
Comparative genome analysis of patulin-producing Penicillium paneum OM1 isolated from pears
- Published
- Accepted
- Received
- Academic Editor
- Sushanta Deb
- Subject Areas
- Agricultural Science, Genomics, Mycology
- Keywords
- Biosynthetic gene cluster, Genome sequence, Patulin biosynthetic gene, Penicillium paneum OM1, Secondary metabolite
- Copyright
- © 2025 Zhao et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2025. Comparative genome analysis of patulin-producing Penicillium paneum OM1 isolated from pears. PeerJ 13:e19848 https://doi.org/10.7717/peerj.19848
Abstract
Background
The filamentous fungus Penicillium paneum (P. paneum) produces patulin as a toxic secondary metabolite (SM) on apples and pears. Little is known about the biosynthetic gene clusters (BGCs) of SMs, including patulin in P. paneum.
Methods
In this study, we sequenced the whole genome of P. paneum (isolate OM1), a patulin producer isolated from pears, and analyzed the genome sequence to identify its SM BGCs and compare its patulin BGC with those in other patulin-producing strains. In addition, we investigated the genes that encode carbohydrate-active enzymes (CAZymes) in P. paneum OM1, which play important roles in the degradation of plant cell walls, and analyzed the phylogenetic relationships among P. paneum OM1 and other closely related Penicillium species.
Results
The genome of P. paneum OM1 was estimated to be approximately 27.16 Mb with four chromosomes. Gene Ontology analysis using 7,098 functionally annotated proteins showed that genes involved in fungal defense mechanisms, such as SM biosynthesis, are enriched in the genome of P. paneum OM1. Of the 7,098 functionally annotated proteins from the genome, we identified 370 putative CAZymes. A phylogenetic analysis revealed that P. paneum OM1 has an evolutionarily close relationship with Penicillium chrysogenum (isolate Wisconsin 54-1255, a penicillin-producing strain) and Penicillium digitatum (isolate Pd1, a citrus fruit pathogen). We also identified a total of 33 SM BGCs, including a patulin BGC in P. paneum OM1. Moreover, the functional conservation analyses on all 15 patulin biosynthetic genes showed that each gene in P. paneum OM1 shares a high degree of sequence identity (above 73% identity) at both nucleotide and amino acid levels with the corresponding genes in four other patulin-producing Penicillium strains, while it shares a relatively low degree of identity (0-93%, identity, 0 and 60% as medians for amino acid sequence identity) with those in two non-patulin producing Penicillium species.
Conclusions
Our study improves understanding about BGCs of SMs, including patulin in P. paneum OM1, which causes blue mold rot on pome fruits. These data could provide the genetic basis of patulin biosynthesis in P. paneum OM1 to develop effective strategies for reducing patulin contamination on pome fruits.
Introduction
Penicillium species are ubiquitous in soil and grow in temperate to cool climates (Cabanes, Bragulat & Castella, 2010). They are well-known fungi in the phylum Ascomycota (Shah et al., 2022). Some of them are economically important in food and drug industries. A member of the genus Penicillium camemberti (P. camemberti) is used for manufacturing cheese (Ropars et al., 2020), while other species including Penicillium digitatum (P. digitatum), which is a postharvest pathogen on citrus fruits, are detrimental fungal pathogens that infect important agricultural crops and cause spoilage (Yang et al., 2019). In addition, some other species such as Penicillium chrysogenum (P. chrysogenum) produce penicillin as a beneficial secondary metabolite (SM) (Peng et al., 2014).
SMs are structurally diverse, low-molecular-mass bioactive compounds. The production of SMs is a characteristic of Penicillium species (Frisvad et al., 2004). In general, SMs are not essential for growth and reproduction of microorganisms, but are responsible for their development and self-protection (Demain & Fang, 2000; Netzker et al., 2015). Genes responsible for SM biosynthesis are mainly organized together in clusters at one or more loci in fungal genomes (Brakhage, 2013). Some Penicillium species such as Penicillium expansum (P. expansum) and Penicillium griseofulvum (P. griseofulvum) are known to produce patulin, a highly toxic SM, and sometimes they can cause blue mold rot on pome fruits such as apples and pears (Morales et al., 2008; Spadaro et al., 2011).
Patulin is a mycotoxin found on mainly fruits and fruit-based products including apples, pears, peaches, cherries and their products (Bacha et al., 2023; Mahato et al., 2021; Moake, Padilla-Zakour & Worobo, 2005). Consumption of patulin-contaminated pome fruits poses a serious health risk to humans. It can cause severe acute and chronic toxicities, resulting in gastrointestinal, immunological, and neurological diseases (Puel, Galtier & Oswald, 2010; Tannous et al., 2018). Thus, it led to limitation on the level of patulin in apple juice by many regulatory agencies worldwide (European Commission, 2023; US Food and Drug Adiministration, 2005; Kang et al., 2010).
Since the patulin biosynthetic gene clusters (BGCs) have been identified in Aspergillus clavatus (A. clavatus) NRRL 1 and P. expansum NRRL 35,695 (Artigot et al., 2009; Tannous et al., 2014), they were found in many different patulin-producing Penicillium species (Ballester et al., 2015; Banani et al., 2016; Li et al., 2015; Wu et al., 2019). The patulin BGCs are conserved in the patulin producers such as P. expansum and P. griseofulvum (Ballester et al., 2015; Banani et al., 2016; Li et al., 2015).
On the other hand, unlike P. expansum, Penicillium paneum (P. paneum) as a patulin producer has been primarily found in grains or grain-derived products (Boysen et al., 1996; Nielsen et al., 2006). Only a few previous studies showed that patulin-producing P. paneum strains were isolated from pome fruits (Labuda et al., 2005; Wang et al., 2018; Yin et al., 2017). Previously, we isolated a patulin producer P. paneum (strain OM1) from pears and reported the optimal conditions for its patulin production under different physicochemical parameters (Zhao et al., 2024). Considering that P. paneum is frequently isolated from apples, pears, and grains, it is of great interest to study SM BGCs in P. paneum OM1. In addition, to date, little is known about SM BGCs including the patulin BGC in P. paneum. Thus, in the current study, in order to gain more detailed information on genetic diversity and patulin biosynthesis, the whole genome of P. paneum OM1 was sequenced and the comparative genome analysis of SM BGCs including the patulin BGC was performed using its newly sequenced genome and publicly available genomes of other closely related fungal species. Also, carbohydrate-active enzymes (CAZymes), which are involved in the breakdown of plant cell walls and the host-pathogen interactions, were compared between P. paneum OM1 and the related fungal strains. Furthermore, the phylogenetic relationships were analyzed in P. paneum OM1 and other related fungal species.
Materials & Methods
Fungal strains and culture conditions
P. paneum OM1, a patulin-producing strain isolated from a pear as described previously (Zhao et al., 2024), was used for whole-genome sequencing. For genomic DNA isolation, fungal spores (107) were inoculated into 100 mL of YES (2% yeast extract, 15% sucrose, 0.1% MgSO4 7H2O) and incubated at 20 °C for 5 days under static conditions as described previously (Zhao et al., 2024). For total RNA isolation, the fungal strain was cultured in 100 mL of YES without shaking and 100 mL of potato dextrose broth (PDB; MB cell, Seoul, Korea) with shaking at 20 °C for 5 days.
Genomic DNA extraction for whole genome sequencing
Genomic DNA was isolated from a filtered mycelium by eGnome Co. Ltd. (Seoul, Korea). Briefly, a frozen mycelium was ground into a fine powder in liquid nitrogen using a mortar and pestle. Then, high molecular weight (HMW) genomic DNA was isolated from the powdered mycelium using a modified CTAB method (Porebski, Bailey & Baum, 1997) with 2% polyvinylpyrrolidone (PVP; 1% with MW 10,000 and 1% with MW 40,000) (Sigma-Aldrich, Burlington, MA, USA). The quality and quantity of the extracted HMW genomic DNA were assessed by absorbance ratios of optical density (OD) OD260 nm/OD230 nm and OD260 nm/OD280 nm, respectively, using Synergy HTX Multi-Mode microplate reader (Biotek, Rochester, VT, USA), Quant-iT PicoGreen dsDNA assay kit (Invitrogen, Waltham, MA, USA), and Agilent 4150 TapeStation system (Agilent Technologies, Santa Clara, CA, USA). The isolated genomic DNA was stored at −20 °C until use.
Genome sequencing and assembly
The genome of P. paneum OM1 was sequenced at eGnome Co. Ltd. (Seoul, Korea) using MinION Mk1B (Oxford Nanopore Technologies, Oxford, UK) as described previously in Lee et al. (2022) with slight modifications. For long-read sequencing, short DNA fragments (<10 kb) were selectively removed using the Short Read Eliminator Kit (Circuomics, Baltimore, MD, USA). Subsequently, the sequencing library was prepared using ONT Ligation Sequencing Kit V14 (SQK-LSK114; Oxford Nanopore Technologies) according to the manufacturer’s instructions. Briefly, genomic DNA was repaired using NEBNext FFPE DNA Repair Mix (New England Biolabs, Ipswich, MA, USA) and NEBNext Ultra II End Repair/dA-Tailing Module (New England Biolabs). The end-prepped DNA was then ligated with Nanopore adapters using the NEBNext Quick Ligation Module (New England Biolabs). Next, the DNA sample was purified using AMPure XP beads (Beckman Coulter, Brea, CA, USA). The final DNA library was loaded onto a MinION flow cell (FLO-MIN114, R10.4.1; Oxford Nanopore Technologies) for sequencing on a MinION Mk1B platform using MinKNOW software (v22.12.7).
