hypothalamic-pituitary-adrenal (HPA) axis, thereby lessening glucocorticoid-induced osteoblast apoptosis. In peripheral contexts, neuropeptides such as calcitonin gene-related peptide (CGRP) and Semaphorin 3A (Sema3A) promote osteogenesis. Moreover, androgens are shown to enhance neuromuscular junction functionality, providing additional mechanical stress that contributes to the maintenance of bone mass. This review aims to elucidate the intricate mechanisms governing bone homeostasis and proposes a novel therapeutic framework for the management of osteoporosis induced by androgen deficiency. By emphasizing the regulatory roles of androgens within the context of immune-neural interactions, this work provides valuable insights that may inform future treatment strategies. The target audience includes endocrinologists, rheumatologists, osteoporosis researchers, and clinicians involved in metabolic bone disease management.
Introduction
Osteoporosis, caused by an imbalance in bone conversion between resorption and formation, is a key health problem worldwide. Hormone deficiencies are closely linked to primary osteoporosis, disrupting the delicate balance of bone remodeling. The decline in hormone levels following menopause in women, as well as the reduction of estrogen and androgens in older men can lead to the development of osteoporosis (Almeida et al., 2017). Estrogen plays a vital role by directly influencing osteoclasts, osteoblasts, and osteocytes, primarily acting as an anti-resorptive agent. It reduces the number and function of osteoclasts through pathways such as the receptor activator of nuclear factor-kappa B ligand (RANKL) system (Rozenberg et al., 2020). By promoting the production of osteoprotegerin, a decoy receptor for RANKL, estrogen prevents RANKL from activating osteoclasts. Additionally, estrogen helps inhibit osteoblast apoptosis by regulating the c-Jun N-terminal kinase and activating the Src/Shc/extracellular signal-regulated kinase (ERK) pathway, along with enhancing Wnt/β-catenin signaling to boost osteoblast differentiation. These mechanisms collectively highlight estrogen’s positive effects on osteoblast proliferation and function (Lu & Tian, 2023). Despite this understanding, there remains a relative scarcity of information regarding the downstream targets and the pathophysiology associated with androgen deficiency-induced bone loss, an essential aspect of bone metabolism that primarily impacts individuals with testosterone deficiency (Chang et al., 2013). This gap in knowledge is crucial for understanding bone metabolism and developing targeted therapies for those affected.
Men at greatest risk include those with hypogonadism, patients undergoing androgen deprivation therapy (such as prostate cancer patients), and older men (Casado et al., 2022). In women, those with polycystic ovary syndrome (PCOS) often demonstrate hyperandrogenemia, which influences bone metabolism (Schmidt et al., 2012). Traditionally, it was thought that androgens act directly on bone via the androgen receptor (AR) or are converted into estrogens that influence bone indirectly by activating estrogen receptor subtypes (ERα or ERβ). Both AR and ERα pathways play a crucial role in periosteal bone expansion, with AR signaling being the principal mechanism for normal trabecular bone development in males (Hsu, Chen & Chen, 2024; Vescini et al., 2021). AR is expressed in a wide range of cells that are essential for bone homeostasis, particularly in mesenchymal stem cells (MSCs) found in the bone marrow and periosteum, as well as osteoblasts, osteoclasts, chondrocytes, adipocytes, endothelial cells, lymphocytes, and monocytes. Activation of the AR by androgens facilitates the proliferation, differentiation, and survival of osteoblasts while concurrently suppressing the activity of osteoclasts through modulation of the receptor activator of nuclear factor kappa-B ligand/osteoprotegerin (RANKL/OPG) signaling axis. This dual mechanism highlights the significant contribution of androgens to the maintenance of bone density and structural integrity (Kalyanaraman et al., 2025). However, ongoing research highlights the significant roles of the immune and nervous systems in the pathophysiology of androgen deficiency-induced osteoporosis.
Beyond classical endocrine pathways, emerging evidence underscores the existence of a sophisticated immune-neuro-skeletal network that collaboratively regulates bone homeostasis. This review elucidates the complex interplay within this network under androgen regulation, with a particular emphasis on its sexual dimorphism. Androgens modulate bone remodeling not only by direct actions on bone cells but also by fine-tuning immune responses and neural signaling. Notably, their effects exhibit significant gender differences, influencing immune cell populations—such as macrophage polarization, T-cell differentiation, and B-cell function—and neural pathways—including the hypothalamic-pituitary-adrenal (HPA) axis and sensory neuropeptide secretion—in a sex-specific manner. Key molecular players, such as Protein Kinase C-delta (PKC-δ) and aromatase, further illustrate the divergent mechanisms of androgen action in males versus females. In states of androgen deficiency, the disruption of this sexually dimorphic tripartite cross-talk leads to dysregulated bone remodeling, accelerated bone loss, and the development of osteoporosis. By integrating the latest advances in gender-specific mechanisms, this review aims to provide a holistic perspective for deciphering the pathogenesis of androgen deficiency-induced osteoporosis and identifying novel, sex-stratified therapeutic targets.
Androgens regulate immune cells through complex processes. They promote an anti-inflammatory macrophage phenotype, reduce the production of pro-inflammatory mediators, and lower Toll-like receptor (TLR) expression, which subsequently affects RANKL secretion and indirectly modulates osteoclast activity (Ainslie et al., 2024; Lampiasi, Russo & Zito, 2016). Furthermore, androgens regulate various T-cell subpopulations, and the ability of these T-cells to produce RANKL varies. By influencing the differentiation of cells, particularly CD4+ T cells, including Th1, Th2, Th17, and regulatory T cells (Tregs), androgen deficiency can lead to an imbalance between pro-inflammatory and anti-inflammatory T-lymphocytes and their associated cytokines. This imbalance can amplify the inflammatory response and alter RANKL expression levels, consequently impacting bone metabolism (Bhattacharya et al., 2023; Moulana, 2023). Additionally, immune cells such as neutrophils, mast cells, and B lymphocytes—under the influence of androgens—also produce RANKL and other cytokines that play a role in regulating bone homeostasis (Ainslie et al., 2024; Mackey et al., 2020). RANKL binds to the RANK receptor on the surface of osteoclasts, initiating bone resorption effects. Once activated, osteoclasts attach to the bone surface and release proteases and protons that dissolve minerals in the bone while breaking down the extracellular matrix. The primary proteases involved in collagen matrix breakdown are cysteine proteases and the matrix metalloproteinase (MMP) family (Elahmer et al., 2024).
Moreover, the nervous system plays a significant role in the development of osteoporosis linked to androgen deficiency. Both male and female bone metabolism is profoundly affected by androgen interactions with the HPA axis, along with the dopaminergic pathways within the central nervous system. In females, the hypothalamic-pituitary-gonadal (HPG) axis further contributes to bone regulation (Goel et al., 2014; Kauffman, 2024). Androgen deficiency activates the HPA axis, resulting in elevated glucocorticoid (GCs) secretion, which inhibits critical pathways such as the phosphoinositide 3-kinase/Akt/semaphorin 3A (PI3K/Akt/Sema3A) and activates c-Jun N-terminal kinase (JNK) and proline-rich tyrosine kinase 2 (Pyk2), ultimately leading to osteoblast apoptosis and diminished bone formation. Additionally, GCs can enhance the RANKL/OPG ratio, thereby promoting osteoclastogenesis (Chen et al., 2023; Goel et al., 2014; Wang, Liu & He, 2020a; Xing, Feng & Zhang, 2021). Furthermore, androgens influence the activity of dopaminergic neurons. In rat studies, dopamine has been shown to play a critical role in regulating bone formation by inhibiting the osteogenic differentiation of bone marrow mesenchymal stem cells (rBMSCs) (Kuang et al., 2022). Sensory nerves in the peripheral nervous system control the balance of bone production and resorption by releasing neurotransmitters and neuropeptides that interact with bone cells, and androgens regulate the regeneration of sensory neurons. Neuropeptides released by sensory neurons, such as substance P (SP), calcitonin gene-related peptide (CGRP), and semaphorin 3A (Sema3A), can directly stimulate osteoblasts, promoting osteogenic activity (Chen et al., 2024; Ward et al., 2021). Additionally, experiments in mice suggest that androgens may extend motor neuron axons and enhance skeletal muscle contraction capability, which generates mechanical stress in the muscles and stimulates bone tissue. The interplay between muscle and bone, facilitated by biochemical factors, also influences the osteoporosis process (Bargiota et al., 2020; Li et al., 2019; Ward et al., 2021).
In summary, the immune and nervous systems are critical in the development of osteoporosis resulting from androgen deficiency, and their interactions with androgens create a complex regulatory network. A comprehensive understanding of androgen deficiency-induced osteoporosis’s pathophysiology and the formulation of effective treatment strategies depend on examining the molecular mechanisms that underlie this regulatory network. By incorporating the latest research advancements, we aim to elucidate the molecular mechanisms through which androgens regulate bone metabolism via the synergistic effects of the immune and nervous systems, providing new theoretical foundations for the clinical management of androgen deficiency-induced osteoporosis.
Survey methodology
Literature search strategy
This systematic review employed a comprehensive search strategy across electronic databases including PubMed, Web of Science, and Scopus to identify relevant studies published between January 2000 and May 2025. Search terms combined Medical Subject Headings (MeSH) and free-text keywords related to four core themes: (1) androgen signaling (e.g., “androgen deficiency,” “testosterone,” “androgen receptor,” “hypogonadism”); (2) bone metabolism (e.g., “osteoporosis,” “osteoblast,” “RANKL”); (3) mechanistic pathways (e.g., “immune regulation,” “macrophage polarization,” “neuro-skeletal axis”); and (4) signaling systems (e.g., “erythropoietin and erythropoietin receptor (EPO/EPOR) signaling,” “HPA axis”). Boolean operators (AND/OR) were utilized to link concepts, such as pairing androgen deficiency with osteoporosis while integrating immune or nervous system components. To ensure literature saturation, reference lists of included articles were manually screened for additional studies.
