
Circular RNAs in endometriosis analyzed through RNA sequencing and bioinformatics for expression profile
- Published
- Accepted
- Received
- Academic Editor
- Rodrigo Nunes-da-Fonseca
- Subject Areas
- Bioinformatics, Genomics, Gynecology and Obstetrics, Women’s Health
- Keywords
- Circular RNA, Endometriosis, Bioinformatics, RNA sequence
- Copyright
- © 2025 Yue et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2025. Circular RNAs in endometriosis analyzed through RNA sequencing and bioinformatics for expression profile. PeerJ 13:e19298 https://doi.org/10.7717/peerj.19298
Abstract
Background
Endometriosis severely affects women’s physical and mental health; it is particularly important to find targets for the treatment and diagnosis of endometriosis.
Method
This research aimed to investigate the circRNA expression pattern in endometriosis, a type of non-coding RNA that can modulate parental gene expression by acting as miRNA sponges. Through high-throughput sequencing, we analyzed the circRNA expression profile in endometriosis patients in comparison to individuals without the condition.
Results
We detected 371 circular RNAs (circRNAs) showing increased expression and 308 circRNAs displaying decreased expression levels. To validate these findings, we employed quantitative real-time PCR (qRT-PCR) to confirm the expression of the top three differential expressed circRNAs listed in circBase. We inferred potential roles of these differentially expressed circRNAs in endometriosis development by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis. Moreover, by examining the circRNA-microRNA-target gene network, we uncovered a plausible mechanism. Specifically, interactions involving the markedly upregulated hsa_circ_000005 and significantly downregulated hsa_circ_000011 with miR-5787 may influence downstream targets, potentially contributing to the pathogenesis of endometriosis. Our study offers a foundational and crucial circRNA expression profile within the framework of endometriosis.
Introduction
Endometriosis, an inflammatory benign condition, is defined by the presence of endometrial stroma and glands in locations outside their normal position, predominantly found in the pelvic peritoneum, ovaries, and rectovaginal septum (Leone Roberti Maggiore et al., 2024). Around 10% is the estimated prevalence of endometriosis (Nnoaham et al., 2011); however, due to differences in study populations and methodologies, estimates vary significantly. According to data from European databases based on population studies (ranging from 0.8% to 1.8%) (Ballard et al., 2008; Abbas et al., 2012), the reported prevalence of diagnosed endometriosis is 10.8 per 1,000 (95% CI [10.5–11.0]) (Eisenberg et al., 2018). In China, there has been a noteworthy decrease in the age-standardized death rate (ASDR) and age-standardized disability-adjusted life years (DALY) rate associated with endometriosis. The average annual percent change (AAPC) values for ASDR were −4.7 (95% confidence interval CI [−5.10 to −4.30]), while for DALY rate, they were −1.2 (95% CI [−1.20 to −1.10]) (Wang et al., 2022). However, endometriosis can have various adverse health effects on individuals such as chronic pelvic pain (Sachedina & Todd, 2020) and infertility (Bonavina & Taylor, 2022). Due to the nonspecific symptoms of endometriosis and the lack of non-invasive detection methods (Mangeshikar et al., 2024), diagnosis and treatment can be delayed by 5 to 10 years (Arruda et al., 2003; Watanabe, 2024; Singh et al., 2023). Endometriosis imposes both a substantial economic burden at the national level and badly affects the quality of life of women (Della Corte et al., 2020). However, the pathogenic mechanisms of endometriosis are still unknown.
Endometriosis is indeed a heritable, complex, and chronic disease that primarily affects women of reproductive age (Abe et al., 2014). The exact pathogenesis of endometriosis remains unclear. Prior research indicated that the etiology and pathogenesis of endometriosis involve contributions from both genetic and epigenetic factors (Koninckx et al., 2019). Studies have indicated that endometriotic lesions were monoclonal (Thomas & Campbell, 2000). Molecular investigations into loss of heterozygosity in endometriotic lesions have identified potential sites of tumor suppressor genes. These sites may play a role in the advancement of endometriotic implants towards endometrioid ovarian cancers (Bischoff & Simpson, 2000). Epigenetic factors are thought to influence the development of endometriosis. Differences in the levels of DNA methyltransferases, estrogen and progesterone receptors, microRNAs, and histone deacetylases have been noted between endometriosis and normal endometrial tissue (Grimstad & Decherney, 2017). Endometriosis shares several biological behaviors with tumors, leading some researchers to describe it as a “benign tumor-like condition” (Moraru et al., 2023). Studying molecular abnormalities in ectopic endometrium (EC), along with paired eutopic endometrium (EU) and normal endometrium from women unaffected by endometriosis, could provide insights into the mechanisms of endometriosis and the abnormal growth of EU outside the uterus (Istrate-Ofiţeru et al., 2024). In epithelial-mesenchymal transition (EMT), epithelial cells undergo a biological process of transformation, losing their typical characteristics and adopting mesenchymal properties. In the context of diseases like cancer and endometriosis, aberrant EMT can contribute to disease progression and metastasis (Makker & Goel, 2016). Therefore, it is crucial to identify potential molecular markers for diagnosis and regulatory factors that drive the progression of endometriosis, especially when it may be influenced by epithelial mesenchymal transition.
Circular RNAs (circRNAs) have been identified as crucial players in controlling gene expression (Hansen et al., 2013). circRNAs possess the capability to function as ceRNAs by sequestering miRNAs via their miRNA-binding motifs, thereby engaging in miRNA sequestration. This ceRNA mechanism involves circRNAs sequestering miRNAs, thereby regulating the availability of miRNAs to target mRNA transcripts (Thomson & Dinger, 2016). In prior research, it was demonstrated that circRNAs exhibit varied expression patterns in endometriosis, potentially linking them to the disease’s pathogenesis (Shen et al., 2018). In an independent study, it was observed that circ_0004712 and miR-148a-3p play a pivotal role in the E2-mediated epithelial-to-mesenchymal transition (EMT) process occurring during the progression of endometriosis. The underlying molecular machinery is hypothesized to be intimately associated with the β-catenin signaling cascade (Chen et al., 2020). Hence, circRNAs are deemed crucial biological controllers in unraveling the molecular intricacies of endometriosis, aiding in the discovery of efficient diagnostic indicators or treatment targets (Balci, 2022).