The raw reads were processed using Porechop (v0.2.4) (Wick et al., 2017) to remove adapters and demultiplex them after basecalling with Guppy (v6.4.4+e3004fa). NanoFilt (v2.8.0) (De Coster et al., 2018) was used to remove low-quality reads (quality score <7 and read length <2,000 bp). Quality control checks on both raw and filtered sequencing data were performed using NanoPlot (v1.41.0) (De Coster & Rademakers, 2023).
De novo genome assembly was performed using Flye (v2.9.2-b1786) with the parameters “–nano-hq mode, –asm_coverage 100, and –genome -size 30M” (Kolmogorov et al., 2019). The quality of the genome assembly was evaluated using Quality Assessment Tool for Genome Assemblies (QUAST; v5.1.0rc1) (Gurevich et al., 2013). The completeness of the genome assembly was assessed by analyzing Benchmarking Universal Single-Copy Orthologs (BUSCO) (v5.4.4) scores against the fungi_odb10 database under the genome mode (Manni et al., 2021). The draft assembled genome sequence was polished to remove systematic errors using Homopolish (v0.3.4) (Huang, Liu & Shih, 2021) and genome assembly data of 9 different P. paneum strains, which were retrieved from the National Center for Biotechnology Information (NCBI). However, the draft genome assembly was used as the final genome because the BUSCO value of the polished genome assembly was lower than that of the draft assembly.
Total RNA isolation and RNA-Seq analysis
After P. paneum OM1 was cultured in YES and PDB media as described above, total RNA was extracted using Qiagen RNeasy Mini Kit (Qiagen, Hilden, Germany) following a procedure provided by the manufacturer. Briefly, a filtered mycelium was ground into a fine powder in liquid nitrogen using a mortar and pestle, and approximately 100 mg of the ground mycelium was re-suspended in 450 µL of Buffer RTL containing 4.5 µL of β-mercaptoethanol. Then, on-column DNase digestion was performed with RNase-Free DNase Set (Qiagen) by the procedure provided by the manufacturer.
The RNA purity and integrity number (RIN) was determined by absorbance ratios of OD260 nm/OD230 nm and OD260 nm/OD280 nm, respectively, using Agilent 4200 TapeStation system (Agilent Technologies). The RNA concentration was measured using Quant-it RiboGreen RNA Assay Kit (Invitrogen). High-quality RNA samples were used for RNA-sequencing (RNA-Seq) library construction using NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs) along with NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs) for mRNA enrichment according to the manufacturer’s instructions. Briefly, mRNA was purified from the total RNA using the oligo d(T)-coupled magnetic beads method.
For cDNA library construction, the purified mRNA was fragmented to an average insert size of 200 bp, and cDNA was then synthesized with these fragments. The cDNA fragments were end-repaired, ligated with adapters, and subsequently amplified with Illumina index primers by PCR. The final RNA-Seq library was evaluated for size distribution of the cDNA fragments using Agilent 4200 TapeStation system and sequenced on Illumina NovaSeq 6000 (Illumina, San Diego, CA, USA) with the parameter “100 bp paired-end reads”.
The quality of the Illumina RNA-Seq data was assessed using FastQC (v0.12.1; https://github.com/s-andrews/FastQC/). The paired-end reads from RNA-Seq libraries were trimmed using Trimmomatic (v0.39) (Bolger, Lohse & Usadel, 2014) with the options “TRAILING:20 and MINLEN:75”.
Gene annotation
Genes in the assembled draft genome were predicted using FunGAP pipeline (v1.0.1) (Min, Grigoriev & Choi, 2017) with a guide by clean RNA-Seq reads. The predicted genes were annotated by a homology-based search against Swiss-Prot database using DIAMOND (v2.1.9) with the options “—very-sensitive and e-value 0.00001” (Buchfink, Reuter & Drost, 2021). tRNA and rRNA were predicted using tRNAscan-SE (v2.0.9) (Chan et al., 2021) and RNAmmer (v1.2) (Lagesen et al., 2007), respectively.
For functional annotation using Gene Ontology (GO), protein sequences of the predicted genes were aligned against UniProt database using DIAMOND BLASTP (v2.9.0) with the following option “e-value cutoff of 1e−5” (Ashburner et al., 2000; Bateman et al., 2015). The results were used to determine protein families using Pfam database (Mistry et al., 2021) from InterPro (Paysan-Lafosse et al., 2023).
CAZymes were identified by analyzing the protein sequences against CAZymes database using HMMER (v3.4) with the parameters “e-value 1e−18 and coverage >0.35” (Yin et al., 2012). For CAZymes analysis, CAZyme data for P. expansum T01 and P. digitatum PHI26 were obtained from a previous study (Li et al., 2015).
Identification of SM BGCs
The antiSMASH (v7.0.0) program with default settings was used to identify SM BGCs in the assembled genome of P. paneum OM1 (Blin et al., 2017).
Phylogenetic analysis
Gene family clustering analysis was performed on proteins in P. paneum OM1 and seven other fungal strains (P. expansum MD-8, P. expansum ATCC 24692, P. expansum d1, P. griseofulvum PG3, P. digitatum Pd1, P. chrysogenum Wisconsin 54-1255, and A. clavatus NRRL 1) using OrthoFinder (v2.2.5) with default parameters (Emms & Kelly, 2019), and 4,778 single-copy gene families were obtained. The protein sequences of these single-copy gene families were aligned using MUSCLE (v5.1.linux64) with default settings for the multiple sequence alignment (Edgar, 2004). The aligned sequences were then trimmed using Gblocks (v0.91b) with the parameter “-t=p” to remove poorly aligned regions (Castresana, 2000). The resulting trimmed sequences were concatenated to a single sequence of 2,262,742 amino acids. To construct the phylogenetic tree, RAxML (v8.2.12) was used with the parameter “-p 12345 -m PROTGAMMAAUTO -f a -N 100 -x 12345 -o A.clavatus” based on the maximum likelihood method (Stamatakis, 2006). A. clavatus NRRL 1 was selected as an outgroup. All of the nodes in the tree had a bootstrap value of 100.
Genome synteny analysis
The genome data of seven other fungal strains were used as query sequences and aligned to the genome of the reference strain P. paneum OM1 using NUCmer (v3.1) with default parameters. The resulting whole genome alignment was filtered using delta-filter with the parameter “-i 30 -1 1000 -1”. The show-coords program was employed to obtain the coordinates of all collinear relationships. These coordinates of the collinear relationship were then inputted into tbtools, and the Advanced Circos function was utilized to generate a circos plot which illustrates the collinear relationships.
Multiple sequence comparison and functional conservation analysis of patulin BGCs
For multiple comparison of patulin BGCs in P. paneum OM1 and eight other fungal strains, P. expansum NRRL 35695 was selected as a reference strain, and protein sequences of the gene clusters in P. expansum NRRL 35695 and eight other fungal strains were aligned using DIAMOND (v2.0.13.151) with a blastp command and default parameters (Buchfink, Reuter & Drost, 2021). A Python program with SVG scripts was employed to determine the precise location and transcriptional orientation of each gene in patulin BGCs. The schematic diagram of patulin BGCs was generated using R programming.
The functional conservation of each gene in patulin BGCs from P. paneum OM1 and eight other fungal strains was assessed by multiple sequence alignments using UniProt. The similarity between nucleotide or protein sequences of each gene in patulin BGCs was determined by the percentage of the sequence alignments. The diagram of the functional conservation analysis was generated using RStudio (v4.3.0) with default settings. The number of introns within each patulin biosynthetic gene was analyzed using the Softberry program (FGENESH, a gene finder for eukaryotes) (Solovyev et al., 2006).
Data availability
Nucleotide sequence data of P. paneum OM1 from the Whole Genome Shotgun project have been deposited at DDBJ/ENA/GenBank under the accession number JAZGPW000000000. The version described in this article is JAZGPW010000000.
The genome and proteome data of P. expansum MD-8 (NCBI GenBank accession number: GCA_000769745.1), P. expansum ATCC 24692 (NCBI GenBank accession number: PRJNA196076), P. expansum NRRL 35695 (NCBI GenBank accession number: KF899892), P. expansum d1 (NCBI GenBank accession number: GCA_000769735.1), P. griseofulvum PG3 (NCBI GenBank accession number: GCA_001561935.1), P. digitatum Pd1 (NCBI GenBank accession number: GCA_000315645.2), P. chrysogenum Wisconsin 54-1255 (EMBL accession number: AM920416–AM920464), and A. clavatus NRRL 1 (NCBI GenBank accession number: GCA_000002715.1) were retrieved from the NCBI (https://www.ncbi.nlm.nih.gov), the genome portal (MycoCosm) of the US Department of Energy (DOE) Joint Genome Institute (JGI) (https://mycocosm.jgi.doe.gov/mycocosm/home), and the EnsemblFungi (https://www.fungi.ensembl.org/index.html).