Inclusion and exclusion criteria
Studies were selected based on their focus on molecular pathways linking androgen deficiency to bone loss through immune or neural mechanisms. Inclusion encompassed original research (animal models, in vitro experiments, clinical trials) and reviews reporting key pathways such as RANKL/OPG, Janus kinase 2/Signal transducer and activator of transcription 5 (JAK2/STAT5), or neuropeptide interactions. Publications were restricted to English language. Exclusion criteria eliminated studies unrelated to bone metabolism or androgen signaling, along with case reports and conference abstracts lacking full datasets.
Quality assessment and data synthesis
Two authors independently screened titles/abstracts and reviewed full texts, resolving discrepancies through consensus. Data extraction captured study design, model systems, mechanistic insights, and key findings. Methodological rigor was evaluated using AMSTAR-2 for systematic reviews and ARRIVE 2.0 guidelines for animal studies, ensuring robust evidence synthesis.
Data availability
As a review synthesizing published literature, no new raw data were generated. All extracted information is fully referenced within the manuscript. Detailed search strategies are provided in File S1. The key molecules discussed in this review are summarized in Table 1.
| Category | Key molecule/pathway | Cellular source/target | Function in bone metabolism |
|---|---|---|---|
| Signaling | RANKL/RANK/OPG | Macrophages, B cells, Th1, Th2, Th17, Activated neutrophils, Osteoblasts | RANKL binds RANK on osteoclasts, promoting differentiation & activation; OPG is a decoy receptor that inhibits this. |
| EPO/EPOR | Kidney, Osteoblasts, Osteoclasts, Immune cells | Dose-dependent: Physiological EPO promotes osteogenesis; High EPO activates JAK2/STAT5 signaling, leading to osteoclastogenesis. | |
| JAK2/STAT5 | Osteoclast precursors, Immune cells | Downstream of EPOR, mediates EPO-induced osteoclast differentiation. | |
| Protein Kinase C-delta (PKC-δ) | Osteoclasts | Conditional knockout increases AR transcription and reduces osteoclast function (male-specific). | |
| NFATc1, c-Fos | Osteoclasts | Master regulators of osteoclast differentiation and function. | |
| PI3K/Akt | Osteoblasts | Promotes cell survival and differentiation; inhibited by excess GCs. | |
| Wnt/β-catenin | Osteoblasts, MSCs | Critical pathway for osteoblast differentiation and bone formation. | |
| Aromatase | Various tissues | Converts androgens to estrogens. Critical for bone protection in females. | |
| Immune system | TLR4/MyD88/NF-κB | Macrophages, Neutrophils | TLR4 activation leads to MyD88/NF-κB signaling, resulting in M1 polarization and pro-inflammatory cytokine release. |
| IFN-γ | Th1 cells | Inhibits RANKL signaling during osteoclast differentiation. | |
| IL-4 | Th2 cells | Promotes Th2 polarization; reduces RANKL expression. | |
| IL-6, TNF-α | M1 Macrophages, T cells | Pro-inflammatory cytokines that enhance osteoclast activity and bone resorption. | |
| IL-10 | M2 Macrophages, Tregs | Anti-inflammatory cytokine; inhibits osteoclastogenesis. | |
| IL-12 | Antigen-presenting cells | Promotes Th1 polarization; can inhibit RANKL-induced osteoclastogenesis. | |
| IL-17 | Th17 cells | Stimulates osteoblasts/synoviocytes to produce RANKL; promotes osteoclastogenesis. | |
| IL-21/IL-23 | T cells | Axis modulates inflammatory cytokines and RANKL expression in T cells. | |
| Central nervous system | CRH, ACTH, GCs | HPA Axis | GCs inhibit osteoblast differentiation (via suppression of PI3K/Akt/Sema3A) and promote apoptosis. |
| CGRP | Sensory nerves | Inhibits osteoclastogenesis; promotes osteoblast differentiation via cAMP/PKA/CREB. | |
| Substance P (SP) | Sensory nerves | Low dose promotes osteogenesis via Wnt/β-catenin; high dose is pro-inflammatory and increases RANKL. | |
| Oxytocin (OT) | Pituitary gland | Binds receptors on bone cells, stimulates bone formation and inhibits resorption. | |
| Prolactin (PRL) | Pituitary gland | Increases bone resorption and decreases formation, reducing BMD. | |
| FSH, LH | Pituitary gland | FSH stimulates osteoclast activity; LH/FSH stimulate ovarian sex hormone production. | |
| Peripheral nervous system | Semaphorin 3A (Sema3A) | Sensory nerves, Osteoblasts | Binds Nrp1, promotes osteoblast differentiation and inhibits osteoclastogenesis. |
| Dopamine (D1–D5 receptors) | CNS, Osteoblasts, Osteoclasts | D2-like receptors inhibit osteoclastogenesis (via reducing cAMP); D1-like receptors may promote osteoblast function. | |
| ACh | Motor neurons | Binds AChR at NMJ, triggers muscle contraction, generating mechanical load on bone. | |
| Norepinephrine (NE) | Sympathetic nerves | Binds β2-adrenergic receptor on osteoblasts and immune cells, influencing bone remodeling. |
Regulation of bone metabolism by androgens via the immune system
The regulation of bone metabolism is a complex process influenced by various hormonal and cellular factors. A pivotal player in this regulatory network is the AR, which is expressed across multiple bone cell types, including osteoblasts, osteoclasts, osteocytes, and chondrocytes. Notably, research reveals that AR expression is considerably higher in osteoblast precursor cells and mature osteoblasts, particularly in male specimens, where levels are approximately tenfold greater than those found in females. This elevated expression underscores the critical role androgens play in bone physiology, influencing both bone formation and resorption (Kalyanaraman et al., 2025).
Androgens exert their effects through classical nuclear mechanisms and nonclassical membrane-associated pathways, principally functioning within the cytoplasm of osteoblasts. In the classical pathway, the binding of androgens to AR facilitates the transcription of target genes through interaction with androgen response elements (AREs). Concurrently, membrane-bound AR activates significant signaling cascades such as SRC, ERK, and AKT via the nitric oxide/cyclic guanosine monophosphate/protein kinase G2 (NO/cGMP/PKG2), enhancing β-catenin expression and fostering osteoblast proliferation, differentiation, and survival (Kalyanaraman et al., 2025). Interestingly, AR expression is observed to be lowest during the initial stages of osteoblast proliferation, gradually increasing as differentiation progresses, ultimately reaching a peak in mature mineralized cultures (Wiren, Chapman Evans & Zhang, 2002).
Androgens also serve to inhibit bone resorption in osteoblasts by reducing the ratio of RANKL to OPG (Russell et al., 2015). However, it is important to note that osteoclasts may not express functional AR, suggesting that androgens do not have a direct anti-resorptive effect on these cells. Studies involving osteoclast-specific AR knockout (ocl-ARKO) mice have shown that bone mass, microstructural integrity, and responses to orchiectomy and androgen replacement therapy are comparable to those observed in control groups. This indicates that AR is not a primary target for the inhibition of bone resorption in osteoclasts; rather, it appears to modulate the bone microenvironment, particularly the RANKL/OPG balance, through interactions with bone marrow stromal cells (BMSCs) (Sinnesael et al., 2015). Notably, a sexually dimorphic mechanism has been identified wherein conditional knockout of PKC-δ specifically in osteoclasts led to enhanced androgen receptor transcription and expression, resulting in decreased osteoclast function and increased bone mass exclusively in male mice (Li et al., 2020). This indicates that while the AR pathway within osteoclasts is typically inactive, it can be potently engaged in a male-specific context (Li et al., 2020).
In addition to their direct effects on bone cells, androgens also influence bone metabolism indirectly by modulating immune system function. Substantial evidence suggests that androgens can affect the development and activation of immune cells, with alterations in RANKL expression levels having a direct impact on osteoclast differentiation and activity (Gubbels Bupp & Jorgensen, 2018). Consequently, androgens may be vital for maintaining bone homeostasis by indirectly reducing osteoclast activity through the inhibition of RANKL production in immune cells. Moreover, the role of EPO in this context warrants further investigation, as it may provide additional insights into the interplay between androgens, bone metabolism, and immune regulation. In summary, androgens play a fundamental role in the regulation of bone metabolism, influencing not only the activity of bone cells but also the immune system’s contributions to bone homeostasis.
Interplay of androgens and erythropoietin in bone metabolism via immune regulation
EPO is a critical hormone produced by fetal liver cells and adult kidneys that plays an essential role in stimulating erythropoiesis through its interaction with the EPOR. Clinically, EPO is utilized to address anemia associated with chronic kidney disease, myelodysplastic syndromes, and various cancers. The binding of EPO to EPOR facilitates the formation of a homodimer, which is capable of activating several downstream signaling pathways, including JAK2, mitogen-activated protein kinase (MAPK), and PI3K, culminating in the phosphorylation of STAT5 (Cantarelli, Angeletti & Cravedi, 2019). Recent studies indicate that EPO’s effects extend beyond erythropoiesis, with significant implications for bone metabolism and homeostasis (Rauner et al., 2021).
In the context of bone tissue, EPO and its receptor play a fundamental role by directly influencing osteoblasts and osteoclasts in a dose-dependent manner. Osteoclast precursors express elevated levels of EPOR, in contrast to mature osteoclasts that exhibit minimal expression. EPO activates the JAK2/STAT5 pathway within these precursors, promoting their differentiation into mature osteoclasts (Rauner et al., 2021). Moreover, osteoprogenitor cells and mature osteoblasts also express EPOR, which may exacerbate bone resorption through modulation of the RANKL/OPG axis (Awida et al., 2022). Interestingly, low doses of EPO do not significantly increase the number of osteoclast precursors but inhibit osteoblast precursor proliferation and mineralization capacity. Conversely, high doses of EPO notably boost osteoclast precursor numbers, while neither stimulating osteoblast precursors nor prominently altering the RANKL/OPG ratio (Kolomansky et al., 2020). In young murine models, physiological levels of EPO may enhance osteoblast differentiation and promote bone formation through several signaling pathways, including mTOR and Ephrin B2/EphB4, while also stimulating endothelial cells to enhance vascularization, thereby supporting bone repair (Suresh, Lee & Noguchi, 2020).