Currently, there is limited understanding regarding the expression levels and role of circRNAs in endometriosis. Within our research, we examined the expression patterns of circRNAs in endometriosis and healthy endometrium samples, emphasizing circRNAs that hold significant relevance in endometriosis. Specifically, leveraging publicly available databases, We underscored the clinical significance of circular RNAs (circRNAs) and corroborated their expression levels in endometriosis patients through qRT-PCR validation.
Methods
Participants and sample collection
In this study, a total of three individuals with endometriosis (case group) aged between 25 to 35 years in the proliferative phase, alongside three individuals with normal endometrium devoid of endometriosis (control group) aged between 26 to 38 years in the proliferative phase, were included. Tissue specimens from snap-frozen cyst walls of ovarian endometriosis and corresponding normal endometrial tissues were collected for RNA sequencing analysis and qRT-PCR, samples are quickly frozen in liquid nitrogen after collection and stored in liquid nitrogen tanks. All participants had consistent menstrual cycles and had abstained from hormonal therapy for a period of six months prior to the procurement of the samples. Additionally, individuals in the control group showed no signs of endometrial tumors and lacked a histological diagnosis of adenomyosis. Specimen diagnoses were confirmed through histopathology, and menstrual cycle phases were verified using both the last menstrual period and histological assessments. Authorization for the conduct of this research was provided by the institutional ethical review committee of the first People’s Hospital of Wuhu City (approval number: YYLL20220004). Full and voluntary written consent was obtained from participants before all tissue samples were obtained, and the study strictly adhered to the ethical guidelines set out in the Declaration of Helsinki and its subsequent revisions or similar ethical codes. The statistical power of this experimental design, calculated in RNASeqPower is 0.90.
RNA isolation and quality control
The frozen tissue samples underwent total RNA extraction employing the TRIzol reagent (Invitrogen, Waltham, MA, USA) in accordance with the manufacturer’s prescribed protocol. Subsequently, the RNA concentration in each sample was quantified utilizing the NanoDrop 2000 spectrophotometer (NanoDrop, Thermo Fisher Scientific, Waltham, MA, USA). Furthermore, the integrity of the extracted RNA was validated through 1% formaldehyde-based denaturing gel electrophoresis.
circRNA sequencing
The responsibility of circular RNA (circRNA) sequencing was assigned to the Gene Denovo Company, situated in Guangzhou, China. The initial phase involved preparing circRNA sequencing libraries, which kicked off with isolating total RNA from individual endometriosis tissue samples. Prior to library construction, the total RNA mixture underwent depletion of both ribosomal RNAs (rRNAs) and linear RNAs. This was accomplished using the Ribo-Zero Gold rRNA Removal Kit sourced from Illumina (San Diego, CA, USA) and the Ribonuclease R Kit procured from Epicentre (Madison, WI, USA), respectively. Following this, double-stranded cDNA was synthesized employing the polymerase chain reaction (PCR) technique. Subsequently, this cDNA underwent a series of processing steps, including adapter ligation, A-tailing, and end-repair, utilizing the NEBNext poly(A) mRNA Magnetic Extraction Module from New England Biolabs (Ipswich, MA, USA). Then, a 12-cycle PCR amplification was carried out, and the resulting products were purified with AMPure XP beads manufactured by Beckman (Brea, CA, USA). The quality assessment of the sequencing libraries was performed using the DNA 1000 Assay Kit from Agilent Technologies. The circRNA sequencing process was executed on the Illumina NovaSeq 6000 platform.
The raw sequencing data underwent processing to exclude those adapter reads that were of low quality, resulting in clean reads. These cleaned reads were then aligned to the rRNA database using Bowtie2. The reads that successfully mapped to rRNA were subsequently discarded, and the remaining reads were mapped to the reference genome through the utilization of TopHat2 version 2.0.3.1214. Finally, the unmapped reads were selected for the purpose of identifying circRNAs.
Differentially expressed circRNA analysis
Following QC procedures, the raw sequencing data underwent filtration to remove concatenated sequences and excessively short fragments. Following the trimming process, the refined dataset was aligned to the reference genome (GRCh37/hg19) by employing the STAR software tool. This alignment step involved mapping the cleaned and processed data to the specified reference genome, utilizing the capabilities of STAR. The subsequent step involved differential expression analysis of the circRNA-seq data, which was executed within the R statistical environment (version 4.2.0) utilizing the edgeR package. CircRNAs that demonstrated a significant fold change (either ≥ 2.0 or ≤ 0.5), a p-value of less than 0.05, and an FDR q-value of less than 0.05, when compared between the case and control groups, were designated as differentially expressed circRNAs. Annotation of these circRNAs was achieved by cross-referencing with circBase, with those that remained unannotated being categorized as novel circRNAs. Unmapped sequencing data to the reference genome were further scrutinized for circRNAs using CIRCexplorer2 software to detect reverse splicing events. Visualization of these differentially expressed circRNAs was achieved using CIRCOS visualization software in circular format.
qRT-PCR
To confirm the expression levels of the circRNAs identified through sequencing, qRT-PCR validation was undertaken. A total of six circRNAs, consisting of the three most upregulated and three most downregulated circRNAs annotated in circBase, were chosen for validation. The primers required for this validation process were procured from Sangon Biotech, located in Shanghai, China. For the purpose of qRT-PCR analysis, The sample is ground in liquid nitrogen and placed in Trizol for RNA extraction, the entire process is carried out in an environment containing DNase and RNase, including subsequent experiments. RNA concentration is measured using a Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA). cDNA synthesis was carried out using the All-in-One First-Strand cDNA Synthesis Kit offered by GeneCopoeia Inc (Santa Cruz, CA, USA), with one µg of total RNA as the starting material, the total reaction system is 20 µl, which includes one µg RNA, four µl RT mix, one µl Priming oligonucleotide, and then filled up to 20 µl with DEPC water. The machine program is set as follows: 25 °C for 10 min, 55 °C for 40 min, and 85 °C for 5 min. Subsequently, the qRT-PCR assays were conducted utilizing the All-in-One qPCR Mix from GeneCopoeia Inc (Rockville, MD, USA) on the ABI 7500HT qRT-PCR System manufactured by Applied Biosystems (Foster City, CA, USA). The thermal cycling protocol for the qRT-PCR encompassed an initial denaturation step at 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 5 s and annealing/extension at 60 °C for 30 s, the reaction system is as follows, with a total volume of 20 µl, including 10 µl of SYBR Green mix, one µl of 10 µM primers each, two µl of cDNA, and finally topped up with DEPC water to 20 µl. The threshold cycle (Ct) value represented the point at which a positive amplification signal was detected. GAPDH served as the internal reference gene for normalization. Expression levels were quantified employing the 2-ΔΔCt method, all checks repeated three times, and the primer sequences are presented in Table 1.