Results & Discussion
Genome sequencing and assembly
The whole genome of P. paneum OM1 was sequenced using the MinION long read sequencing platform. A total of 190,226 reads with an average length of 18,086 base pairs (bp) were obtained as raw sequencing data. After adapter sequences were removed from raw reads and the low-quality reads were filtered, the filtered data yielded 179,499 reads with an average length of 19,064 bp.
Genome assembly was performed using Flye software. A total of six contigs were obtained, including the largest contig spanning 7,558,500 bp and the smallest contig spanning 27,875 bp (Table S1). After the draft genome was polished using Homopolish, the completeness of the genome assembly was slightly reduced from 98.8% to 98.7% complete BUSCO value. Thus, the draft genome assembly before the polishing step was chosen as the final assembly.
Quality control checks on Illumina RNA-Seq data using FastQC produced a total of 40,966,193 reads from fungal samples cultured in PDB medium, whereas it produced a total of 35,520,322 reads from fungal samples cultured in YES medium. After adapter sequences were trimmed from the RNA-Seq data and the low-quality reads were filtered, 39,114,789 read pairs (95.48% of the initial reads) were retained from the data derived from the fungal samples cultured in PDB medium, while 34,265,235 read pairs (96.47% of the initial reads) were retained from the data derived from the fungal samples cultured in YES medium. These read pairs were used for transcript assembly.
The genome assembly was annotated using FunGAP with a guide by the RNA-Seq reads. After 49 rRNAs and 187 tRNAs were predicted, a total of 10,679 putative protein-coding genes were identified in the assembled genome (Table S2). Of these protein-coding genes, 7,136 genes (66%) were functionally annotated using the Swiss-Prot database and DIAMOND tool.
Genome sequencing statistics for P. paneum OM1 and seven other fungal strains
The genome sequencing data of P. paneum OM1 and seven other fungal strains (six closely related Penicillium strains and one Aspergillus strain) were compared and analyzed. The genome size of the eight fungal strains ranged from 26.05 Mb (P. digitatum Pd1) to 32.45 Mb (P. expansum ATCC 24692) (Table 1). It showed that P. paneum OM1 genome is relatively small (27.16 Mb), which is consistent with the data of nine other P. paneum strains (26.55–27.79 Mb) retrieved from the Genome database at the NCBI. It is also in line with the genome size in the range of 25.4–46.5 Mb in Penicillium species (Petersen et al., 2023). The total length of the four largest scaffolds (equivalent to four major contigs (contig 8, contig 6, contig 9, and contig 10)) is 27.1 Mb, which covers approximately 99.8% of the P. paneum OM1 genome (Table S1 and Table 1). It is known that Penicillium species usually have four to eight chromosomes (Fierro et al., 1993; Yuen et al., 2003), which is in agreement with the P. paneum OM1 genome composed of four chromosomes in our study.
|
Genome feature |
P. paneum OM1 | P. griseofulvum PG3 | P. expansum d1 | P. expansum MD-8 | P. expansum ATCC 24692 | P. digitatum Pd1 | P. chrysogenum Wisconsin 54-1255 | A. clavatus NRRL 1 |
|---|---|---|---|---|---|---|---|---|
| Sequencing technology | Oxford nanopore | Illumina | Illumina | Illumina | Illumina | Roche 454 | NR | NR |
| Genome size (Mb) | 27.16 | 29.14 | 32.07 | 32.36 | 32.45 | 26.05 | 32.22 | 27.86 |
| Sequencing coverage (x) | 127 | 270 | 605 | 473 | 119 | 24 | 9.8 | 1 |
| Number of contigs | 6 | 92 | 270 | 377 | 136 | 561 | 242 | 231 |
| Number of scaffolds | 4 | 92 | 249 | 345 | 12 | 53 | 49 | 143 |
| Number of scaffolds (≥ 100 kb) | 4 | 14 | 80 | 89 | 8 | 26 | 1NR | 16 |
| N50 (bp) | 7,466,314 | 2,267,136 | 360,559 | 376,691 | 5,330,000 | 1,533,507 | 1NR | 2,493,640 |
| GC (%) | 48.8 | 47.3 | 47.6 | 47.5 | 47.4 | 48.5 | 48.9 | 49.2 |
| Number of protein-coding genes | 10,679 | 9,629 | 11,048 | 11,060 | 11,807 | 8,946 | 7,328 | 9,121 |
Notes:
The genome assembly data of seven fungal strains were obtained from the information publicly available on NCBI, MycoCosm, and EnsemblFungi.
NR indicates Not Reported.
The results of genome assembly can be evaluated based on the number of contigs and scaffolds as well as the N50 value (Seppey, Manni & Zdobnov, 2019). Of these eight fungal strains, P. paneum OM1 exhibited the highest N50 value (7,466,314 bp), which indicates the high quality of the genome assembly (Table 1).
The GC content of the genome is another interesting genome feature because it can influence genome stability, structure, and functionality (Smarda et al., 2014). The GC content of the genomes in the eight fungal strains ranged from 47.3 to 49.2% (Table 1). It showed that P. paneum OM1 genome has 48.8% of the GC content, which is in line with the data of nine other P. paneum strains (48.7–49.0%) retrieved from the Genome database at the NCBI.
The number of protein-coding genes provides important information about the genetic diversity and functional potential of fungal species. As shown in Table 1, the number of predicted protein-coding genes ranged from 7,321 to 11,807 across the eight different fungal strains. Although P. paneum OM1 has a smaller genome size (27.16 Mb) than the three P. expansum strains (32.07–32.45 Mb), it contains a similar number of predicted protein-coding genes (10,679) to those of the three P. expansum strains (11,048–11,807). It suggests that the high quality of genome sequencing data was generated from P. paneum OM1. Of the 10,679 predicted protein-coding genes, 7,136 were functionally annotated using the Swiss-Prot database (Table S2).
Gene Ontology categories
All 7,136 functionally annotated proteins, which were identified in the newly sequenced genome of P. paneum OM1, were analyzed using the UniProt database along with Pfam domains to assign Gene Ontology (GO) terms. Of the 7,136 protein sequences, 7,098 (99.5% of all functionally annotated proteins) were able to be successfully assigned to three GO categories (biological processes, cellular components, and molecular functions categories) (Fig. 1). However, since some protein sequences corresponded to multiple GO annotation entries, a total of 18,804 entries were generated: 4,233 biological processes, 5,201 cellular components, and 9,370 molecular functions (Fig. 1 and Table S3).
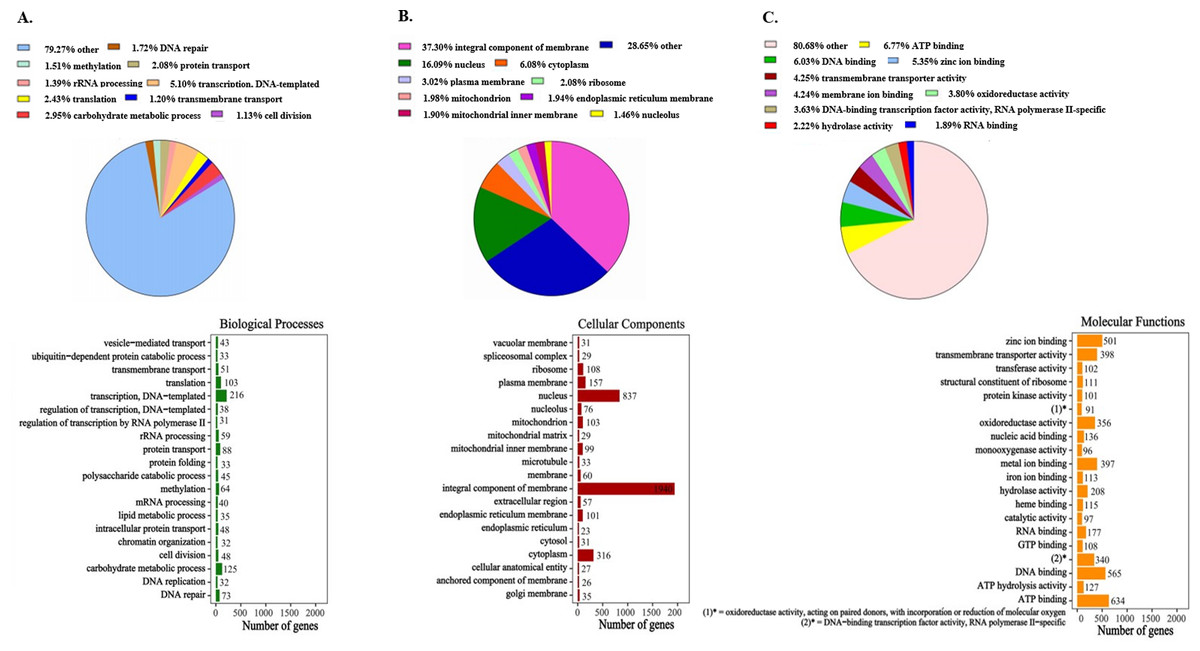
Figure 1: Gene Ontology (GO) classification of protein families in P. paneum OM1.