The clinical application of local EPO has demonstrated efficacy in promoting fracture healing, suggesting potential benefits in bone formation at physiological doses. These findings underscore the notion that the role of EPO in bone metabolism is nuanced and context-dependent, necessitating a careful assessment of its clinical use against the backdrop of potential pro-hematopoietic effects and risks for bone loss (Suresh, Lee & Noguchi, 2020). Androgens, particularly testosterone, exert significant regulatory effects on the EPO/EPOR system. In vitro investigations suggest that testosterone may enhance EPO bioactivity by elevating EPOR expression rather than directly increasing EPO levels. Membrane-active androgens can modulate EPOR transcription, thereby amplifying the efficacy of EPO signaling (Maggio et al., 2013). Additionally, some evidence suggests that supraphysiologic doses of anabolic androgenic steroids, particularly testosterone, may elevate serum EPO levels by inhibiting hepatic clearance or directly stimulating renal EPO production (Heiland et al., 2023). EPOR is not limited to osteoblasts and osteoclasts; it is also expressed on a variety of immune cells, including monocyte-macrophages, B cells, T cells, neutrophils, mast cells, and dendritic cells, suggesting the potential for androgens to influence the EPO/EPOR system and its impact on bone homeostasis, either directly or indirectly via immune modulation (Cantarelli, Angeletti & Cravedi, 2019; Wiedenmann et al., 2015). For instance, EPO has been shown to alter macrophage polarization, reducing the recruitment and differentiation of pro-inflammatory M1-like macrophages while promoting the characteristic anti-inflammatory M2 macrophages (Horwitz et al., 2022; Yang et al., 2021). Furthermore, EPO enhances the secretion of pro-angiogenic factors from bone marrow macrophages, ultimately affecting the bone marrow microenvironment (Eggold & Rankin, 2019). B cells also contribute significantly to bone homeostasis through EPOR signaling. EPO stimulation facilitates the upregulation of RANKL expression in B cells, fostering osteoclast differentiation and enhancing bone resorption. Notably, B cells, especially Pro-B cell subpopulations, have the ability to transdifferentiate into functional osteoclasts in vitro and in vivo, a process dependent on CD115 expression, which is upregulated by EPOR. Bone loss is significantly mitigated in conditional knockout models of EPOR in B cells, underscoring the critical regulatory role of B-cell-mediated EPO/EPOR signaling in bone remodeling (Deshet-Unger et al., 2020).
EPO exerts primarily quantitative and indirect anti-inflammatory effects on neutrophils while demonstrating a bidirectional regulatory function on T cells, suppressing effector T-cell activity to mitigate inflammation and promoting regulatory T cells to enhance immune tolerance (Cantarelli, Angeletti & Cravedi, 2019). Additionally, while mouse bone marrow-derived mast cells express EPOR, most cell-surface receptors are under-expressed, resulting in EPO conveying anti-inflammatory signals without impacting mast cell differentiation, proliferation, or apoptosis (Wiedenmann et al., 2015). EPO further targets immature dendritic cells via EPOR, enhancing their immunostimulatory capacity and amplifying the inflammatory response in conjunction with TLR-4 signaling (Rocchetta et al., 2011).
In conclusion, the EPO/EPOR system serves a multifaceted role in bone homeostasis, its actions meticulously governed by androgens while establishing a dynamic interplay with the immune system. Future research endeavors may aim to explore intervention strategies targeting the EPO/EPOR system, potentially paving the way for innovative therapeutic approaches to address metabolic bone diseases such as osteoporosis.
The role of androgens in regulating osteoclast activity via RANKL expression by immune cells
Androgens modulate osteoclast activity primarily by regulating RANKL production from various immune cells, as summarized in Fig. 1. RANKL, also referred to as tumor necrosis factor superfamily member 11 (TNFSF11), is a critical component of the tumor necrosis factor (TNF) superfamily. In conjunction with osteoprotegerin (OPG) and receptor activator of nuclear factor-kappaB (RANK), which belong to the TNFR superfamily, RANKL plays a significant role in bone physiology. RANKL forms a homotrimer that binds to the RANK receptor, whereas RANK and OPG are monomers and homodimers, respectively (Ono et al., 2020).
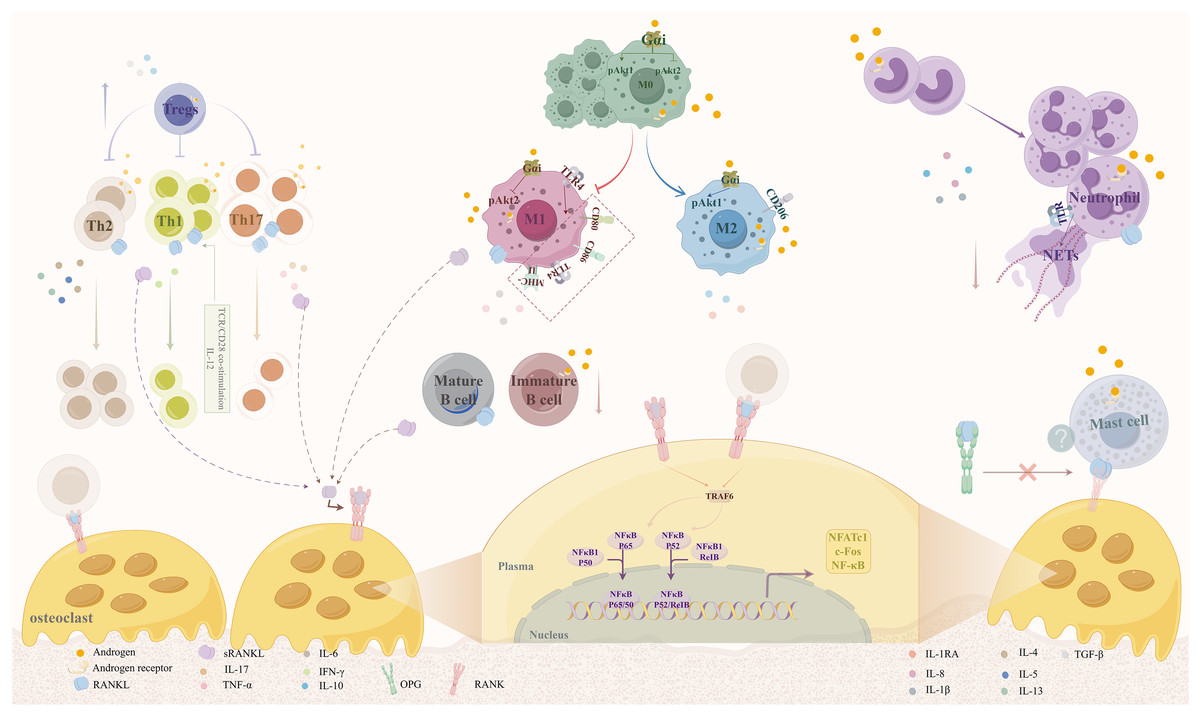
Figure 1: Mechanisms by which androgens regulate osteoclast activity via RANKL expression in immune cells.
Androgens act through AR expressed on various immune cells, including macrophages, B cells, T cells, neutrophils, and mast cells, to modulate RANKL secretion. This regulation occurs via both genomic and non-genomic signaling pathways. By promoting anti-inflammatory phenotypes (e.g., M2 macrophage polarization, Th2 response) and suppressing pro-inflammatory responses (e.g., M1 polarization, Th1/Th17 differentiation), androgens reduce RANKL production, thereby inhibiting osteoclast differentiation and bone resorption. Abbreviations: RANKL, receptor activator of nuclear factor kappa-B ligand; AR, androgen receptor; OPG, osteoprotegerin.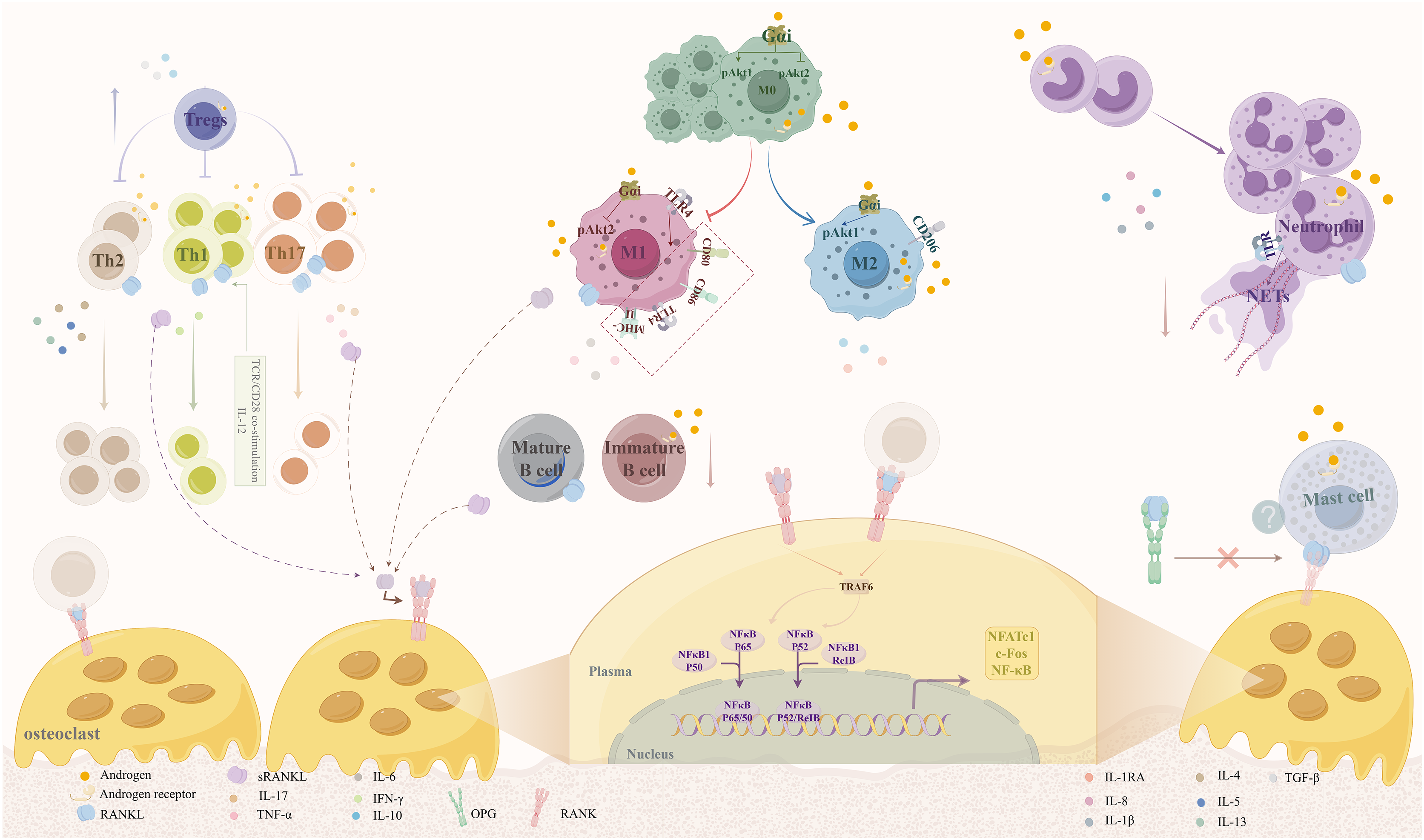{kind=link}
The RANK/RANKL/OPG signaling pathway plays a crucial role in the regulation of osteoporosis and bone metabolism. When RANK, which is expressed on osteoclast precursors, binds to RANKL, it induces trimerization and activation of the RANK receptor. The intracellular domains of RANK contain binding sites for the bridging protein TNF receptor-associated factor (TRAF), which is instrumental in the signaling cascade that follows RANK activation. Upon activation of RANK, several signaling complexes are recruited, particularly TRAF6. This recruitment focuses on activating various kinases, leading to the nuclear translocation and activation of key transcription factors, including nuclear factor of activated T-cells 1 (NFATc1), c-fos, and nuclear factor κ B (NF-κB). These factors are essential regulators of the osteoclast-specific transcriptional program, driving the differentiation and activity of osteoclasts (Takegahara, Kim & Choi, 2022). Excessive RANKL expression can result in heightened osteoclast activity, resulting in an imbalance between bone resorption and formation, which ultimately contributes to bone loss. Recent studies have highlighted new insights into this pathway and its implications for therapeutic strategies in osteoporosis management.