| circRNA | Primer sequence | Position |
|---|---|---|
| hsa_circ_000005 | F:5′-AGCTGAGGAGGAGGAAGGA-3′ | chr5:124036706-124036962 |
| R:5′-CCGTCTTCCCTCTTCTCT-3′ | ||
| hsa_circ_000002 | F:5′-CGCTAAGCAGTAGGAAGCA-3′ | chr10:126631025-126631876 |
| R:5′-CAGTCTTCCCTCTTCCGT-3′ | ||
| hsa_circ_000016 | F:5′-ACCTGATGAGGAGGAAACA-3′ | chr11:33307958-33309057 |
| R:5′-GCCTCTTCCCTCTTCCCG-3′ | ||
| hsa_circ_000011 | F:5′-TGGTAAGGAGGAGGAAAGC-3′ | chr7:128845043-128846428 |
| R:5′-CGGATGTTCCGTCTTCTCT-3′ | ||
| hsa_circ_000010 | F:5′-ACCTGATGAGCAGGATGCA-3′ | chr10:125120834-125121663 |
| R:5′-TTGTCTTCCCTCTTCGGC-3′ | ||
| hsa_circ_000021 | F:5′-ATGTAAGCAGGAGGAACAA-3′ | chr15:41961025-41962156 |
| R:5′-CGCTATTCTCGCTTCTCG-3′ | ||
| GAPDH | F:5′-AACTGAGGAGGAGCGAAGA-3′ | chr12:6493356-6531955 |
| R:5′-CCGTGTCACTTCGTCAGT-3′ |
Notes:
- F
-
forward
- R
-
reverse
GO and KEGG pathway analysis
Utilizing the DAVID tool, we delved into the potential functions and roles of the differentially expressed circRNAs and their corresponding parental genes. We executed a Gene Ontology (GO) enrichment analysis, encompassing biological processes (BP), cellular components (CC), and molecular functions (MF), drawing upon data from the Gene Ontology database. Additionally, to identify the significantly enriched pathways associated with the parental genes of these circRNAs, we performed a KEGG pathway enrichment analysis, leveraging the extensive information contained within the KEGG database.
CircRNA-miRNA-mRNA network analysis
To delve into the modus operandi of circular RNAs (circRNAs) serving as decoys for microRNAs (miRNAs), thereby exerting an indirect influence on the synthesis of their respective messenger RNAs (mRNAs), we utilized miRanda software (accessible at http://mirtoolsgallery.tech/mirtoolsgallery/node/1055) and RNAhybrid software (located at https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid/) to anticipate the miRNAs and mRNAs that are the targets of the most perturbed circRNAs. Subsequent to this anticipation, we employed Cytoscape software (version 3.9.0, hosted at https://cytoscape.org/) to visually represent the intricate interplay within the intricate circRNA/miRNA/mRNA networks, reshuffling word order and incorporating synonyms for clarity.
Statistical analysis
Data analysis was conducted utilizing SPSS Statistics (Version 20, Chicago, IL, USA). The continuous data were averaged, and both addition and subtraction operations were applied. Additionally, the standard deviation was calculated. To examine the disparities between two groups, a t-test was executed. The outcomes unveiled a noteworthy distinction between the two groups (p < 0.05).
Results
Quality control of circRNA sequencing
Using high-throughput deep sequencing, we first examined the circRNA expression profiles in six paired tissues collected from patients with ovarian endometriosis and control subjects. Among the host genes of these candidate circRNAs, 97.5% originated from exonic regions, while the remainder were located in intronic and unannotated regions (Fig. 1A). Next, we conducted Pearson correlation analysis to assess the correlation among all samples. As illustrated in Fig. 1B, we observed highly correlated gene expression patterns of circRNAs in both the control group (C1, C2, C3) and the case group (M4, M5, M6), respectively. The correlation heat map of circRNA expression levels across different samples also revealed a strong correlation between the internal samples of the control group and the case group (Fig. 1C). Subsequently, PCA analysis was performed, revealing a clear separation between the case group and the control group (Fig. 1D).