Assignment of GO terms for (A) biological processes, (B) cellular components, and (C) molecular functions. In the right boxes, only top 20 protein families are shown in each category.{kind=link}
The GO annotation results showed that proteins involved in several functions are enriched in the genome of P. paneum OM1, such as transcription (DNA-templated transcription in the biological process category, DNA binding in the molecular function category), energy utilization (ATP binding in the molecular function category), and carbohydrate metabolism (carbohydrate metabolic process in the biological process category) (Fig. 1). In particular, in the molecular function category, metal ion (including zinc ion and iron ion) binding and heme binding activities are significantly enriched in the genome with oxidoreductase and monooxygenase activities, which are cofactors-binding tailoring enzymes involved in the biosynthesis of several SMs (Cresnar & Petric, 2011; Petersen et al., 2023) (Fig. 1C). The high enriched heme binding activity is also related to combating plant defense mechanisms along with the iron ion binding activity, which acts as siderophores for iron sequestration (Wu et al., 2019).
CAZymes in P. paneum OM1
CAZymes, which are involved in the degradation of plant cell walls, are of great importance for the pathogenicity of plant pathogens. These enzymes play pivotal roles in the synthesis and modification as well as degradation of carbohydrates in plant cell walls. To gain insights into these enzymes, 7,136 putative proteins, which were functionally annotated in the genome of P. paneum OM1, were analyzed against the CAZyme database using HMMER. Moreover, a comparative analysis was performed by comparing the CAZyme data with those of P. expansum T01 and P. digitatum PHI26 (Li et al., 2015).
In total, 370, 640, and 394 putative CAZymes were identified in P. paneum OM1, P. expansum T01, and P. digitatum PHI26, respectively (Fig. 2, Table S4). The number of CAZymes in P. paneum OM1 was lower than that in P. expansum T01, whereas it was similar to that in P. digitatum PHI26. It may reflect that the genome size of P. paneum OM1 (27.16 Mb) is similar to that of P. digitatum PHI26 (25.96 Mb), but is smaller genome size than P. expansum T01 (33.52 Mb) (Table 1).
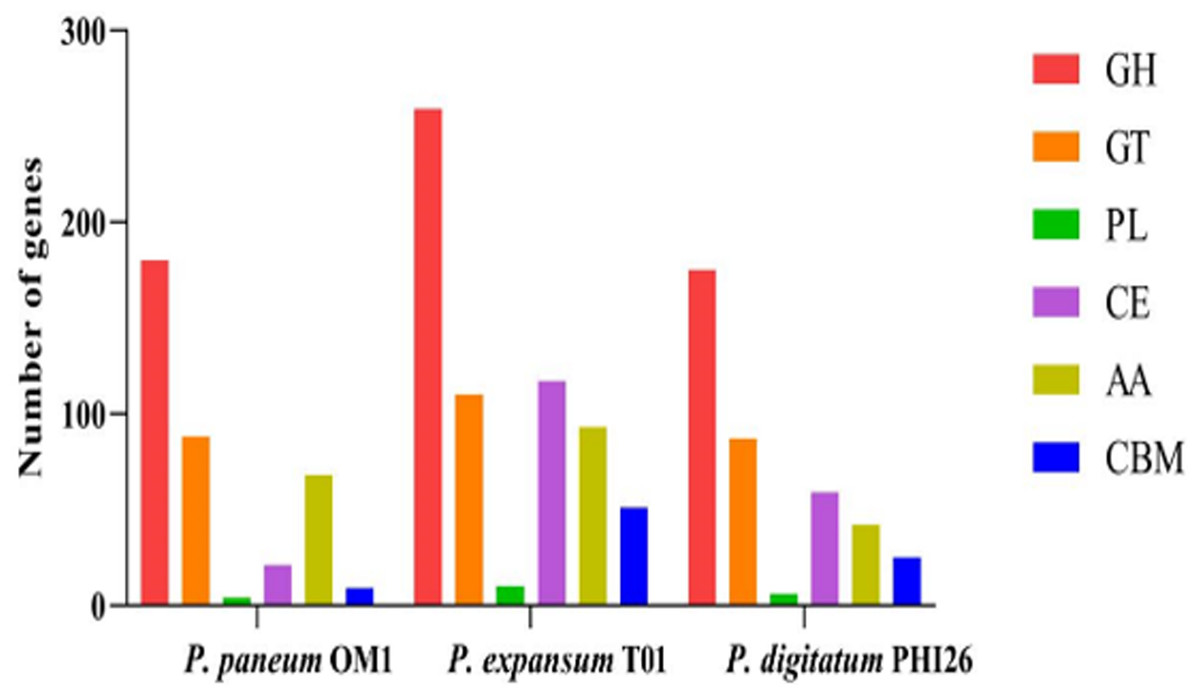
Figure 2: Comparison of genes encoding CAZymes in P. paneum OM1 and two closely related fungal species.
Abbreviations: GH, glycoside hydrolase; GT, glycosyl transferase; PL, polysaccharide lyase; CE, carbohydrate esterase; AA, auxiliary activity; CBM, carbohydrate-binding module.{kind=link}
Figure 2 shows that P. paneum OM1 possesses 180 glycoside hydrolases (GHs), which catalyze the breakdown of complex carbohydrates into smaller carbohydrates or monosaccharide units. However, P. paneum OM1 has a significantly lower number of GHs than P. expansum T01 (259 GHs) but a similar number to that of P. digitatum PHI26 (175 GHs). This difference may suggest that P. paneum OM1 has a relatively weaker ability than P. expansum T01 in the utilization of carbohydrates under specific environmental or nutritional conditions, such as host plants. It also indicates that the ancestors of P. paneum OM1 and P. digitatum PHI26 may have occupied similar carbohydrate-rich niches.
In addition, P. paneum OM1 contains 88 glycosyl transferases (GTs), which is similar to the numbers found in P. expansum T01 (110 GTs) and P. digitatum PHI26 (87 GTs) (Fig. 2). These enzymes have the capacity to transfer sugar moieties to diverse substrates, resulting in the synthesis of intracellular carbohydrates and the formation of novel carbohydrate linkages. Since GTs are mainly involved in the basal activities in carbohydrate utilization of fungal cells, previous studies described that the number and type of GTs tend to be conserved in fungi (Li et al., 2015; O’Connell et al., 2012). A similar number of relatively abundant GTs in the three fungal strains might suggest that these species have robust abilities in carbohydrate synthesis and modification, which potentially play an important role in their interaction with host plants.
Moreover, it was observed that P. paneum OM1 has only four polysaccharide lyases (PLs), while P. expansum T01 and P. digitatum PHI26 have ten and six PLs, respectively (Fig. 2). These PLs are known to degrade cell wall components such as pectin for maceration of pome fruit tissues (Yao, Conway & Sams, 1996). Also, in this study, 21 carbohydrate esterases (CEs) were identified in P. paneum OM1, whereas 117 and 59 CEs were found in P. expansum T01 and P. digitatum PHI26, respectively (Fig. 2). These CEs catalyze hydrolysis of ester bonds in carbohydrates, resulting in their active participation in carbohydrate metabolism. P. paneum OM1 has a relatively lower number of PLs or CEs than P. expansum T01. It may suggest that P. paneum OM1 has lower abilities than P. expansum T01 in breakdown of specific carbohydrates such as acidic carbohydrates, or removal of O- or N-acylation of carbohydrates.
In P. paneum OM1, 68 auxiliary activities (AAs) were detected. The number of AAs in P. paneum OM1 was similar to those in P. expansum T01 (93 AAs) but higher than that in P. digitatum PHI26 (42 AAs) (Fig. 2). The existence of the relatively high AAs in P. paneum OM1 may strengthen the fungal capacity to degrade complex carbohydrates, thereby resulting in its wide ecological adaptability.
Lastly, nine carbohydrate-binding modules (CBMs) were also identified in P. paneum OM1. This fungal strain possesses lower number of CBMs than P. expansum T01 (51 CBMs) and P. digitatum PHI26 (25 CBMs) (Fig. 2). These modules bind to carbohydrates and often interact with other carbohydrate enzymes, which assist in their catalytic activity or localization. The lower number of CBMs in P. paneum OM1 may suggest that it has low abilities in recognition and metabolism of specific carbohydrates. However, P. paneum OM1 might have the reduced number of specialized CBMs for adaptation to other types of carbohydrates.
Overall, our data showed that P. paneum OM1 has the lower number of GHs, PLs, CEs, AAs, and CBMs than P. expansum T01, which may have resulted from the smaller genome size of the former than the latter. Thus, P. paneum OM1 may encounter more challenges in the effective utilization of complex polysaccharides than P. expansum T01, potentially limiting its competitive ability in specific environments. Nevertheless, P. paneum OM1, which possesses a similar number of putative CAZymes to P. digitatum PHI26, may have other distinct strategies for effective utilization of complex polysaccharides.