Furthermore, the immune system plays a pivotal role in the regulation of bone metabolism through the actions of cytokines and the secretion of RANKL by immune cells. Certain immune cell types, including specific T-lymphocyte subtypes, B-lymphocytes, M1 macrophages, mast cells, and neutrophils, have been shown to secrete RANKL, influencing bone metabolism. Additionally, ARs present in the cellular matrix or on immune cell surfaces may mediate the effect of androgens on RANKL secretion, thus indirectly affecting bone mass (Fischer & Haffner-Luntzer, 2022; Gubbels Bupp & Jorgensen, 2018).
Androgens also serve as crucial immunomodulatory factors by binding to the AR, which is expressed in various immune cells, including macrophages and neutrophils, as well as in bone marrow stromal cells (Mantalaris et al., 2001). Through genomic pathways, androgens regulate immune cell function by inducing a conformational change in the AR, allowing it to translocate into the nucleus. Within the nucleus, AR binds to the ARE in target genes, thus regulating gene transcription and influencing immune cell activity (Li & Al-Azzawi, 2009; Mantalaris et al., 2001). Additionally, androgens can exert rapid non-genomic effects that activate traditional second messenger signaling pathways, including increased intracellular calcium concentrations and the activation of kinases, including kinase A (PKA), protein kinase C (PKC), and MAPK. These events occur within seconds and do not require transcription and translation processes (Li & Al-Azzawi, 2009).
Overall, androgens and their receptors exhibit distinct regulatory mechanisms that significantly affect immune cell behavior. Notably, in male mice, androgens inhibit the generation of B lymphocytes in the bone marrow by acting on the AR present in osteoblast lineage cells (Wilhelmson et al., 2015). The proper balance of androgens is essential for maintaining bone homeostasis; deficiencies or abnormalities in AR function can lead to bone loss and osteoporosis. The potential of selective androgen receptor modulators (SARMs) for the treatment of osteoporosis and related disorders presents a promising avenue for future research and therapeutic intervention (Xie et al., 2022).
Macrophage polarization
Macrophage polarization plays a crucial role in maintaining bone tissue homeostasis through its influence on osteoblast differentiation, bone formation processes, and bone matrix mineralization. This phenomenon is integral to understanding conditions such as osteoporosis.
Mechanisms of M1/M2 polarization
There are two main phenotypes of macrophages: M1 macrophages, which stimulate pro-osteoclasts through the release of cytokines that enhance the Th1 response, thereby increasing RANKL expression and contributing to bone resorption, and M2 macrophages, which counteract inflammation through the secretion of anti-inflammatory mediators, such as interleukin 1 receptor antagonist (IL-1RA), leading to a reduction in RANKL expression and a decrease in bone resorption (Lampiasi, Russo & Zito, 2016).
It is important to highlight that macrophage polarization is a dynamic process, influenced by the local microenvironment, allowing for a transition between M1 and M2 phenotypes (Muñoz et al., 2020). In the context of bone metabolism, M1 macrophages promote bone resorption by differentiating into osteoclasts and releasing a high concentration of inflammatory cytokines. Conversely, M2 macrophages foster bone formation through the release of osteogenesis-related factors that encourage the differentiation of MSCs into osteoblasts and enhance their mineralization capacity. A disruption in the polarized state of macrophages can lead to immune dysfunction and chronic inflammation, thus disturbing the balance of cytokines within the bone microenvironment and impacting overall bone homeostasis. This disruption is closely associated with the pathophysiology of osteoporosis and other related bone disorders (Fischer & Haffner-Luntzer, 2022; Hu et al., 2023).
While the relationship between macrophage polarization status and RANKL secretion remains a topic of ongoing research, there is evidence suggesting a significant interplay that regulates bone metabolism. Recent studies indicate that RANKL can induce M1-like macrophage polarization, potentially acting as a transitional phase for osteoclast precursors. The polarization towards M1 macrophages enhances RANKL secretion, which promotes osteoclast differentiation through RANK signaling. Furthermore, it has been observed that the expression of RANKL does not typically occur in physiological settings such as the bone microenvironment, emphasizing its dependence on macrophage polarization (Muñoz et al., 2020). Of considerable interest is the finding that M1 macrophages can inhibit RANKL-induced osteoclastogenesis, mediated by IFN-gamma and IL-12, which promote osteoclast apoptosis and inhibit NFATc1 expression (Yamaguchi et al., 2016).
Regulation of polarization by androgens
Androgens have been identified as protective agents for bone mass, primarily by mitigating osteoclast activation through the inhibition of pro-inflammatory macrophage polarization, modulation of the RANKL/OPG balance, and influencing sex-specific immune responses. Gender differences in macrophage responses reveal that androgens exert more potent anti-inflammatory effects in males. They down-regulate TLR4 expression in macrophages and attenuate their reactivity to inflammatory stimuli, such as bacterial lipopolysaccharides (LPS) (Lorenzo, 2020). Studies confirm that macrophages express androgen receptors, and androgens exhibit immunosuppressive effects by modulating macrophage polarization under various pathological conditions (Sciarra et al., 2023).
For example, in patients with PCOS, androgens significantly increase the M1/M2 ratio, which exacerbates local inflammatory responses by elevating the presence of pro-inflammatory M1-type macrophages while inhibiting the polarization of anti-inflammatory M2-type macrophages (Salehi et al., 2023). In a study utilizing a male mouse model, it has been demonstrated that testosterone plays a regulatory role in macrophage polarization. This regulation occurs through the activation of membrane surface G protein-coupled receptors (GPCR), which engage inhibitory regulatory G protein (Gαi) and Akt signaling pathways. As a result, testosterone significantly downregulates the expression of M1 marker genes, including NOS2, IL-6, and TNF-alpha. Additionally, it enhances the IL-4-induced expression of M2 marker genes, such as Arg1, IL-10, and CD206 (Ren et al., 2017). Similarly, in models of myocarditis, androgen receptor activation has been shown to promote M1-type macrophage polarization and escalate inflammatory levels. Currently, there is a lack of additional studies investigating the regulation of bone metabolism by androgen receptors through macrophage polarization in models of myocarditis or systemic inflammation. Notably, ASC-J9, an androgen receptor degradation enhancer, has been found to decrease M1 polarization and encourage M2 reprogramming, which in turn reduces the inflammatory response (Ma et al., 2019).
Additionally, TLR4 serves as a key regulator of M1 pro-inflammatory macrophage polarization. As a pivotal pattern recognition receptor of the innate immune system, TLR4 predominantly recognizes damage-associated and pathogen-associated molecular patterns, such as LPS. Activation of TLR4 initiates downstream signaling pathways, such as MyD88-dependent NF-kappaB activation, increasing the expression of pro-inflammatory factors and M1 markers, including CD80, CD86, MHC-II, and TLR4 itself. In vitro studies have further demonstrated that testosterone specifically down-regulates TLR4 expression, suggesting that androgens may partially inhibit M1 macrophage polarization. Mouse experiments have indicated that gonadectomy significantly increases TLR4 expression in male macrophages, resulting in an enhanced pro-inflammatory response to infection (Cutolo et al., 2022).
Clinical implications
In summary, the collective findings underscore the significant role androgens play in inflammatory diseases by regulating the balance of macrophage polarization. However, it is essential to recognize that the precise effects of androgens may vary based on the specific tissue environment and clinical context.
B-lymphocyte
Osteoblast lineage cells significantly contribute to B-cell development during the pro-B phase by secreting essential cytokines such as IL-7, stem cell factor (SCF), and stromal-derived factor 1 (SDF-1). The expression of AR in osteoblasts has been shown to inhibit the differentiation of pro-B cells from common lymphoid progenitors. Additionally, the presence of AR in B cell progenitors negatively impacts further differentiation along the B cell lineage. While B-cells are capable of promoting bone resorption through the secretion of RANKL, the direct involvement of B-cells in reducing trabecular bone density in osteoblast-specific AR knockout (O-ARKO) models requires further investigation (Gubbels Bupp & Jorgensen, 2018; Wilhelmson et al., 2015).