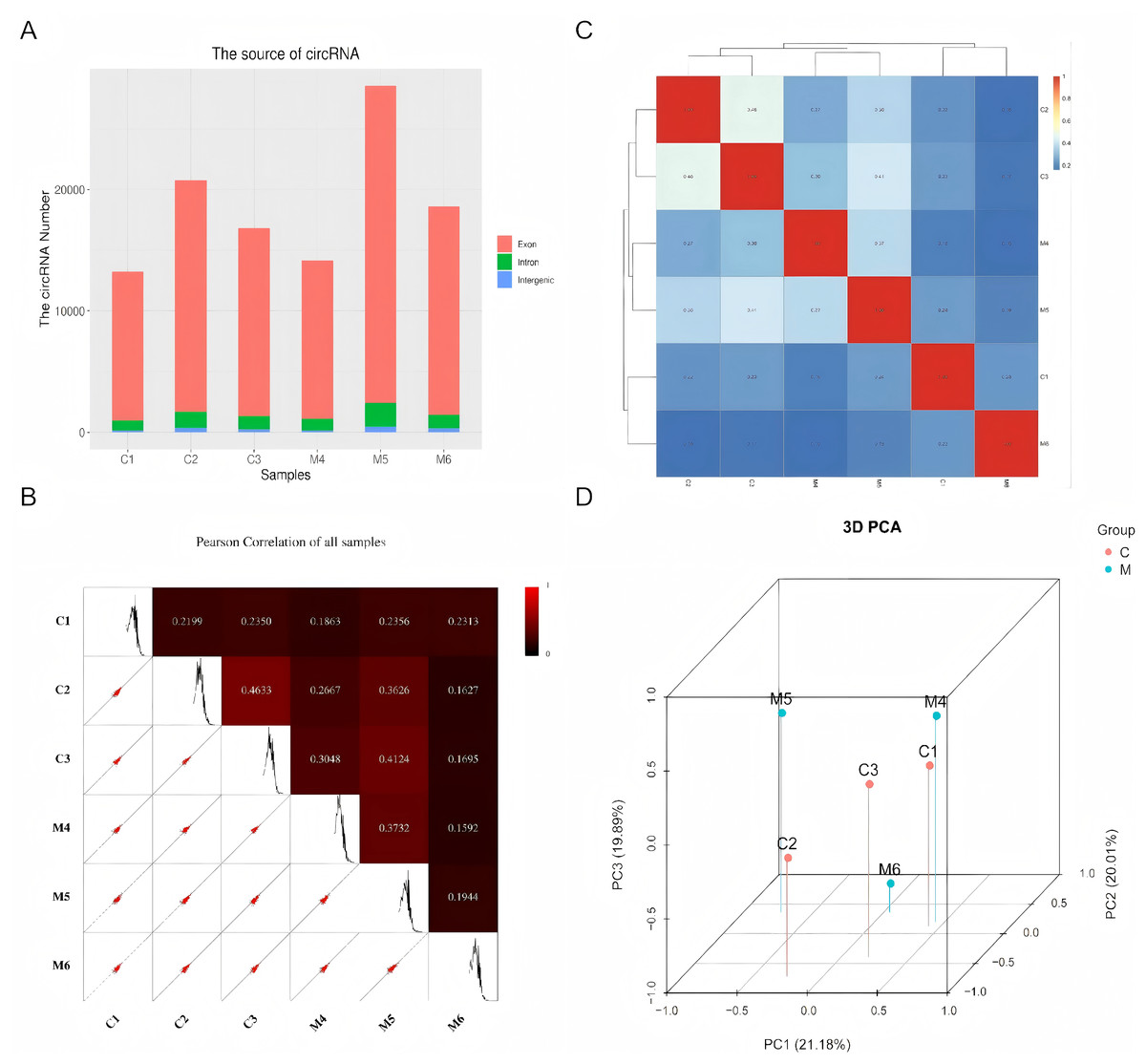
Figure 1: Characteristics of the identified circRNAs in ovarian endometriosis and healthy controls.
(A) Genomic origin of the identified circRNAs. (B) Sample correlation by Pearson correlation analysis. (C) Heatmap of sample correlation by Spearman correlation analysis. (D) 3D-PCA plot of control and case groups.{kind=link}
CircRNA expression profile in endometriosis
A scatter plot (depicted in Fig. 2A) was employed to compare the circRNA expression profiles between the ovarian endometriosis group and a cohort of three corresponding normal tissues. Statistically significant differentially expressed circRNAs were identified based on stringent criteria: fold changes ≥ 2.0 or ≤ 0.5, p-values < 0.05, and FDR q-values < 0.05. This analysis revealed a total of 679 circRNAs exhibiting marked differential expression, with 371 circRNAs being significantly upregulated and 308 circRNAs being noticeably downregulated. These findings were further illustrated in volcano plots (Fig. 2B) and a clustered heatmap (Fig. 2C), showcasing the upregulation and downregulation patterns. Additionally, the distribution pattern of these differentially expressed circRNAs across the human chromosomes was visualized through a heatmap (Fig. 2D).
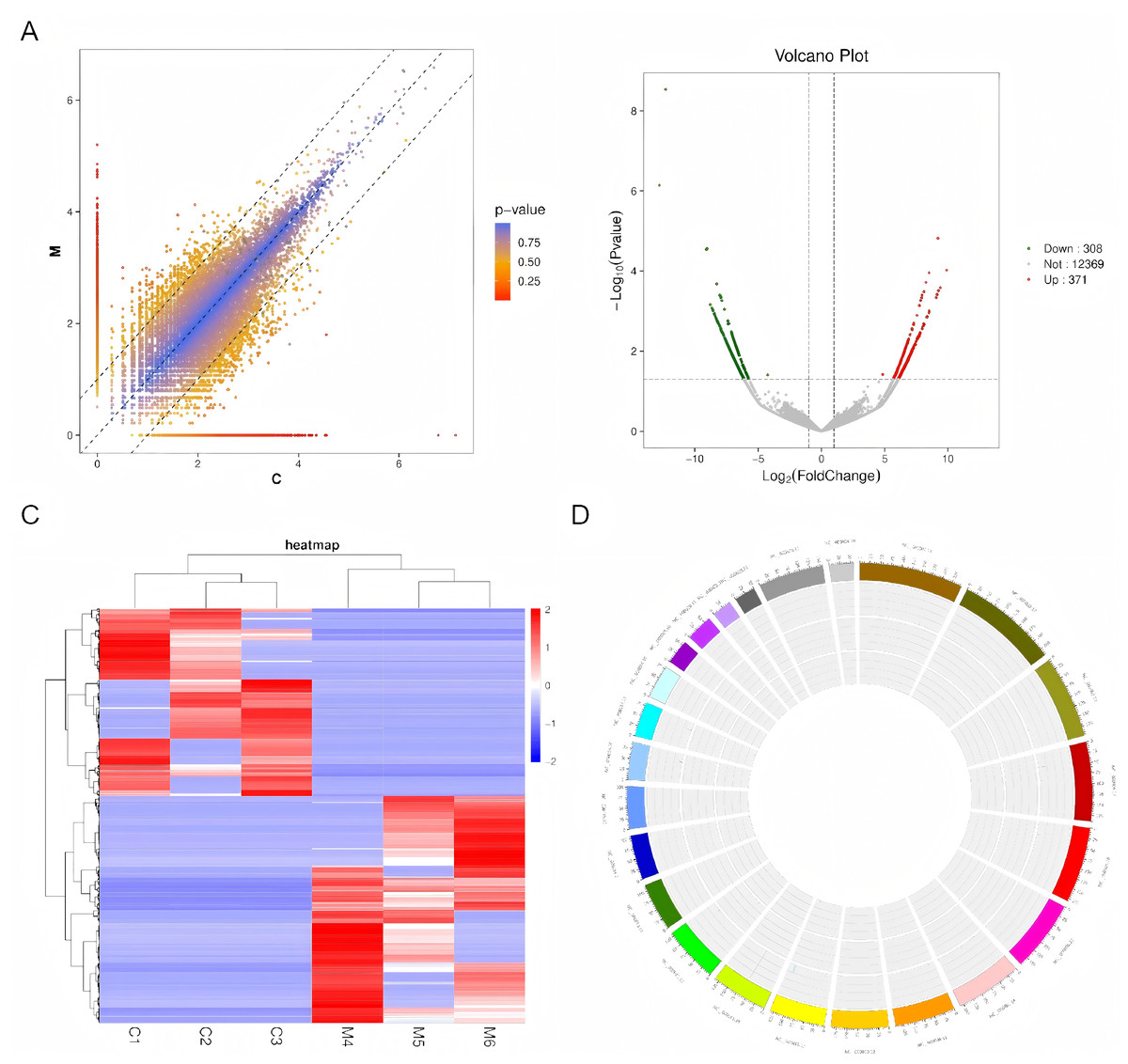
Figure 2: An examination of circRNA expression profiles between ovarian endometriosis and healthy controls.