Phylogenetic analysis of P. paneum OM1
Based on the clustering analysis of gene families from P. paneum OM1 and seven other whole-genome-sequenced fungal strains (six closely related Penicillium strains and one Aspergillus strains), a total of 4,778 single-copy gene families (only one gene in the family of each fungal strain) were identified. A phylogenetic tree for the eight fungal species was constructed using the maximum likelihood method and the single-copy gene families. The phylogenetic analysis revealed that all eight fungal strains belong to the same Eurotiales order (Fig. 3). It also showed that P. paneum OM1 branched off after the divergence of P. chrysogenum Wisconsin 54-1255 (a penicillin-producing strain) but before the divergence of P. digitatum Pd1 (a citrus fruit pathogen). It suggests that P. paneum OM1 has an evolutionarily closer relationship with P. chrysogenum Wisconsin 54-1255 and P. digitatum Pd1 than five other species and that there is a shared ancestral lineage and similar genetic evolution between the three fungal strains. It may also reflect their genomic similarities and shared characteristics in ecological adaptation and metabolic capabilities (McCluskey, Wiest & Plamann, 2010). Moreover, the tree showed that P. paneum OM1 is more closely related to P. digitatum Pd1 than P. expansum ATCC 24692 (Fig. 3). It may have resulted from more similar genome size of P. paneum OM1 (27.16 Mb) to that of P. digitatum Pd1 (26.05 Mb) than that of P. expansum ATCC 24692 (32.45 Mb). Also, P. paneum OM1 has a more distant genetic relationship with P. expansum d1 and P. expansum MD-8 relative to P. expansum ATCC 24692 (Fig. 3). This indicates that they may represent different subtypes or subspecies of P. expansum. The distinct subtypes display variations in biological traits or adaptation to diverse environmental conditions, which is commonly observed in fungi (Callaghan et al., 2004). In addition, P. griseofulvum PG3 exhibits the most pronounced genetic divergence from other Penicillium species (Fig. 3). The analysis showed that P. griseofulvum PG3 branched off before the divergence of P. chrysogenum Wisconsin 54-1255. It indicates that P. griseofulvum PG3 may have followed a distant genetic trajectory during its evolution, such as development of different evolutionary strategies for adaptation to new environments (Giraud et al., 2008). As expected, it showed that A. clavatus NRRL 1, which serves as an outgroup, has far more distant genetic relationship with P. paneum OM1, indicating that it may have experienced a relatively independent evolutionary history by structural changes such as genomic reshuffling during their adaptation to different ecological niches and plant hosts (Leducq, 2014; Stukenbrock, 2013).
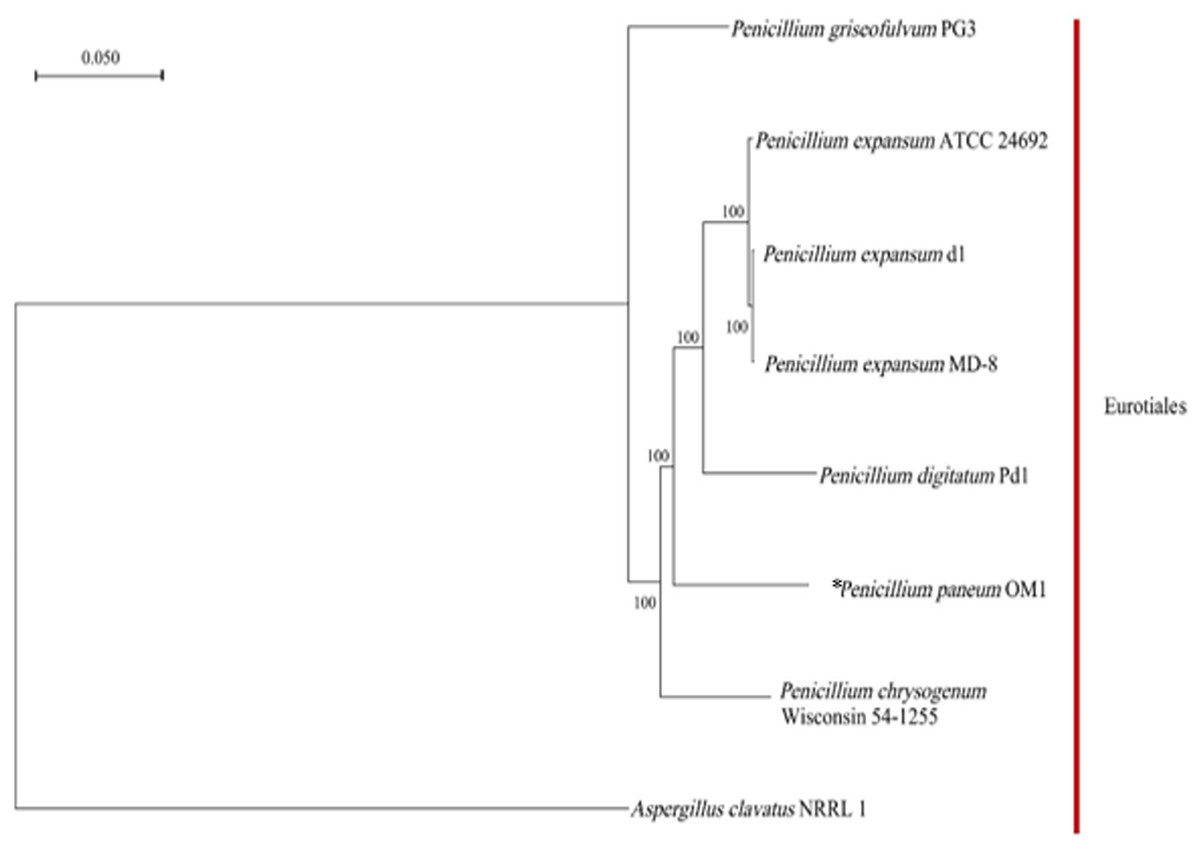
Figure 3: Phylogenetic relationship between P. paneum OM1 and seven other fungal species.
The phylogenetic tree was constructed using RAxML software based on the maximum likelihood method through the clustering analysis of single-copy gene families from eight fungal species. The asterisk (*) represents P. paneum OM1, whose genome was analyzed in this study. A. clavatus NRRL 1 was used as the outgroup. Bootstrap values are indicated on the branches.{kind=link}
Genome synteny analysis of P. paneum OM1 and seven other fungal strains
The analysis of the levels of collinearity among fungal genomes plays a crucial role in understanding the structure and functionality of their genomes. High collinearity due to the high levels of shared genomic regions provides valuable insights into the genetic relationships, functional conservation and resemblance of genes among the fungal species. Conversely, low collinearity implies huge diversity between their genome structures and functions. Thus, in this study the genome synteny analysis was conducted on P. paneum OM1 and seven other fully-sequenced fungal strains. The genomes of six Penicillium strains except for A. clavatus NRRL 1 exhibited similar levels of collinearity (46.28–58.25%) with respect to the genome of P. paneum OM1. Of the seven other fungal strains, the highest conserved collinearity (58.25%) was found between the genomes of P. paneum OM1 and P. expansum ATCC 24692, indicating a strong similarity between their genome structures and functionalities (Fig. 4A). It suggests that they may have undergone similar environmental pressure and selection during their evolutionary history. P. expansum d1 showed the second highest genome similarity (58.0%) to P. paneum OM1, followed by P. expansum MD-8 (57.98%), P. chrysogenum Wisconsin 54-1255 (56.57%), P. griseofulvum PG3 (48.29%), P. digitatum Pd1 (46.28%), and A. clavatus NRRL 1 (0.92%) (Fig. 4A). As expected, it showed very low collinearity between the genomes of P. paneum OM1 and A. clavatus NRRL 1 (Fig. 4). It suggests that there are substantial differences between their genome structures and functionalities due to genetic rearrangement or gene family expansion, resulting in high genetic diversity, which in turn may have led to different environmental adaptation and host specificity. Moreover, these data are slightly different from the phylogenetic tree shown in Fig. 3. The discrepancy might have resulted from methodological differences, such as the use of sequence alignments for the phylogenetic tree construction instead of the use of synteny blocks for the synteny analysis (Drillon et al., 2020).
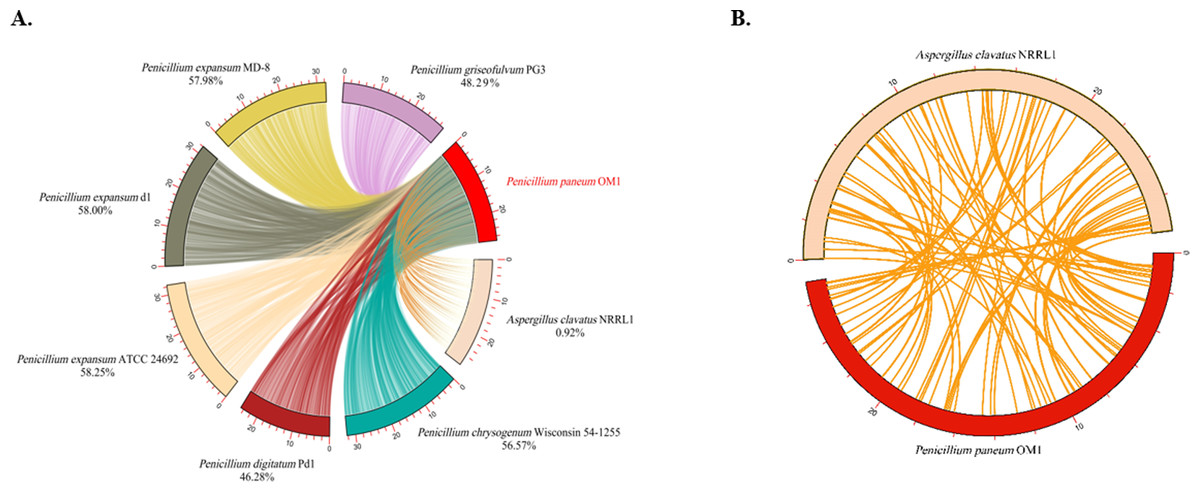
Figure 4: Comparative genome synteny analysis.