Both membrane-bound RANKL and soluble RANKL are produced by B cells. Research utilizing conditional knockout mice with B cell-targeted RANKL deletion has demonstrated a significant reduction in alveolar bone resorption. B cells, upon pathogen stimulation, secrete RANKL, activating osteoclast differentiation and resulting in increased bone resorption. Research has indicated that in patients with rheumatoid arthritis (RA), CD80(+)CD86(+) B cells are more prevalent and exhibit elevated levels of RANKL expression. Furthermore, upon activation through the B cell receptor (BCR) and CD40, converted memory B cells demonstrate high RANKL expression, which is augmented by IFN-gamma and inhibited by IL-21 (Fischer & Haffner-Luntzer, 2022). TLR4 and TLR9 have been shown to potentially reduce RANKL synthesis by inducing apoptosis in RANKL-expressing B cells (Chen et al., 2014).
Several AR-deficient mouse models, including castrated wild-type mice and G-ARKO, Bsp-ARKO, and Tfm mice, have revealed increased B cell populations in their bone marrow and blood (Chang et al., 2013). These findings suggest that androgens may play a role in suppressing B cell proliferation, thereby decreasing RANKL secretion and contributing to the maintenance of bone quality. While mature B lymphocytes do not display AR on their surface, immature B cells, specifically in the pro-B and pre-B phases, do express this receptor. This observation implies that androgens may inhibit the maturation process of B cells by influencing relevant signaling pathways, thereby decelerating their proliferation rate. For instance, male AROM+ mice, characterized by a high estrogen-to-androgen ratio, demonstrate that a reduction in androgen levels diminishes the inhibitory effect on B cell proliferation. Consequently, this allows for the activation and proliferation of autoreactive B cells, potentially increasing the risk of osteoporosis (Sciarra et al., 2023).
In conclusion, the regulation of B cells by androgens is vital for preserving bone health, and fluctuations in androgen levels are closely associated with the etiology of osteoporosis, an area that warrants further investigation.
CD4+T cells Th1, Th2, Th17
The primary subtypes of T cells that secrete RANKL are CD4+ T cells, specifically Th1, Th2, and Th17. While CD8+ T cells exhibit lower levels of RANKL expression, regulatory T cells (Tregs) play a critical role in suppressing RANKL secretion. The relative proportions of Th1 and Th2 cells significantly impact RANKL expression in vivo, with Th1 cells demonstrating a greater tendency to express RANKL compared to Th2 cells. Under TCR/CD28 co-stimulation, Th1 cells show elevated levels of RANKL expression, an effect further enhanced by IL-12-mediated Th1 polarization. Conversely, Th2 polarization mediated by IL-4 results in a reduction of RANKL expression (Chen et al., 2014). Th17 cells significantly influence osteoclast differentiation and bone resorption through both direct secretion of RANKL and the induction of inflammatory factors. Th17 cells express RANKL on their surface, which binds to the RANK receptor on osteoclast precursor cells, activating downstream signaling pathways such as NF-kappaB and NFATc1, thereby driving osteoclast differentiation. Additionally, IL-17 secreted by Th17 cells can stimulate osteoblasts and synoviocytes to produce RANKL, working in concert with pro-inflammatory factors like TNF-alpha and IL-1beta to amplify signals that promote bone resorption (Huang et al., 2022). Another study shows that the RANKL secreted by Th17 cells alone is insufficient to trigger osteoclastogenesis; collaboration with other cell types is necessary for this process. Tregs contribute to the maintenance of immunological balance by limiting the activity of effector T cells. From a bone metabolism perspective, Tregs may help prevent osteoporosis by reducing osteoclastogenesis and RANKL release (Chen et al., 2014; De Martinis et al., 2020).
Androgens have complex and significant effects on T cell subsets. They can modify cytokine release patterns within T cells, promoting Th2-type responses while inhibiting Th1-type differentiation. Additionally, androgens can suppress the proliferation of peripheral T cells and diminish their responsiveness to stimuli. In the context of androgen deficiency, there can be a relative increase in Th1-type T cells, which, due to their strong RANKL expression, disrupt the Th1/Th2 balance (De Martinis et al., 2020; Gubbels Bupp & Jorgensen, 2018). This elevation in RANKL levels can lead to increased osteoclast activity and bone resorption, thereby heightening the risk of osteoporosis. The presence of inflammation further skews the balance towards bone resorption mediated by osteoclasts. Th1 cells are significant producers of IFN-γ, a cytokine that inhibits RANKL signaling during osteoclast differentiation (Wu et al., 2021). The effects of androgens on Treg cells vary by sex. In certain disease models, androgens may impair Treg function in males, while in females, they can enhance the expression of Foxp3, a marker of Tregs, within the total T cell population. Although androgens typically support Treg function in females, excessively low levels may inhibit their ability to effectively regulate RANKL secretion, ultimately contributing to increased osteoclast production and diminished protection against osteoporosis (Gubbels Bupp & Jorgensen, 2018).
In conclusion, T cell subtypes play a crucial role in the pathogenesis of osteoporosis. The differential regulation of RANKL secretion by Th1, Th2, Th17 cells, and Tregs has significant implications for bone metabolic balance and osteoporosis development.
Neutrophil
In the context of inflammatory bone loss disorders, there exists a significant relationship between neutrophils and RANKL secretion. Activated neutrophils utilize membrane-bound RANKL present on their cell membranes to directly facilitate osteoclast differentiation and bone resorption, as opposed to relying on soluble RANKL. Various characteristics can be employed to assess the activation status of mature neutrophils: 1. Protein tyrosine phosphorylation—This serves as a critical indicator of neutrophil activation. 2. Surface markers—Mature neutrophils demonstrate a high expression of the marker CD66b, as evidenced by flow cytometry, whereas immature granulocytes exhibit minimal to no expression of this marker. Upon activation, neutrophils experience alterations in their cytokine production, notably a reduction in IL-8 and IL-1beta levels (Schneider et al., 2024). Upon exposure to inflammatory stimuli, neutrophils form neutrophil extracellular traps (NETs) composed of DNA, histones, neutrophil granule proteins, and other constituents. The formation of NETs is initiated when TLR ligands activate neutrophils. NETs are associated with bone degradation and are produced in elevated concentrations in conditions such as rheumatoid arthritis (Tominari et al., 2024). Research has indicated that NETs enhance the number of osteoclasts and the extent of bone resorption by promoting osteoclast fusion and functional activity. Furthermore, NETs inhibit the release of OPG while stimulating RANKL release from osteoblasts, which indirectly amplifies osteoclast activation and exacerbates bone loss. The interplay between neutrophil membrane-bound RANKL and NETs plays a critical role in promoting osteoclastogenesis and bone resorption, thereby contributing to the development of osteoporosis (Schneider et al., 2024).
Androgens are known to impact neutrophil activity by interacting with the AR present on neutrophils, thereby partially mediating bone loss. The expression pattern of AR is consistent in both men and women, extending across the spectrum of neutrophils from proliferative progenitors to mature cells (Lai et al., 2012; Mantalaris et al., 2001). The influence of androgens on neutrophil function occurs through a dual regulatory mechanism: on one hand, they promote neutrophil differentiation, while on the other, they inhibit the inflammatory activity exhibited by mature neutrophils. Evidence indicates that testosterone can diminish both the antibacterial capacity and the secretion of pro-inflammatory factors by neutrophils, concurrently promoting the anti-inflammatory factor IL-10 (Gubbels Bupp & Jorgensen, 2018; Lai et al., 2012). In the pathophysiology of osteoporosis, such modifications may also lead to a reduction in the expression of neutrophil membrane-bound RANKL, thereby mitigating its activation of osteoclasts and potentially lessening bone loss.
Mast cell
The interaction between mast cells and osteoclasts has garnered considerable attention in the study of bone-destructive conditions such as osteoporosis. In one investigation, human mast cells (HMCs) differentiated from CD34+ stem cells were co-cultured with human monocyte-derived osteoclast precursors. This study revealed that HMCs are capable of expressing membrane-bound RANKL on their surface without the requirement for exogenous RANKL. The presence of membrane-bound RANKL was shown to directly promote osteoclast differentiation and enhance bone resorption activity. Notably, the induction of osteoclastogenesis by HMCs was inhibited upon the introduction of the RANKL inhibitor OPG, thereby affirming the significance of RANKL as a mediating factor in this process (Ng et al., 2022).
Moreover, research involving the human mast cell line LUVA indicated that activation by inflammatory stimuli significantly elevated the expression levels of the RANKL gene, contributing to the mediation of osteoporosis. Collectively, these findings suggest that mast cells may facilitate osteoclast activation through the continuous secretion of RANKL, a key factor in the pathophysiology of osteoporosis. However, it has been observed that the suppression of mast cell-derived RANKL has only a minimal effect on normal physiological bone metabolism (Fischer et al., 2023). Therefore, further comprehensive studies are warranted to fully elucidate the role of mast cell-derived RANKL in osteoporosis. Additionally, the relationship between androgens and mast cells presents a more complex scenario. Previous research has indicated the expression of AR on mast cells, yet the specific effects of androgens on mast cell degranulation and related processes require further investigation (Sciarra et al., 2023).
Androgen regulation of bone metabolism via the central nervous system
Hypothalamic-pituitary-gonadal (HPG) axis
The HPG axis plays a crucial role in the regulation of bone metabolism in females. This regulatory pathway begins with the hypothalamus, which secretes gonadotropin-releasing hormone (GnRH) to control the activity of the pituitary gland. When there is a state of androgen deficiency, feedback mechanisms are activated, leading to an increased release of GnRH. In response to GnRH stimulation, the anterior pituitary gland releases oxytocin (OT), prolactin (PRL), follicle-stimulating hormone (FSH), and luteinizing hormone (LH). The action of FSH and LH on the ovaries stimulates the secretion of various sex hormones, including estrogens, progestins, androgens, and inhibins (Kauffman, 2024; Nicks, Fowler & Gaddy, 2010).