(A) High-throughput sequencing technology was utilized to uncover the circRNA expression landscape in three matched pairs of ovarian endometriosis tissues and their respective healthy controls. (B) Volcano plots were generated to emphasize the differentially expressed circRNAs, where the horizontal dashed line signifies a statistical significance threshold of P = 0.05 (on a –log10 scale). In these plots, red dots signify circRNAs that are significantly upregulated, whereas green dots represent those that are significantly downregulated. (C) A heat map, accompanied by hierarchical cluster analysis, visually represents the expression patterns of all targeted circRNAs. Specifically, red hues indicate elevated relative expression, whereas blue hues correspond to reduced relative expression. (D) Lastly, the chromosomal distribution of these differentially expressed circRNAs is presented in a heatmap, offering insights into their genomic locations.{kind=link}
Validation of differentially expressed circRNAs
To verify the outcomes derived from RNA-sequencing (RNA-seq), we selected the three most prominently increased and decreased circRNAs for quantitative reverse transcription-polymerase chain reaction (qRT-PCR) assessment on an independent cohort with 50 endometriosis patients. Our findings revealed a statistically noteworthy upregulation of hsa_circ_000005, hsa_circ_000002, and hsa_circ_000016 within ovarian endometriosis tissues (Figs. 3A–3C). Conversely, the expression patterns of hsa_circ_000011, hsa_circ_000010, and hsa_circ_000021 exhibited significant downregulation in the identical ovarian endometriosis samples (Figs. 3D–3F), thereby reinforcing the initial RNA-seq discoveries.
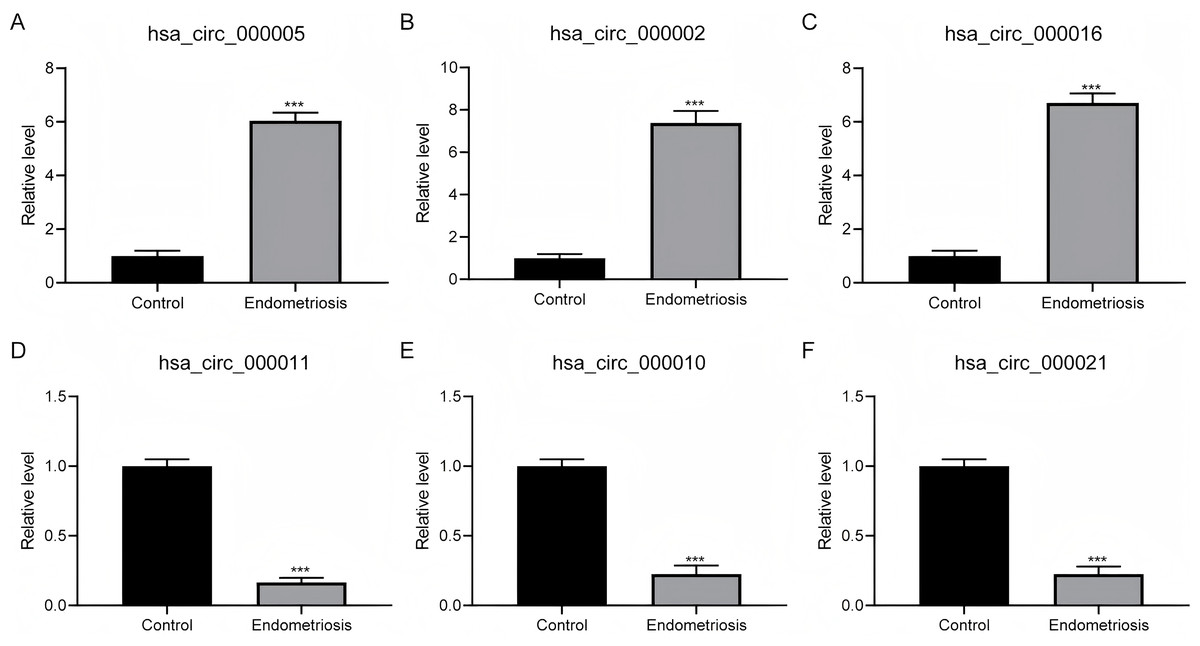
Figure 3: The confirmation of the expression patterns for distinctively expressed circRNAs.