(A) Genome synteny among P. paneum OM1 and seven other fungal strains, and (B) between P. paneum OM1 and A. clavatus NRRL 1. Percentage below species and strain name represents genome similarity with P. paneum OM1.{kind=link}
SM BGCs in P. paneum OM1 and seven other fungal strains
Fungal SMs play key roles in mediating interactions and communication between fungi and plants, as well as contributing to fungal pathogenicity and toxicity (Keller, 2015). Typically, these metabolites can be classified into five major categories (polyketides, non-ribosomal peptides, alkaloids, terpenoids, and ribosomally synthesized and post-translationally modified peptides [RiPPs]), and an SM BGC contains one or more backbone genes that encode for key enzymes in the SM biosynthetic pathway, such as polyketide synthase (PKS), non-ribosomal peptide synthetase (NRPS), dimethylallyl tryptophan synthase (DMATS), terpene synthase, and RiPP precursor synthase, in addition to tailoring enzymes such as transferase, oxidoreductase, and oxygenase (Brakhage & Schroeckh, 2011; Scherlach & Hertweck, 2021; Van der Lee & Medema, 2016). Furthermore, hybrid metabolites such as meroterpenoides, which arise from the fusion of terpenes and polyketides, are frequently observed (Matsuda & Abe, 2016). Thus, to gain insights into the potential for SM production by P. paneum OM1, we analyzed the predicted SM BGCs in the fungal strain using the antiSMASH program and compared them with those in seven other related fungal strains.
In total, 33 SM biosynthetic gene clusters were identified in the genome of P. paneum OM1. These clusters include nine NRPS gene clusters, eleven PKS gene clusters, three terpene synthase gene clusters, one NRPS-type 1 PKS hybrid gene clusters, three Type 1 PKS-NRPS hybrid gene clusters, one fungal RiPP precursor synthase-like gene cluster, two betalactone synthase gene clusters, one NRPS-indole synthase hybrid gene cluster, and one isocyanide synthase gene cluster (Fig. 5, Table 2).
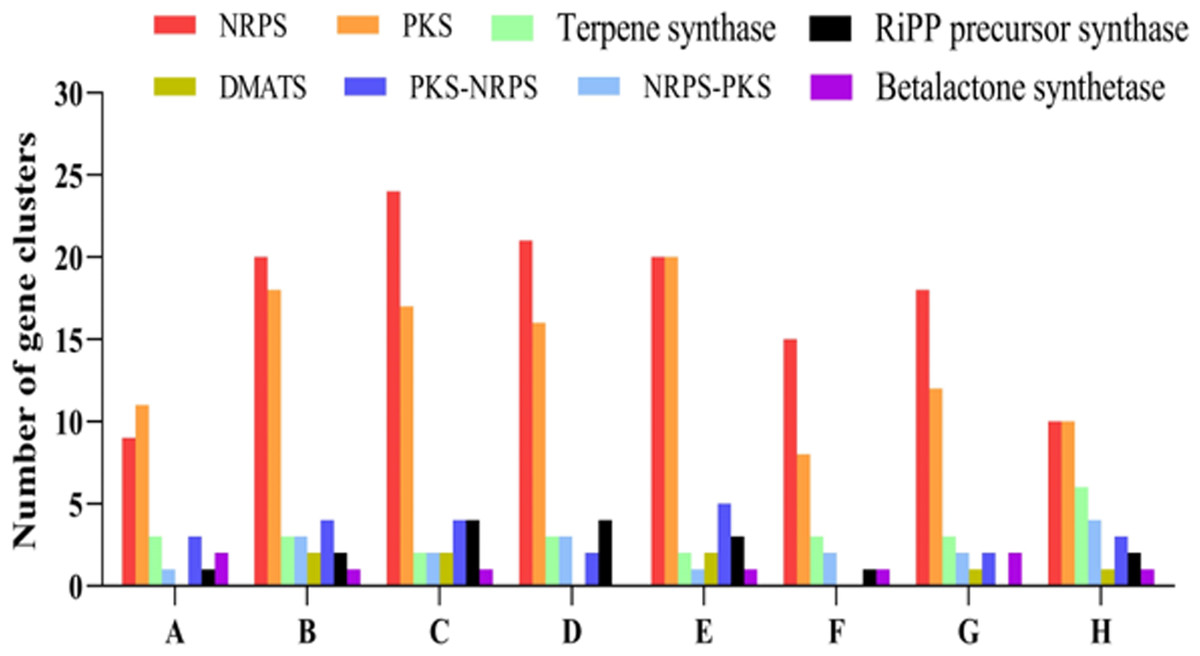
Figure 5: Comparative analysis of different types of predicted secondary metabolites biosynthetic gene clusters (SM BGCs).
The SM BGCs in P. paneum OM1 and seven other fungal strains were identified using antiSMASH (v.7.0.0). A through H indicate P. paneum OM1, P. expansum MD-8, P. expansum ATCC 24692, P. griseofulvum PG3, P. expansum d1, P. digitatum Pd1, P. chrysogenum Wisconsin 54-1255, and A. clavatus NRRL 1. Abbreviations: NRPS, Non-ribosomal peptide synthetase; PKS, Polyketide synthase; DMATS, Dimethylallyl tryptophan synthase; RiPP, ribosomally synthesized and post-translationally modified peptide.{kind=link}
| Type | Number of SM gene cluster | Contig number |
Length (bp) |
Most similar known cluster |
|---|---|---|---|---|
| NRPS | 1 | 10 | 45,286 | metachelin C/metachelin A/metachelin A-CE/metachelin B/dimerumic acid 11-mannoside/dimerumic acid |
| 2 | 8 | 49,474 | – | |
| 58,554 | nidulaninA | |||
| NRPS-like | 4 | 6 | 42,624 | – |
| 43,830 | choline | |||
| 42,181 | – | |||
| 44,283 | – | |||
| 1 | 9 | 44,331 | – | |
| NRPS-indole | 1 | 8 | 52,499 | paraherquamide |
| 1 | 9 | 46,277 | histidyltryptophanyldiketopiperazine/ dehydrohistidyltryptophanyldiketopiperazine/roquefortine D/ roquefortine C/glandicoline A/glandicoline B/meleagrin | |
| NRP-metallophore (NRPS) | 1 | 10 | 58,820 | – |
| NRPS-T1PKS | 1 | 9 | 52,398 | chaetoglobosin A/chaetoglobosin C |
| T1PKS | 3 | 10 | 47,971 | andrastin A |
| 49,366 | – | |||
| 48,164 | – | |||
| 5 | 8 | 47,878 | – | |
| 48,387 | – | |||
| 46,893 | – | |||
| 47,300 | – | |||
| 49,474 | – | |||
| 3 | 9 | 46,052 | YWA1 | |
| 47,407 | trichoxide | |||
| 45,491 | patulin | |||
| T1PKS-NRPS | 1 | 10 | 53,409 | ACE1 |
| 2 | 8 | 52,402 | – | |
| 59,474 | metachelin C/metachelin A/metachelin A-CE/metachelin B/dimerumic acid 11-mannoside/dimerumic acid | |||
| Terpene | 1 | 6 | 21,484 | squalestatin S1 |
| 1 | 8 | 22,538 | – | |
| 1 | 9 | 21,569 | – | |
| Fungal-RiPP precursor synthase-like | 1 | 8 | 60,975 | – |
| Betalactone synthase | 1 | 6 | 23,512 | – |
| 1 | 8 | 30,837 | – | |
| Isocyanide synthase | 1 | 9 | 42,389 | – |
| Total | 33 | – | – | – |
Notes:
- NRPS
-
Non-ribosomal peptide synthetase
- T1PKS
-
type 1 Polyketide synthase
- RiPP
-
Ribosomally synthesized and post-translationally modified peptide
Figure 5 shows that P. expansum ATCC 24692 contains the highest number of SM BGCs (56), followed by P. expansum d1 (54), P. expansum MD-8 (53), P. griseofulvum PG3 (47), P. chrysogenum Wisconsin 54-1255 (40), A. clavatus NRRL 1 (37), and P. digitatum Pd1 (30). This result is in line with genome size data from eight fungal strains shown in Table 1.
Of five major SM classes, NRPS and PKS gene clusters were the most prevalent in all eight fungal strains. In contrast, the number of SM BGCs in the other classes ranged between 0 and 6 (Fig. 5, Table S5). These results are consistent with those from one previous study (Nielsen et al., 2017), in which the authors described that PKS and NRPS gene clusters were the most abundant classes in Penicillium species. Interestingly, no DMAT gene cluster was found in genomes of P. paneum OM1, P. griseofulvum PG3, and P. digitatum Pd1 (Table S5), all of which have a similar genome size (Table 1). The number of SM BGCs may reflect differences in environmental adaptation or survival strategies of the fungi, and competitive dynamics against other microorganisms and plants, suggesting that P. expansum ATCC 24692, P. expansum d1, P. expansum MD-8, and P. griseofulvum PG3 may thrive under diverse or highly competitive environmental conditions.