Androgen deficiency, particularly among hypopituitary women, can vary in its impact on bone density; however, current evidence regarding the significance of androgens in women and the effects of hormone replacement therapy remains limited (Esposito et al., 2024). Beyond their essential involvement in reproductive function, this intricate regulatory network is closely linked to bone metabolism. Disruption at any point within this system can adversely affect the normal progression of bone metabolism, potentially leading to a variety of related disorders. Recent studies have demonstrated that FSH can stimulate osteoclast activity, negatively influencing bone mass. Furthermore, FSH has been found to positively impact adipocyte function, and the blockade of FSH using antibodies against the FSH receptor has resulted in increased bone mass and decreased body fat (Bergamini et al., 2024). Additionally, LH and FSH exert indirect control over bone development by encouraging the ovary to produce sex hormones (Nicks, Fowler & Gaddy, 2010). Oxytocin contributes to bone dynamics by attaching to receptors on bone cells, regulating the function of bone marrow mesenchymal stem cells, osteoblasts, osteoclasts, and other cellular components, thereby stimulating bone formation and inhibiting bone resorption (Wang et al., 2024). Nevertheless, prolactin has detrimental effects on osteoporosis; its multifaceted mechanisms contribute to increased bone resorption and decreased bone formation, reducing bone mineral density and a heightened risk of fractures (di Filippo et al., 2020).
The role of progesterone in bone metabolism remains less well-defined; it may operate through distinct receptors on osteoblasts or by modulating the expression of TGF-beta. Inhibins have been shown to suppress the differentiation of both osteoblasts and osteoclasts, consequently lowering bone turnover and correlating with menopausal bone loss, while activins promote the differentiation of osteoblasts. Estrogen has a significant impact on both osteoblasts and osteoclasts, regulating their proliferation, differentiation, and activity, as well as influencing bone remodeling through the modulation of cytokine production (Nicks, Fowler & Gaddy, 2010).
In summary, the HPG axis plays a delicate and integral role in maintaining the balance of bone formation and resorption through the antagonistic and synergistic interactions of various hormones, a balance that can be disrupted by androgen deficiency. Abnormalities within any component of this system may lead to disorders of bone metabolism, such as osteoporosis. Therefore, a thorough investigation into the individual and collective effects of these hormones is essential to advance our understanding of the mechanisms underlying bone diseases and to develop personalized treatment strategies.
Hypothalamic-pituitary-adrenal (HPA) axis
Androgens bind to pituitary and hypothalamic ARs, exerting significant regulatory effects on the HPA axis. At the hypothalamic level, the activation of the HPA axis is reduced when androgens like testosterone inhibit the production of corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) within small cell neuroendocrine neurons located in the dorsal medial section of the paraventricular nucleus (PVN) (Zuloaga, Lafrican & Zuloaga, 2024). There is a negative correlation between testosterone levels and stress-induced responses involving adrenocorticotropic hormone (ACTH) and corticosterone, as androgens suppress ACTH release in a dose-dependent manner at the pituitary level. When androgen levels drop, the HPA axis gets activated via feedback mechanisms, leading to the release of ACTH from the pituitary gland due to increased CRH and AVP secretion, which then stimulates the adrenal glands to produce high levels of cortisol, such as GCs (Williamson, Bingham & Viau, 2005).
Glucocorticoid-induced osteoporosis (GIO) represents a prevalent form of secondary osteoporosis, contributing to increased morbidity and mortality. The pathogenesis of GIO involves decreased bone formation and increased bone resorption (Wang et al., 2020b). Elevated GCs influence bone formation in two ways: under normal physiological conditions, endogenous GCs support bone formation, while excessive GCs inhibit the PI3K/Akt pathway by downregulating Sema3A expression, hindering osteoblast differentiation (Xing, Feng & Zhang, 2021). Androgens are crucial for maintaining bone health by regulating GC levels and preventing hyperactivation of the HPA axis, thereby protecting bone density. Conversely, when there is a deficiency of androgens, the HPA axis becomes overactive, leading to increased GC levels and consequent bone loss. Interestingly, a retrospective study indicated that androgen excess might protect bone density in women suffering from classical congenital adrenocortical hyperplasia (CAH) who have been exposed to high cumulative doses of GCs (Lee et al., 2022). This body of evidence underscores the pivotal role of androgens in the neuroendocrine-skeletal regulatory system, highlighting their mechanisms as potential key targets in developing therapeutic strategies for bone metabolic disorders.
Regulation of neurotransmitters
Androgens significantly influence bone metabolic balance by modulating the activity of dopaminergic neurons within the central nervous system. The mechanisms through which androgens affect dopamine secretion are characterized by bidirectional regulation and changes in neuronal populations. Specifically, androgens play a crucial role in regulating the burst firing of dopamine neurons by inhibiting prefrontal glutamatergic neurons from projecting to the ventral tegmental area (VTA) and by reducing NMDA receptor-mediated inhibitory control. This regulatory action helps maintain dopamine levels in the prefrontal cortex within an optimal range (Kritzer, Adler & Locklear, 2025). At the structural level, supplementation with testosterone or its metabolite, dihydrotestosterone (DHT), can reverse the effects of androgens that increase neuronal numbers following denervation, thereby preventing the survival of dopamine neurons located in the substantia nigra pars compacta (SNpc) and VTA (Johnson et al., 2010). These processes indicate that androgens dynamically modulate both the activity and survival of dopamine neurons, thereby influencing the functional equilibrium of the dopamine system.
Disruptions in the central dopamine system can significantly impact the processes of bone remodeling through the actions of dopamine receptors expressed in bone tissue. All subtypes of dopamine receptors (D1–D5) are expressed by human monocyte-derived osteoclast precursor cells. In patients with rheumatoid arthritis, stimulation of D2-like receptors has been shown to significantly enhance osteoblast mineralization and promote the formation of bone matrix. Activation of D2-like receptors (D2–D4) dramatically suppresses RANKL-induced osteoclast differentiation and bone resorption by lowering intracellular cAMP levels, thereby reducing TRAP-positive cell numbers, Cathepsin K expression, and the area of bone resorption (Schwendich et al., 2022; Wang et al., 2021). In contrast, D1-like receptors (D1/D5) may indirectly contribute to inflammatory bone loss by influencing Th17 cell differentiation while simultaneously promoting osteoblast function; however, they do not directly impact osteoclast differentiation (Feng et al., 2023a; Schwendich et al., 2022). Additionally, osteoblasts possess the ability to synthesize dopamine independently and utilize both autocrine and paracrine pathways to influence local dopamine receptor activity, creating an autoregulatory network that is integral to bone metabolism (Schwendich et al., 2022).
Through the regulation of dopamine neuron numbers and activity, androgens appear to indirectly support the homeostasis of bone metabolism. These findings underscore the role of the dopamine system in promoting bone formation through receptor-mediated signaling pathways, thereby establishing a novel regulatory network involving the androgen-dopamine-skeletal axis. This framework provides a molecular basis for understanding the mechanisms associated with neuroendocrine-related osteoporosis and the protective effects of androgen replacement therapy on bone health.
Androgen regulation of bone metabolism via the peripheral nervous system
Sensory nerve fibers and neuropeptides
Sensory nerves, which are extensively distributed throughout the periosteum—especially in areas of active bone metabolism—play a crucial role in osteogenesis through the secretion of neuropeptides, including CGRP, SP, and Sema3A (Chen et al., 2024). These neuropeptides can significantly influence the equilibrium of bone remodeling by directly interacting with various bone cells, including osteoblasts, MSCs, and chondrocytes, via specific receptors, or by modulating local inflammation and angiogenesis (Chen et al., 2024; Steverink et al., 2021). The regulation of bone homeostasis by sensory nerves occurs through the secretion of neuropeptides that inhibit osteoclast activity while promoting osteoblast differentiation. CGRP is known to reduce osteoclastogenesis through the inhibition of the RANKL signaling pathway, as well as activate the cAMP/PKA/CREB pathway in osteoblasts to enhance bone formation. Low concentrations of SP increase the proliferative capacity of BMSCs and promote their differentiation into osteoblasts via the Wnt/β-catenin pathway, while also facilitating angiogenesis while high concentrations of over-activated NK1 receptors, triggering the release of pro-inflammatory factors (e.g., IL-6, TNF-alpha), exacerbated local inflammatory responses, and indirectly enhanced osteoclast activity through the up-regulation of RANKL signaling, leading to pathological bone resorption (Chen et al., 2024; Stöckl et al., 2025). Meanwhile, Sema3A serves a vital role in maintaining bone homeostasis by binding to the Nrp1 receptor present on both osteoblasts and osteoclasts, thereby facilitating a dual effect of promoting osteoblast differentiation and inhibiting RANKL-induced osteoclast differentiation (Xu, 2014). Importantly, osteoblasts can also secrete Sema3A; however, the expression of Sema3A originating from osteoblasts is not influenced by androgen signaling (Yamashita et al., 2022).
Research involving a conditional knockout mouse model (SLICK-AR), which specifically deletes the DNA-binding domain of the AR in neurons, has demonstrated that the AR in these neurons directly facilitates sensory neuron axon regeneration through a DNA-binding domain-dependent genomic signaling pathway. It is noteworthy that this action exhibits sex-differentiated characteristics, with female subjects potentially compensating for AR deficiency through non-neuronal AR signaling mechanisms (e.g., glial cells) or ER pathways. Furthermore, aging or decreased testosterone levels may result in reduced nerve regeneration (Ward et al., 2021). Thus, strategically targeting AR signaling may provide synergistic benefits in promoting peripheral nerve regeneration, enhancing functional recovery, and supporting the maintenance of bone metabolic homeostasis.
Synovial tissue plays a critical role by lining the noncartilaginous surfaces of synovial joints, serving to nourish these avascular structures, which are vital for bone formation. Prolonged inflammation within the synovial tissue can result in bone destruction as well as periarticular and systemic osteoporosis (Tanaka, 2019). Androgens have been found to modulate the distribution and activity of sensory nerves through synovial AR, which influences the release of neuropeptides, thereby regulating the equilibrium between bone formation and resorption. An elevated ratio of estrogen to androgen can suppress AR signaling. This suppression may lead to the growth of sensory nerve fibers from inflamed tissue, disrupting the release of neuropeptides such as SP and subsequently amplifying inflammation and exacerbating bone destruction (Liu et al., 2023).