(A–C) A statistically significant surge in the expression abundance of the three most augmented circRNAs (hsa_circ_000005, hsa_circ_000002, and hsa_circ_000016) was observed in ovarian endometriosis tissues in comparison to their counterparts from normal tissues. (D–F) In contrast, a pronounced decline in the expression levels of the three most diminished circRNAs (hsa_circ_000011, hsa_circ_000010, and hsa_circ_000021) was detected in ovarian endometriosis tissues, with a statistically significant difference at *** P < 0.001, when compared to normal tissues.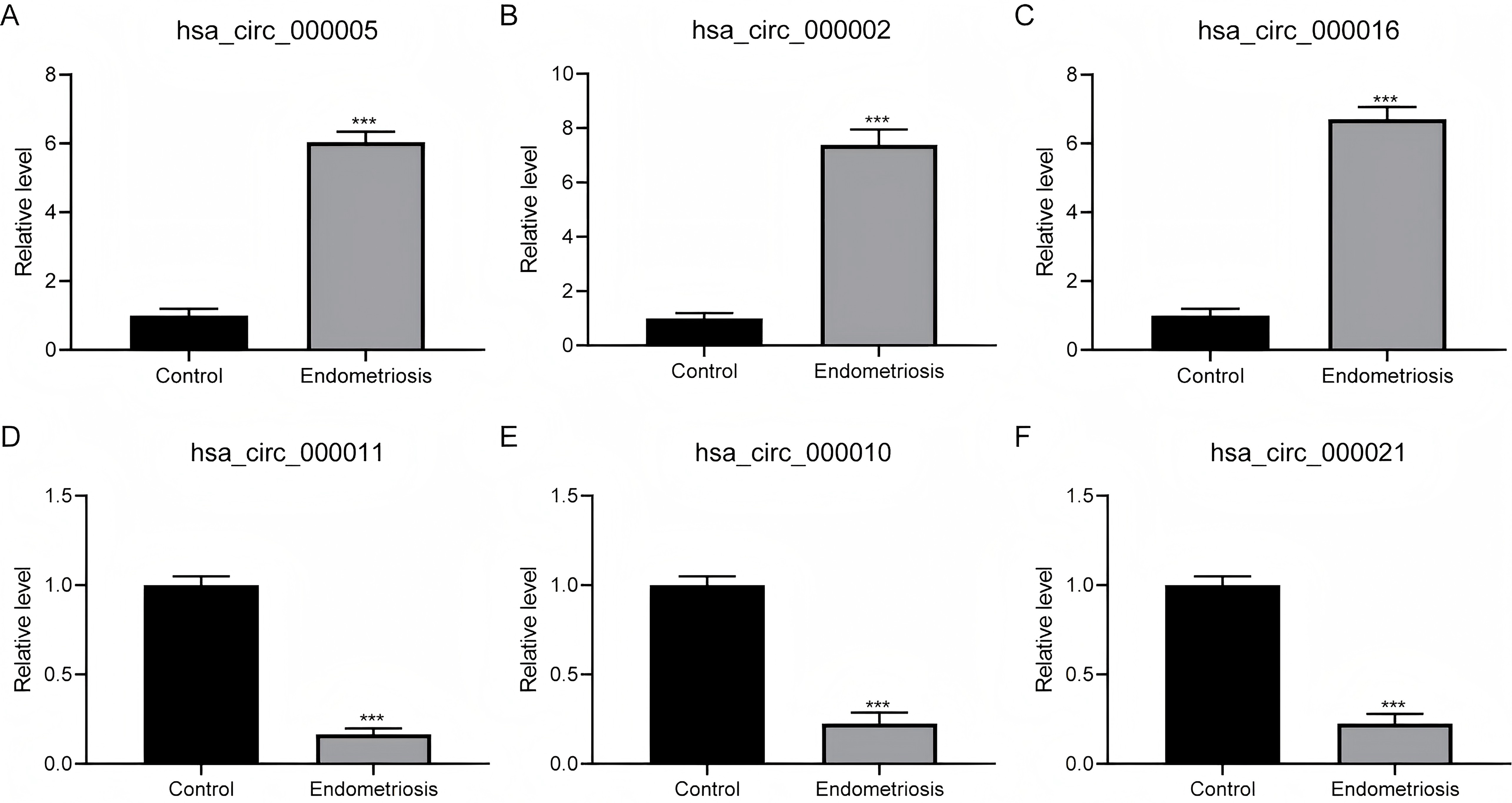{kind=link}
GO and KEGG pathway analyses of changed circRNAs
Using GO and KEGG analyses, the predicted potential function and association of the circRNAs that showed differential expression and their corresponding parental genes were examined. The enriched GO terms for the differentially expressed circRNAs were ranked, highlighting the top eight terms. Notably, the enriched GO terms pertaining to biological processes primarily focused on muscle attachment and the regulation of cAMP-mediated signaling (Fig. 4A). Regarding cellular components, the enriched GO terms were predominantly linked to the cytoskeleton and cytoplasmic vesicles (Fig. 4B). In terms of molecular functions (MF), the enriched GO terms were primarily associated with microtubule binding and the structural constituents of the cytoskeleton (Fig. 4C). Additionally, the enriched scatter diagram illustrated the top eight pathways identified through KEGG pathway enrichment analysis (Fig. 4D), indicating potential associations of the differentially expressed genes with Vitamin B6 metabolism and the synthesis, secretion, and function of Parathyroid hormone.