Intriguingly, our results showed that P. paneum OM1 genome contains a putative squalestatin S1 BGC (Table 2), whose product belongs to terpenes and is a potent inhibitor of squalene synthase with the potential use as an antifungal or cholesterol-lowering agent for the control of cholesterol biosynthesis in mammals and fungi (Bonsch et al., 2016). It also contains a putative andrastin A BGC (Table 2), whose product is a potential anticancer drug candidate since it inhibits farnesyltransferase activity of oncogenic Ras proteins in tumor cells (Rojas-Aedo et al., 2017). In addition, we detected two predicted metachelin C, A, A-CE, and B BGCs (Table 2), which potentially produce the iron-binding siderophore to cope with plant defense mechanisms as described above (Donzelli, Gibson & Krasnoff, 2015).
Some previous studies reported that P. paneum strains produce marcfortine A consistently (Nielsen et al., 2006; O’Brien et al., 2006). However, we did not detect any marcfortine A BGCs but found a BGC of its structurally related paraherquamide, an anthelmintic compound (Table 2) (Zinser et al., 2002). Since marcfortine A was able to be semi-synthetically converted to paraherquamide A (Lee & Clothier, 1997), the marcfortine A BGC may be located near the paraherquamide A BGC to form a single BGC in the genome of P. paneum OM1. It has been reported that in the BGC of roquefortine/meleagrin, which are structurally related to each other, roquefortine C and meleagrin biosynthetic genes are present in a single BGC, in which roquefortine C is produced as an intermediate during meleagrin biosynthesis (Ali et al., 2013).
We also identified two other mycotoxin BCGs (roquefortine C and D, chaetoglobosin A and C) in addition to the patulin BGC in the genome of P. paneum OM1 (Table 2). Previous studies described that P. paneum can produce botryodiploidin and citreoisocoumarins as well as patulin and roquefortine C and D as mycotoxins (Frisvad et al., 2004; Nielsen et al., 2006; O’Brien et al., 2006), but we did not detect botryodiploidin or citreoisocoumarins BGC in the genome of P. paneum OM1.
Multiple sequence comparison and functional conservation analysis of patulin BGCs in P. paneum OM1 and eight other fungal strains
Several previous studies have reported the gene clusters responsible for the biosynthesis of patulin in A. clavatus, P. griseofulvum, and P. expansum (Artigot et al., 2009; Banani et al., 2016; Li et al., 2015). They showed that the patulin BGCs in the fungal strains consist of all 15 genes (patA–patO), of which one gene encodes for a potential transcription factor (patL), three genes for putative transporters (patA, patC, and patM), and eleven genes for biosynthetic enzymes. In this study, we identified the complete and functional patulin BGC in the genome of P. paneum OM1. Thus, a comparison was made between the patulin BGCs from P. expansum NRRL 35695, which was used as a reference strain, and eight other fungal strains including P. paneum OM1. The comparison analysis showed that seven fungal strains including P. paneum OM1 possess all 15 patulin BGCs in the similar size of gene clusters within a 41,717 DNA region, while P. chrysogenum Wisconsin 54-1255 and P. digitatum Pd1 possess only six and eight patulin biosynthetic genes, respectively, in the smaller size of incomplete gene clusters, spanning a 19 kb region (Fig. 6 and Table S6). Moreover, we did not find in P. digitatum Pd1 a gene homologous to the backbone gene (patK) that encodes for key enzymes (PKS) in the patulin biosynthetic pathway although P. chrysogenum Wisconsin 54-1255 has a homologous patK in the incomplete gene clusters (Fig. 6). These data support the fact that five Penicillium strains except two strains (P. expansum MD-8 and A. clavatus NRRL 1) are patulin producers, whereas the other two strains (P. digitatum Pd1 and P. chrysogenum Wisconsin 54-1255) are non-patulin producers. It is known that A. clavatus NRRL 1 is likely to be unable to produce patulin because of a mutation in the promoter or coding sequence of patulin biosynthetic genes although it contains all 15 patulin biosynthetic genes (Tannous et al., 2014). Also, one study from Spain described that patulin production is likely to be impaired in P. expansum MD-8 because of misregulation of the expression of patulin biosynthetic genes due to some mutations in intergenic regions of the patulin BGC (Ballester et al., 2015).

Figure 6: Comparison of patulin BGCs in P. paneum OM1 and eight other fungal strains.
Gene names are shown below the gene cluster. Arrows indicate the direction of transcription of the genes. Homologous genes are represented in the same color across different fungal strains. The asterisk (*) indicates a patulin-producing strain.{kind=link}
In addition, as expected, the organization of the patulin BGCs was highly conserved in P. paneum OM1 and five other Pencillium strains with the exception of A. clavatus NRRL 1, which exhibits rearrangements of patulin biosynthetic genes (Fig. 6). The order of patulin biosynthetic genes within the gene cluster was the same between five Pencillium strains along with non-patulin producing P. expansum MD-8. It also showed that the transcriptional orientation of the patulin biosynthetic genes is conserved between five patulin-producing Pencillium strains (Fig. 6). These data suggest that the patulin-producing strains share the conserved patulin biosynthetic genes in the gene cluster.
We also predicted the number of introns within each patulin biosynthetic gene in P. paneum OM1 using Softberry software and compared them with those from three other fungal strains containing all 15 patulin biosynthetic genes (P. expansum NRRL 35695, P. expansum T01, and A. clavatus NRRL 1). The number of introns in the same patulin biosynthetic genes was similar among the four fungal strains (Table 3). However, the number of introns in patN and patO was lower in P. paneum OM1 than in the other three fungal strains. The number of introns in patD and patK was also lower in A. clavatus NRRL 1 than in three Penicillium strains, including P. paneum OM1.
| Gene | Predicted function | Gene length (bp) | Number of intron | Transcriptional orientation of gene |
|---|---|---|---|---|
| patH | m-Cresol methyl hydroxylase | 1,956 | 4 (4, 4, 4) | – |
| patG | 6-Methyl salicylic acid decarboxylase (Amidohydrolase family protein) |
1,342 | 1 (1, 1, 1) | – |
| patF | Hypothetical protein | 597 | 0 (0, 0, 0) | – |
| patE | Patulin synthase (Glucose-methanol-choline oxidoreductase) | 2,001 | 2 (2, 2, 2) | + |
| patD | Hypothetical protein (alcohol dehydrogenase) | 1,428 | 6 (6, 6, 4) | – |
| patC | MFS transporter | 1,754 | 2 (2, 2, 2) | – |
| patB | Carboxylesterase family protein | 1,906 | 3 (3, 3, 3) | + |
| patA | Acetate transporter | 1,026 | 4 (4, 4, 4) | – |
| patM | ABC transporter | 4,541 | 6 (6, 6, 6) | – |
| patN | Isoepoxydon dehydrogenase | 761 | 1 (2, 2, 2) | + |
| patO | Isoamyl alcohol oxidase | 1,589 | 3 (4, 4, 4) | + |
| patL | C6 transcription activator | 2,972 | 1 (1, 1, 1) | – |
| patI | m-Hydroybenzyl alcohol hydroxylase | 2,287 | 4 (4, 4, 4) | – |
| patJ | Hypothetical protein | 1,086 | 2 (2, 2, 2) | – |
| patK | 6-Methylsalicylic acid synthase | 5,393 | 1 (1, 1, 0) | + |
Notes:
The function of patulin biosynthetic genes was obtained from the information publicly available on NCBI for P. expansum NRRL 35695 and in the previous study (Li et al., 2015) for P. expansum T01, which is shown in the parenthesis.
The number of introns in the parenthesis was obtained from P. expansum NRRL 35695, P. expansum T01, and A. clavatus NRRL 1 in the previous studies (Li et al., 2015; Tannous et al., 2014).
Comprehensive functional conservation analyses based on nucleotide and protein sequences were conducted on 15 patulin biosynthetic genes in the gene clusters from P. paneum OM1 and eight other fungal strains. The results showed that each patulin biosynthetic gene in P. paneum OM1 shares a high degree of sequence identity (above 73% identity) at both nucleotide and amino acid levels with the corresponding gene in four other patulin-producing Penicillium strains, while it shares a low degree of identity (0–93% identity, 0 and 60% as medians for amino acid sequence identity) with those in P. chrysogenum Wisconsin 54-1255 and P. digitatum Pd1 (non-patulin producers) (Fig. 7). Moreover, severe deviations at the nucleotide or protein level were not observed between the patulin BGCs in P. expansum MD-8 (non-patulin producer, above 76% identity) and five other patulin-producing strains including P. paneum OM1. It may strengthen the inability of P. expansum MD-8 to produce patulin by misregulation of the expression of patulin biosynthetic genes as described above. It also revealed that each patulin biosynthetic gene in A. clavatus NRRL 1 shares a relatively low sequence identity (62–89% identity) with the corresponding gene in P. paneum OM1 (Fig. 7). This may explain why no patulin is produced by A. clavatus NRRL 1. Overall, our data demonstrate that the five Penicillium strains including P. paneum OM1 possess highly homologous patulin biosynthetic enzymes to produce patulin.