Furthermore, androgens are recognized for their anti-inflammatory effects. Inflammation can alter bone structure through modified usage and the production of various inflammatory mediators. It is also worth noting that the peripheral nervous system may assume a sensory role in responding to mechanical and inflammatory influences on bone structure (Straub et al., 2013). Targeting the androgen-sensory neuraxis presents a promising avenue for novel therapeutic strategies aimed at treating rheumatoid arthritis and related bone metabolic disorders. It is essential to pursue further research to explore the underlying molecular mechanisms and assess the clinical translational potential of this approach.
Neuromuscular junction
Bone possesses the ability to sense mechanical signals and convert them into biochemical signals, which are crucial for maintaining bone homeostasis (Mei et al., 2024). The neuromuscular junction (NMJ), as a specialized synapse between motor neurons and skeletal muscle fibers, is central to this process. It facilitates the release of acetylcholine (ACh), which activates the postsynaptic membrane acetylcholine receptor (AChR). This activation generates an endplate potential (EPP), leading to action potentials in muscle fibers that enable contractile functions. In recent years, accumulating evidence has highlighted the crosstalk between muscle and bone, with a pooled analysis indicating that osteomuscular dystrophy is associated with an increased risk of fractures, falls, and mortality (Teng et al., 2021). The modulation of muscle contractile activity may significantly influence the pathophysiology of osteoporosis, indirectly affecting bone metabolism. Mechanical stress from muscle activity initiates strain production in bone through mechanical loading, thereby enhancing the osteogenic response. This occurs through the stimulation of nitric oxide (NO) and prostaglandins (PGE2) release from osteoblasts, which synergizes with hormone therapy and promotes the secretion of myofactors (such as FGF-2 and IGF-1) and bone-derived factors (such as osteocalcin). Collectively, these factors regulate the processes of bone formation and resorption (Li et al., 2019; Notelovitz, 2002).
Impairments in the structure or function of the NMJ frequently lead to decreased efficiency in neuromuscular transmission. Such dysfunction can result in reduced mechanical loads on bone, ultimately diminishing the activation of bone formation signaling and increasing bone resorption. Moreover, NMJ dysfunction may contribute to decreased endocrine hormone release, exacerbating the neural-skeletal connection and potentially accelerating bone loss and the development of osteoporosis (Iyer, Shah & Lovering, 2021; Kaji, 2024). Studies indicate that following gonadectomy in adult male rats, testosterone deficiency results in a significant reduction of nicotinic-type AChR tropism, with a 70% decrease in the amplitude of neurally evoked endplate currents and a 1.8-fold reduction in maximal AChR binding in rat aponeurotic muscles, although receptor-binding rate constants remain unaffected. These observations suggest that testosterone is vital for modulating neuromuscular transmission by maintaining the density of postsynaptic membrane AChRs without altering their binding properties (Souccar et al., 1991). Furthermore, androgens have been shown to enhance muscle mass and strength, stimulate bone through mechanical loading, and promote bone formation (Chang et al., 2013; Notelovitz, 2002).
Synergistic mechanisms of immune-neural interactions
The maintenance of skeletal homeostasis is contingent upon the synergistic regulation of the endocrine, immune, and neural systems. Androgens play a critical role in this interaction by inhibiting pro-inflammatory factors such as IL-6 and TNF-alpha, regulating the RANKL/OPG axis, and facilitating immune-neural communication through neurotransmitters. This integrated system collectively reduces osteoclast activity and promotes osteogenic differentiation. An imbalance in this regulatory network can result in heightened bone resorption, ultimately leading to the development of osteoporosis (Banoriya et al., 2025).
ARs are expressed in various immune organs and cells, thereby establishing a foundation for androgen modulation of immune function. This expression significantly impacts the development and functionality of immune cells, while also providing protective effects on the neuromuscular junction, thereby shielding it from inflammation and oxidative damage to maintain normal physiological function (Gubbels Bupp & Jorgensen, 2018). During bone regeneration, a dynamic interaction occurs between peripheral nerves and immune cells. Peripheral nerves release neuropeptides, including CGRP, SP, and norepinephrine (NE), which influence the polarization and functionality of immune cells, particularly macrophages. Conversely, cytokines such as TNF-alpha and IL-1beta produced by immune cells can activate sensory nerves and regulate nerve signaling through feedback mechanisms. This reciprocal communication transpires via common signaling pathways, notably the Wnt/β-catenin and NF-kappaB pathways, which synergistically regulate osteoblast differentiation, osteoclast activity, and angiogenesis, thereby facilitating the establishment and repair of the bone regeneration microenvironment (Minoia et al., 2022; Tao et al., 2023). Additionally, the sympathetic nervous system exerts regulatory effects on immune function by activating adrenergic receptors on immune cells, particularly the beta2-adrenergic receptor, which is widely expressed. However, overactivation of the immune system may exacerbate bone loss. Sensory nerves are also essential for maintaining bone homeostasis; their severance can impede inflammatory regression and alter macrophage polarity and origin (Ahmari, Hayward & Zubcevic, 2020).
Research involving the dorsal root ganglion (DRG) has demonstrated that the modeling of depletion and subsequent repopulation of DRG macrophages reveals the presence of four distinct macrophage subpopulations following nerve injury. Among these populations, self-renewing DRG macrophages are critical for promoting axon regeneration, with no observed gender differences in this role (Feng et al., 2023b). Additionally, the transfer of IL-4/G-CSF polarized bone marrow neutrophils into a central nervous system (CNS) injury model has been shown to trigger substantial axonal regeneration in the optic nerve and spinal cord (Jerome et al., 2024). These findings illuminate the intricate relationship between nerves, immunity, and bone, thereby providing new insights and potential directions for the treatment of related diseases.
Osteoporosis induced by androgen deficiency exhibits sexual dimorphism
The effects of androgens on osteoporosis exhibit significant gender dimorphism. A large-scale study of American adolescents provides direct evidence for this disparity: in males, serum testosterone levels show a stable positive correlation with bone mineral density (BMD), confirming its fundamental role in maintaining skeletal health. However, in females, this association exhibits a complex inverted U-shaped relationship, indicating that moderate testosterone levels benefit female bones, but beyond a specific threshold, its protective effect diminishes or even vanishes (Xu et al., 2022). This clinical observation raises questions about the pathophysiological mechanisms underlying the distinct etiologies of osteoporosis in men and women. Androgen deficiency has been widely recognized as the primary cause of male osteoporosis. In contrast, the role of androgens in female skeletal health is more complex and nuanced. One hypothesis suggests that despite androgen deficiency in women, adequate androgens primarily promote bone health by converting into estrogens, while potentially exerting auxiliary effects through AR. However, this protective effect has an upper limit—excessively high androgen levels (as seen in PCOS patients) disrupt normal bone metabolism, consistent with observations that high androgen concentrations diminish protective effects (Zhang et al., 2024). Crucially, mechanistic research provides definitive evidence for this complex interplay. One pivotal study employing an aromatase knockout (ArKO) mouse model dissected the mechanisms underlying this “androgenic osteoporosis” paradox. This model, which features elevated endogenous androgen levels that cannot be converted into estrogen, creates a high-androgen, low-estrogen environment. Histomorphometric analysis revealed that while both male and female ArKO mice developed osteoporosis due to estrogen deficiency, their bone remodeling patterns showed fundamental sex-specific differences. In males, the critically low estrogen levels led to suppressed osteoblast activity and low-turnover bone loss, despite the presence of high androgen levels. In females, the model resulted in a high-turnover state. This evidence conclusively demonstrates that the skeletal outcome is not solely determined by androgen levels but is profoundly dependent on their aromatization into estrogen, thereby explaining the gender dimorphism observed in both clinical and pathological settings (Oz et al., 2000).
Sex differences in immune responses
Sex hormones are key regulators of immune system and skeletal function, and alterations in their levels—such as age-related declines—constitute major risk factors for osteoporosis. In males, androgens influence immune responses through AR signaling or aromatization into estrogens, thereby maintaining bone homeostasis. Overall, declining androgen levels activate the immune system, thereby driving bone resorption. In women, skeletal immune regulation is more complex, involving a sharp drop in estrogen and relative changes in androgen levels. Together, these factors induce a chronic, low-grade inflammatory state that drives bone loss. Sex hormones (estrogens, progestogens, and androgens) act as key immunomodulators, regulating innate immune cell functions through their nuclear receptors and inducing epigenetic remodeling, thereby participating in the regulation of sexual dimorphism in bone metabolism. Androgens exert overall anti-inflammatory and immunosuppressive effects, indirectly inhibiting osteoclast activity by suppressing macrophage cytokine release, downregulating TLR4 expression, and reducing leukotriene synthesis, thereby protecting male bones. Similarly, progesterone exhibits potent immunosuppressive properties, which contribute to maintaining bone immune homeostasis. Estrogen’s effects are more complex and concentration-dependent: At physiological levels, it moderately enhances immune responses. At high concentrations, it exhibits anti-inflammatory effects by suppressing Th1/Th17 cells and enhancing Treg function, potentially providing indirect skeletal protection. Postmenopausal estrogen decline leads to increased inflammation, contributing to osteoporosis. These sex hormone-mediated differences in the immune microenvironment partially explain why postmenopausal women, whose immune state shifts toward pro-inflammation, are more susceptible to osteoporosis. In contrast, men enjoy relative bone protection due to the sustained immunosuppressive effects of androgens, highlighting the central role of immune mechanisms in sex hormone-related bone metabolic disorders (Forsyth et al., 2024; Shepherd et al., 2020). Androgens regulate Tregs by directly acting on the Foxp3 gene. Within Treg cells, upon activation, androgen receptors bind directly to this specific region of the Foxp3 gene, thereby initiating gene transcription. This effect exhibits specificity in terms of gender and physiological state: androgens significantly increase Foxp3+ Treg numbers only in female T cells during the ovulatory phase, with no such effect observed in male T cells or female T cells in other menstrual cycle phases. Tregs are potent immunosuppressive cells that effectively inhibit the activation and function of effector T cells, macrophages, and osteoclast precursors. By upregulating Tregs, androgens indirectly suppress osteoclast generation and activity, thereby reducing bone resorption and providing protective effects for maintaining bone density. Persistently elevated androgen levels in males may keep the Treg-skeleton axis in a stable, “locked-in” protective state over the long term, contributing to the generally lower incidence of osteoporosis in men compared to women of the same age. The androgen surge during female ovulation may dynamically maintain immune and skeletal homeostasis throughout the reproductive cycle by transiently and potently expanding Tregs to counterbalance the pro-inflammatory tendencies potentially induced by estrogen at this time. Postmenopausally, with declines in both androgen and estrogen, this dynamic protective mechanism of Tregs weakens, potentially exacerbating bone loss (Walecki et al., 2015).