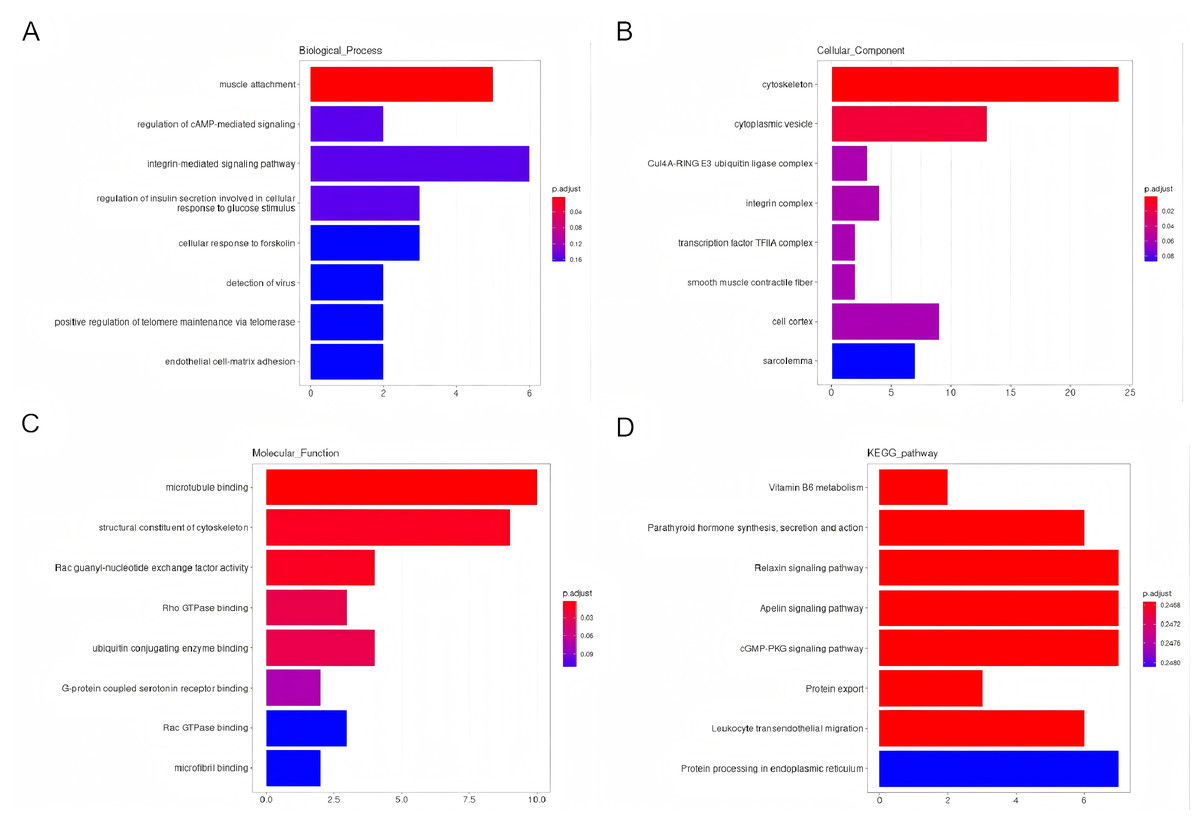
Figure 4: An investigation into the Gene Ontology (GO) categories and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with the parental genes of the differentially expressed circRNAs.
Biological processes (BP), cellular compartments (CC), and molecular functionalities (MF) (A–C). Furthermore, a KEGG pathway analysis was conducted to gain insights into their functional pathways (D).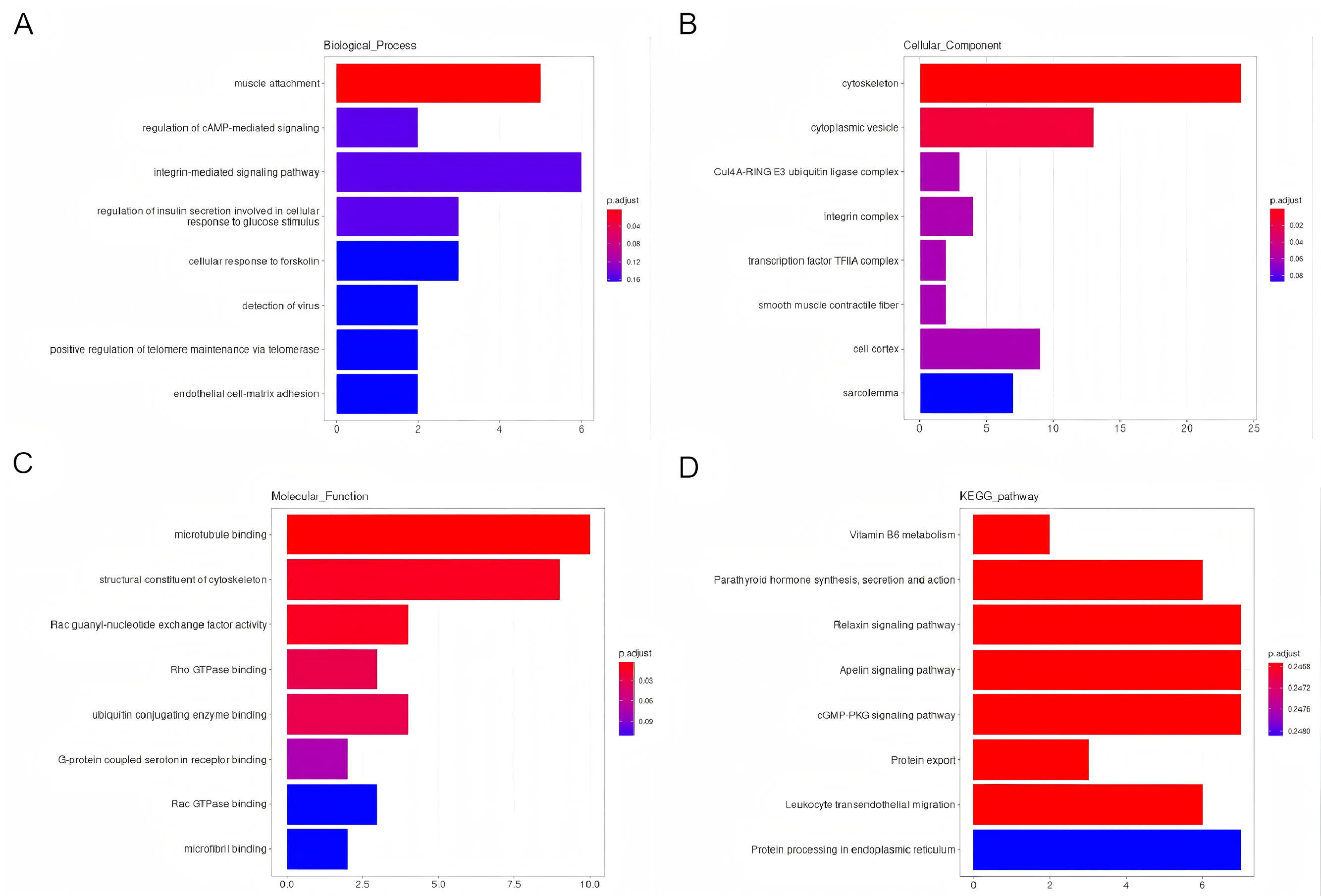{kind=link}
CircRNA-miRNA-mRNA network
The circRNA–miRNA–mRNA regulatory network was analyzed using miRanda and RNAhybrid software. The most upregulated circRNA, hsa_circ_000005, and the most downregulated circRNA, hsa_circ_000011, were prioritized in the network. Both circRNAs were predicted to interact with multiple miRNAs, with at least four miRNAs binding to each circRNA. The potential functional network involving hsa_circ_000005 and hsa_circ_000011 is illustrated in Fig. 5.