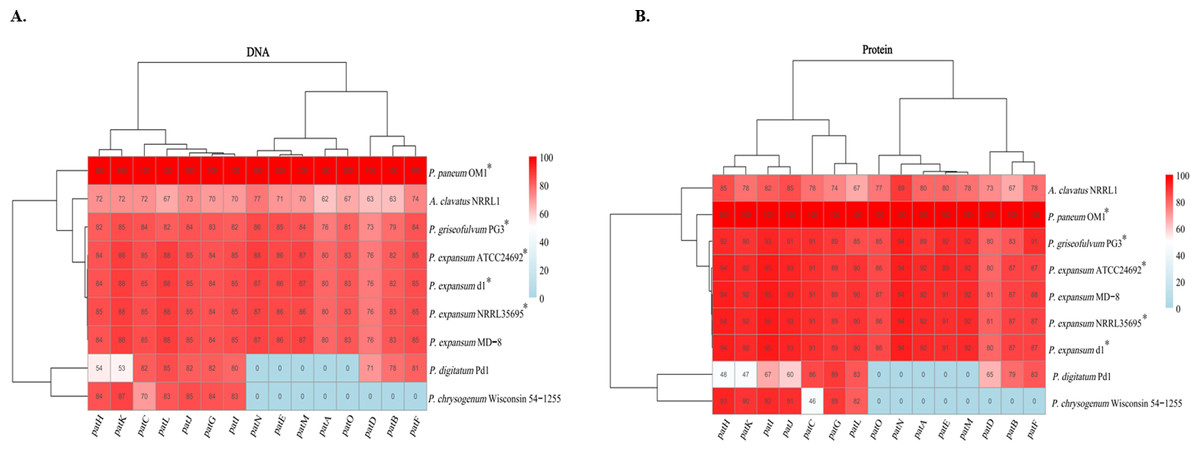
Figure 7: Functional conservation analysis of patulin BGCs in P. paneum OM1 and eight other fungal strains.
The percent identity among (A) the nucleotide sequences and (B) protein sequences of the homologous genes is shown as indicated by the color key when it is based on 100% sequence identity of each gene in P. paneum OM1. The asterisk (*) indicates a patulin-producing strain.{kind=link}
It has been reported that some Penicillium species, including P. expansum, P. griseofulvum, P. chrysogenum, can cause blue mold rot on pome fruits such as apples and pears (Morales et al., 2008; Sanderson & Spotts, 1995; Spadaro et al., 2011). Of these Penicillium species, P. expansum and P. griseofulvum are known to produce patulin, a polyketide-derived mycotoxin, on pome fruits (Morales et al., 2008; Spadaro et al., 2011). Previously, we reported a patulin producer P. paneum OM1 isolated from pears (Zhao et al., 2024). Little is known about SM BGCs, including a patulin BGC in P. paneum. The genome sequence analysis of P. paneum can be used to assist the discovery of molecular mechanisms that are responsible for production of SMs in Penicillium species. Thus, to gain insight into SM biosynthesis in P. paneum OM1, we sequenced its whole genome and analyzed SM BGCs, including the patulin BGC in this study.
The genome sequence analysis of P. paneum OM1 revealed that the fungal strain possesses four chromosomes as described above. It also produced two small sizes of contigs (33.2 kb of contig 11 and 27.9 kb of contig 14) in addition to four major contigs (four chromosomes). The BLAST-based analysis showed 46.1% of sequence alignment between the contig 14 and the mitochondrial chromosome of Penicillium polonicum (P. polonicum), suggesting that it is a mitochondrial chromosome of P. paneum OM1. However, the contig 11 did not align well with any mitochondrial chromosome from Penicillium species, but it aligned with chromosome 2 of Penicillium roqueforti (P. roqueforti) by 13.5%. Thus, we assume that it is a plasmid-related fragment from P. roqueforti or an unassembled fragment from P. paneum OM1.
Our data showed that P. paneum OM1 has a similar genome size (27.16 Mb) to P. griseofulvum PG3 (29.14 Mb) or P. digitatum Pd1 (26.05 Mb, a citrus fruit pathogen but non-patulin producer), but smaller genome size than the three P. expansum strains (32.07–32.45 Mb). A previous study suggested a relationship between the genome size of fruit pathogens and their host range, noting that P. expansum, with a large genome, can infect a wide variety of hosts beyond pome fruits, while P. digitatum, with a smaller genome, has a more limited host range, (Ballester et al., 2015). Another study also described that Fusarium culmorum (F. culmorum) and Fusarium graminearum (F. graminearum), which possess a small genome size (38.2 and 36.2 Mb, respectively), have a narrow range of plant hosts, while Fusarium oxysporum (F. oxysporum), fungi with larger genome (59.9 Mb), has a remarkably broad range of hosts (Schmidt et al., 2018). In addition, fungal pathogens produce a number of enzymes to break down polysaccharides such as pectin and utilize them as nutrients (Marcet-Houben et al., 2012). Moreover, they can produce bioactive SMs including patulin as a cultivar-dependent aggressiveness factor on plant hosts (Ballester et al., 2015; Li et al., 2015; Snini et al., 2016). Considering the relatively small genome size of P. paneum OM1, along with its low number of genes encoding CAZymes (as virulence factors) and backbone genes for SM production, it may suggest that this fungus can infect a more limited host range compared to P. expansum. However, patulin production by P. paneum OM1 may enhance its competitive advantage over other microbes in certain ecological niches, compared to P. digitatum Pd1, which has a similarly sized genome but does not produce patulin.
As described above, numerous fungal pathogens can produce bioactive SMs on plant hosts to facilitate fungal development and as a defense mechanism (Keller, Turner & Bennett, 2005). Fungal SM BGCs are typically clustered around one or more backbone genes that encode for key enzymes such as PKS, NRPS, DMAT, and RiPPs precursor synthase, all located at specific loci in the genome (Andersen et al., 2013). In this study, we identified all 15 patulin biosynthetic genes in the gene cluster from P. paneum OM1. Previously, we reported that the expression of four patulin biosynthetic genes (the early biosynthetic genes (patA, patK), middle biosynthetic gene (patN), and late biosynthetic gene (patE) in the pathway) in P. paneum OM1 was up-regulated to produce patulin in a patulin conducive medium (Zhao et al., 2024). It suggests that all 15 patulin biosynthetic genes are functional and potentially responsible for patulin biosynthesis in the fungal strain. In contrast, two non-patulin producers (P. chrysogenum Wisconsin 54-1255 and P. digitatum Pd1) contain only several genes of the 15 patulin biosynthetic genes, which are part of the gene cluster, and they are located in the same order as in patulin producers. This may suggest that these non-patulin producers lost their ability to synthesize patulin by differential gene loss.
Interestingly, two non-patulin-producing species possess a part of the complete patulin biosynthetic gene cluster found in seven other fungal strains. As described above, eight patulin biosynthetic genes homologous to the corresponding genes in P. paneum OM1 were identified in the genome of P. digitatum Pd1. It is in line with two previous studies, in which authors reported the same eight patulin biosynthetic genes in one gene cluster from the genomes of two different P. digitatum strains (PHI26 and Pd1) (Ballester et al., 2015; Li et al., 2015). However, this differs from two other studies, which reported eight homologous patulin biosynthetic genes located within one gene cluster, and two additional homologous genes (patH and patK) located in a separate cluster in the genome of P. digitatum PHI26 (Marcet-Houben et al., 2012; Tannous et al., 2014). This discrepancy might be explained by the use of different cut-off values in percent identity for selecting homologous proteins in the comparative analyses. Also, as described above, six patulin biosynthetic genes homologous to the corresponding genes in P. paneum OM1 were found in the genome of P. chrysogenum Wisconsin 54-1255. This is consistent with a previous study from Spain (Ballester et al., 2015), but differs from another study (Tannous et al., 2014), which reported that the genome of P. chrysogenum ATCC 28089 contains an additional gene (patC). This difference in the obtained results in the listed research may be due to the use of different strains or to the use of a different percent identity cut-off for selecting homologous proteins in the latter study.
In the current study, antiSMASH program identified multiple putative BGCs in P. paneum OM1, including BGCs of squalestatin S1 and andrastin A as potentially beneficial SMs. However, 13 of the 33 bioinformatically detected SM BGCs were predicted to produce unknown SMs. Future investigation of these unknown SMs will provide new insights into the function of the SM BGCs and the mechanism of the SM biosynthesis in P. paneum OM1.
Conclusions
In this study, we provide fundamental information on (1) the molecular basis of patulin biosynthesis in P. paneum OM1 by comparison of patulin biosynthetic genes in the strain with those in other fungal strains, (2) 33 predicted SM BGCs and 15 patulin biosynthetic genes in its genome, (3) 370 genes that encode CAZymes for its pathogenicity, and (4) genome-wide phylogenetic relationships among P. paneum OM1 and other closely related Penicillium species. These findings have expanded our knowledge about BGCs of SMs including patulin in P. paneum OM1. Our data could help explore the biosynthesis of other potentially beneficial SMs in addition to patulin in Penicillium species to find uncovered novel natural products. Moreover, our results could facilitate studies on finding potential strategies to reduce patulin contamination on fresh fruits such as pears.