Sex differences in neuroendocrine regulation
Beyond immune regulation, the nervous system’s control over bone metabolism also exhibits significant sex differences, with androgens playing distinct roles in males and females. In males, high levels of androgens exert a direct effect via AR on corticotropin-releasing factor (CRF) neurons widely distributed throughout the limbic system (e.g., bed nucleus of the stria terminalis, amygdala). This action constitutes a potent “tonic inhibition” of the CRF system, stably reducing the basal activity and stress responsiveness of the HPA axis, thereby maintaining GCs at physiologically low levels. When androgen deficiency occurs (e.g., castration, aging), this inhibition is lifted. This leads to excessive activation of neural circuits involved in stress and anxiety—particularly CRF neurons in the bed nucleus of the stria terminalis and amygdala—which ultimately drives increased release from CRF neurons in PVN via not-yet-fully-elucidated neural connections. This results in HPA axis hyperactivation and pathologically elevated GCs levels. Persistently elevated GCs then reduce bone formation by inhibiting osteoblast activity and promoting osteoblast apoptosis, ultimately triggering or exacerbating osteoporosis. This pathway is particularly pronounced in males, as their CRF neurons express AR at significantly higher levels than females, constituting a key mechanism for the neuroendocrine regulation of osteoporosis gender dimorphism (Rybka et al., 2023).
Discussion
Osteoporosis, a complex metabolic bone disease, arises from the intricate interplay of multiple biological systems, particularly under the influence of androgen deficiency. This review elucidates the molecular mechanisms through which androgens affect bone metabolism via immune and neural pathways, emphasizing the critical role of the “immune-neuro-skeletal” regulatory network in maintaining bone homeostasis.
While numerous reviews have addressed the role of androgens in bone metabolism, the present article distinguishes itself through its integrative focus on the immune-neural regulatory network in the context of androgen deficiency. Previous reviews have often treated immune and neural pathways as separate entities, with limited discussion of their cross-talk. In contrast, we systematically synthesize evidence linking androgen signaling to both immune cell behavior (e.g., macrophage polarization, T-cell differentiation, EPO/EPOR signaling) and neural modulation (e.g., HPA axis, sensory neuropeptides, neuromuscular junction), thereby proposing a novel “immune-neuro-skeletal” axis. This integrative perspective not only clarifies how androgens coordinate multi-systemic control over bone homeostasis but also identifies potential synergistic therapeutic targets—such as CGRP analogs, Sema3A agonists, or Th17 inhibitors—that have not been collectively emphasized in earlier works. By foregrounding the crosstalk between immunity and innervation, this review offers a more holistic framework for understanding and treating androgen deficiency-induced osteoporosis, paving the way for future interdisciplinary research.
Androgens significantly modulate bone metabolism by influencing immune cell functionality, predominantly through the EPO signaling pathway and the regulation of RANKL secretion. Notably, androgens exhibit immunomodulatory effects that inhibit the activity of pro-inflammatory immune cells and RANKL secretion while promoting the differentiation of anti-inflammatory phenotypes. This creates an “immune checkpoint” mechanism that protects against osteoporotic changes. Furthermore, the bi-directional regulation of the androgen-EPO axis underscores the necessity to consider dosage dependence in clinical treatment, highlighting the complexity of androgen action.
However, it is essential to acknowledge the existing contradictions in the literature regarding the osteoprotective effects of androgens. These discrepancies may arise from sex- and tissue-specific responses to androgens, such as the exacerbation of localized inflammation in conditions like PCOS while simultaneously offering protective effects on bone density. Differences in the regulation of Treg cells between genders further complicate the understanding of androgen’s effects. The variability in tissue microenvironments suggests that androgen action is highly context-dependent, necessitating a nuanced approach in therapeutic applications.
In addition to their immune regulatory role, androgens confer multifaceted protection for bone health through neuromodulation. This involves both central and peripheral nervous system mechanisms. Androgens inhibit the activation of the HPA axis, thereby minimizing glucocorticoid-induced inhibition of osteoclasts. Concurrently, they enhance NMJ function, increasing mechanical stress on bones through sustained ACh receptor density and muscle contractility. A deterioration of NMJ efficiency due to androgen deficiency leads to diminished mechanical loading and accelerates disuse osteoporosis.
It is also crucial to recognize the interconnectedness of immune and neural pathways in bone metabolism. Neuropeptides, such as CGRP and Sema3A, secreted by sensory nerves exert direct inhibitory effects on osteoclast activity, while sympathetic nervous system activation modulates immune cell functionality via β2-adrenergic receptors. This interplay forms a closed “immune-neural-skeletal” feedback loop that is essential for bone health.
The findings presented herein validate the hypothesis that androgens directly influence RANKL secretion through androgen receptors on immune cells. Evidence of macrophage polarization imbalances and altered T-cell subpopulations further supports the role of androgens in regulating osteoclast activity. Moreover, the dual regulatory effect of EPO signaling is apparent, with physiological levels promoting osteoblast differentiation and supraphysiological levels triggering osteoclastogenesis. This evidence substantiates the deeper connections between androgen deficiency and disrupted bone-immune crosstalk. Despite these advancements, several areas warrant further exploration. In particular, the molecular mechanisms underlying gender-specific variations in Treg cell regulation by androgens in women require more comprehensive elucidation. Furthermore, the debate surrounding the role of mast cell-derived RANKL in osteoporosis indicates that specific immune cell functions may be significantly influenced by the tissue microenvironment. These lingering questions underscore the importance of continued research in this domain.
The integrative analyses offered in this review pave the way for potential targeted therapies for androgen-induced osteoporosis. Strategies such as targeting immune regulatory pathways—specifically the TLR4/MyD88 pathway or Th17 differentiation inhibitors—and exploring neuroprotective agents, including CGRP analogs or Sema3A agonists, may represent viable therapeutic options. However, translating these findings into clinical practice presents challenges. Notably, androgen replacement therapy poses potential risks, such as increased prostate cancer incidence, necessitating the development of tissue-specific androgen receptor modulators. Furthermore, the complexity of immune-neural interactions necessitates therapeutic regimens that are both systemic and precisely tailored.
Despite the comprehensive synthesis presented herein, several limitations in the existing literature warrant acknowledgment. First, the majority of mechanistic insights are derived from animal models, particularly rodent studies, which may not fully recapitulate the complexity of human bone immunology and neuroendocrinology. Translational challenges arise from interspecies differences in immune cell populations, hormonal regulation, and bone turnover rates. Second, the gender-specific effects of androgens on immune and neural pathways remain inadequately characterized. While androgens exert distinct effects in males and females, most preclinical studies are conducted in male animals, limiting the generalizability of findings to females or transgender individuals undergoing hormone therapy. Furthermore, the interplay between androgens, aging, and comorbidities—such as chronic inflammation or metabolic syndrome—adds another layer of complexity that is often unaddressed in controlled experimental settings. Future research should prioritize human cohort studies, integrated multi-omics approaches, and sex-stratified analyses to bridge these gaps and validate the proposed immune-neural regulatory axis in diverse populations.
While this review synthesizes current evidence on the immune-neural regulatory network in androgen deficiency-induced osteoporosis, several critical scientific questions remain unresolved and warrant urgent investigation:
Sex-specific regulation of tregs cells by androgens: The differential effects of androgens on Tregs between males and females remain poorly understood. Future studies should elucidate how androgen signaling modulates Treg function in a sex-dependent manner and its implications for bone immune homeostasis.
Dose-dependent mechanisms of EPO/EPOR Signaling in Bone Immunity: The dual role of EPO in promoting osteogenesis at physiological levels while inducing osteoclastogenesis at high doses highlights the need to delineate the precise dose-response relationship and downstream signaling mechanisms in bone-immune cross-talk.
Neuro-immune crosstalk in the bone microenvironment: The molecular pathways mediating communication between sensory neuropeptides and immune cells in bone remain largely unexplored. Investigating these interactions could reveal novel therapeutic targets for osteoporosis.
Tissue-specific androgen action in inflammatory bone loss: the paradoxical effects of androgens—protective in bone yet pro-inflammatory in certain contexts like PCOS—underscore the need to understand how tissue microenvironment influences AR signaling and immune cell behavior.
Addressing these questions will not only advance our mechanistic understanding but also facilitate the development of targeted, sex-specific, and context-aware therapeutic strategies for osteoporosis induced by androgen deficiency.
Conclusion
In conclusion, this review synthesizes compelling evidence that androgen deficiency contributes to osteoporosis not only through direct effects on bone cells but, more significantly, by disrupting a sophisticated immune-neuro-skeletal regulatory network. We have delineated the key mechanisms by which androgens, via this network, maintain bone homeostasis: by modulating immune cell function (e.g., promoting M2 macrophage polarization, regulating T-cell subsets, and fine-tuning the EPO/EPOR axis) to suppress RANKL-driven osteoclastogenesis, and by regulating neural pathways (e.g., inhibiting the HPA axis and enhancing sensory neuropeptide signaling and neuromuscular junction function) to promote bone formation and mitigate resorption.
The identification of this integrated axis, with its inherent sexual dimorphism, provides a novel and holistic framework for understanding the pathophysiology of androgen deficiency-induced osteoporosis. It moves beyond traditional, siloed perspectives and highlights the critical cross-talk between different physiological systems. This refined understanding opens promising avenues for future research and paves the way for developing innovative, multi-targeted, and sex-stratified therapeutic strategies aimed at restoring balance within the immune-neuro-skeletal network to combat bone loss effectively.
Supplemental Information
Detailed literature search strategy for Immune-Neural Regulatory Mechanism of Osteoporosis Induced by Androgen Deficiency.
The comprehensive search methodology, including databases, date ranges, keyword concepts, and screening process, used to identify studies exploring the interplay between androgen signaling, bone metabolism, immune regulation, and signaling pathways such as erythropoietin and neuropeptides.