Figure 5: Analysis of the circRNA-miRNA-mRNA network focused on the top upregulated hsa_circ_000005 and top downregulated hsa_circ_000011.
{kind=link}
Discussion
Endometriosis, a intricate gynecological condition, is marked by the occurrence of tissue resembling endometrium outside the uterus, frequently resulting in infertility and chronic pelvic pain (Chauhan et al., 2022). The molecular mechanisms underlying endometriosis pathogenesis remain incompletely understood, prompting investigations into various aspects of gene expression regulation, including the role of circular RNAs (circRNAs). This study undertook an extensive analysis of circRNA expression patterns in patients with ovarian endometriosis compared to control subjects, with the goal of uncovering the possible functions of circRNAs in the advancement and evolution of this disorder.
Our study builds upon prior research efforts that have explored the involvement of circRNAs in endometriosis (Wang et al., 2019). While the specific circRNAs identified in our study may differ, the overarching findings align with some previous reports. For instance, the upregulation of circRNAs associated with muscle attachment and cAMP-mediated signaling pathways is consistent with studies implicating dysregulation in cellular processes and signaling cascades in endometriosis pathophysiology (Abera, 2009). Furthermore, the downregulation of circRNAs linked to microtubule binding and cytoskeletal components mirrors observations in other studies highlighting alterations in cytoskeletal dynamics in endometriosis (Szymański et al., 2024).
Our study adds to the expanding evidence that highlights the complex regulatory functions of circRNAs in influencing gene expression through interactions with miRNAs, particularly in the context of circRNA-miRNA-mRNA network analysis (Tong et al., 2022). By focusing on validated circRNAs, such as hsa_circ_000005 and hsa_circ_000011, and investigating their potential interactions with miRNAs, we provide insights into specific regulatory networks that may be perturbed in ovarian endometriosis. This complements existing literature that has demonstrated the importance of circRNA-mediated regulatory networks in various disease contexts, including gynecological disorders (Huang, Zhou & Li, 2019).
The differential expression of circRNAs identified in our study, including both upregulated and downregulated candidates, suggests a potential role for these molecules in the molecular landscape of ovarian endometriosis. The dysregulation of circRNAs associated with specific biological processes and molecular functions highlights the complexity of gene expression regulation in this condition. Notably, the enrichment of circRNAs involved in pathways such as vitamin B6 metabolism and parathyroid hormone function underscores the potential impact of circRNAs on key physiological processes implicated in endometriosis pathogenesis (Pabona et al., 2012; Vysotskyi, Khramova & Kachanova, 2015; Widasari et al., 2020).
The corroboration of differentially expressed circRNAs via qRT-PCR bolsters our findings, validating the RNA sequencing outcomes and underscoring the pivotal role of the identified circRNAs in ovarian endometriosis. The functional annotation of these circRNAs through GO and KEGG pathway analyses provides valuable insights into the potential biological roles and regulatory networks associated with these molecules. The identification of specific pathways and functions enriched in differentially expressed circRNAs offers a starting point for further mechanistic investigations and targeted studies.
The findings from this study provide compelling evidence for the involvement of specific circular RNAs (circRNAs) in the pathogenesis of ovarian endometriosis, a condition characterized by the abnormal growth of endometrial tissue outside the uterus. The upregulation of hsa_circ_000005, hsa_circ_000002, and hsa_circ_000016, along with the downregulation of hsa_circ_000011, hsa_circ_000010, and hsa_circ_000021, suggests a differential regulation of circRNAs that may contribute to the molecular mechanisms driving endometriosis. The GO analysis reveals that the differentially expressed circRNAs are involved in processes such as muscle attachment and cAMP-mediated signaling, both of which are crucial in cellular interactions and tissue remodeling during endometriosis progression. The association of these circRNAs with cellular components such as the cytoskeleton and cytoplasmic vesicles points to a potential role in regulating cellular structure and dynamics, which could affect cell migration and tissue invasion, key features of endometriosis. Additionally, the molecular functions related to microtubule binding and structural components of the cytoskeleton highlight the importance of cytoskeletal regulation in disease pathogenesis, as alterations in cytoskeletal integrity can facilitate the aberrant movement of endometrial cells. The KEGG pathway enrichment further suggests that the differentially expressed circRNAs might influence pathways related to vitamin B6 metabolism and parathyroid hormone signaling, which could have significant implications for the metabolic and endocrine dysregulation observed in endometriosis. The circRNA-miRNA-mRNA interaction analysis identified potential regulatory networks involving hsa_circ_000005 and hsa_circ_000011, which may modulate the expression of key mRNAs involved in these pathways, adding another layer of complexity to the regulation of endometriosis at the post-transcriptional level. By validating these findings through qRT-PCR, this study reinforces the potential of circRNAs as biomarkers and therapeutic targets for ovarian endometriosis. These results highlight the importance of circRNAs in the regulation of key signaling pathways that influence the pathogenesis of endometriosis and suggest that targeting specific circRNA-miRNA-mRNA networks could offer novel therapeutic strategies for this debilitating condition.
Moving forward, it will be crucial to delve deeper into the functional roles of the identified circRNAs in ovarian endometriosis. Experimental investigations, such as knockdown or overexpression studies, can clarify the precise impacts of these circRNAs on cellular mechanisms pertinent to the development of endometriosis. Furthermore, investigating the clinical significance of circRNA profiles as indicators for diagnosing or predicting outcomes in endometriosis could lead to tailored treatment strategies and enhanced patient results.
Conclusions
In conclusion, our study offers a thorough examination of circRNA expression patterns in ovarian endometriosis, providing insights into the potential functions of circRNAs in the molecular pathways implicated in this condition. By integrating quality control measures, differential expression analyses, validation studies, functional annotations, and network analyses, we have uncovered novel insights into the regulatory landscape of circRNAs in endometriosis. Our findings contribute to the broader understanding of endometriosis pathophysiology and offer a foundation for future research endeavors aimed at translating these molecular insights into clinical applications.