
Transcription factor EB, a promising therapeutic target in cardiovascular disease
- Published
- Accepted
- Received
- Academic Editor
- Vladimir Uversky
- Subject Areas
- Molecular Biology, Cardiology, Internal Medicine
- Keywords
- Cardiovascular disease, TFEB, Lysosome, Autophagic flux
- Copyright
- © 2024 Yan et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2024. Transcription factor EB, a promising therapeutic target in cardiovascular disease. PeerJ 12:e18209 https://doi.org/10.7717/peerj.18209
Abstract
Cardiovascular disease (CVD) remains the major cause of morbidity and mortality around the world. Transcription factor EB (TFEB) is a master regulator of lysosome biogenesis and autophagy. Emerging studies revealed that TFEB also mediates cellular adaptation responses to various stimuli, such as mitochondrial dysfunction, pathogen infection and metabolic toxin. Based on its significant capability to modulate the autophagy-lysosome process (ALP), TFEB plays a critical role in the development of CVD. In this review, we briefly summarize that TFEB regulates cardiac dysfunction mainly through ameliorating lysosomal and mitochondrial dysfunction and reducing inflammation.
Introduction
Cardiovascular diseases (CVDs) are a range of disorders that affect both the blood vessels and heart. They are a major global threat and one of the leading causes of mortality and morbidity worldwide, placing a heavy burden on patients and their families. Common CVDs include acute myocardial infarction (AMI), heart failure, atrial fibrillation (AF), and atherosclerosis.
Transcription factor EB (TFEB) is a member of the MiT/TFE bHLH-LZ subfamily (Steingrímsson, Copeland & Jenkins, 2004). It is considered a major transcriptional regulator of autophagy and lysosomal biogenesis (Xu & Ren, 2015). Recent studies have shown that TFEB binds directly to CLEAR elements on lysosomal genes, promoting the expression of the entire network of genes in their promoters that contain CLEAR-regulated motifs (the CLEAR network) (Palmieri et al., 2011; Sardiello et al., 2009). In resting cells under nutrient-rich conditions, TFEB is primarily located in the cytoplasm and is inactive (Sardiello et al., 2009; Settembre et al., 2011). However, under conditions of starvation, bacterial infection, lysosomal dysfunction, or other stress processes, TFEB quickly translocates to the nucleus and activates the transcription of its target genes, promoting organismal homeostasis (Martina et al., 2012). TFEB is increasingly believed to regulate homeostasis in the cardiovascular system and has a protective effect against CVD, such as AMI, atherosclerosis, and cardiotoxicity (Chen et al., 2022b; Li et al., 2022; Haas et al., 2022). This article reviews the research progress of TFEB in CVD and discusses the related molecular mechanisms.
Survey Methodology
To identify the pertinent literature, we conducted a PubMed search using the following keywords: (Transcription factor EB) and (Cardiovascular disease)/(Transcription factor EB) and (Angiocardiopathy). We then proceeded to a title and abstract screening and elimination process, which excluded articles not related to CVD, in order to ensure the comprehensiveness and accuracy of this review.
TFEB and atherosclerosis
Atherosclerosis is a progressive and inflammatory vascular disease caused by lipid dysregulation. It is characterized by the abnormal accumulation of lipids and immune cells within the vessel wall (Arida et al., 2018; Ammirati et al., 2015). This accumulation ultimately leads to severe clinical complications of arterial disease, such as AMI and stroke (Zhao et al., 2019; Ching et al., 2011). Atherosclerosis is a complex pathophysiological process that involves multiple cell types, including macrophages (Moore, Sheedy & Fisher, 2013), endothelial cells (Sun et al., 2021), and vascular smooth muscle cells.
Numerous studies have confirmed the involvement of TFEB in the development of CVD. Vascular dysfunction, in particular endothelial dysfunction, is a key factor in CVD. Furthermore, overexpression of TFEB in endothelial cells (EC) has been shown to significantly promote EC migration and tube formation. Endothelial-specific TFEB overexpression in mice demonstrated enhanced capillarisation and augmented blood perfusion following ischemic injury (Fan et al., 2018). Moreover, the restoration of autophagic flux through TFEB overexpression diminished oxidative stress and enhanced endothelium-dependent relaxation in aortic endothelial cells of diabetic mice (Zhao et al., 2022). Similarly, Lu et al. (2017) demonstrated that laminar shear stress, one of the crucial processes in the atherosclerotic process, can prevent atherosclerosis by increasing the abundance of TFEB in endothelial cells. In vitro experiments have demonstrated that the overexpression of TFEB in endothelial cells effectively inhibits the inflammatory response, while the down-regulation of TFEB exacerbates it. This effect may be attributed, in part, to the reduction of oxidative stress by TFEB (Lu et al., 2017). TFEB increases the abundance of antioxidant genes, such as heme oxygenase 1 (HO1) and superoxide dismutase 2 (SOD2), which reduces intracellular reactive oxygen species (ROS) (Fig. 1A) (Lu et al., 2017).
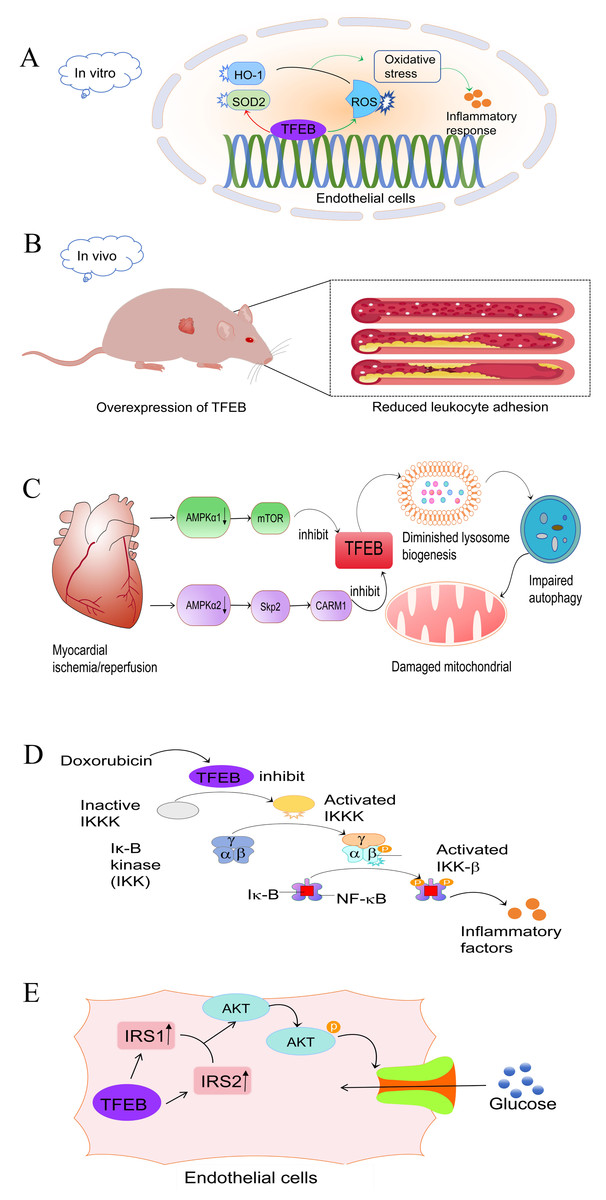
Figure 1: Role and mechanism of TFEB in CVD.
(A) In endothelial cells, TFEB has an antioxidant effect by activating SOD2 and HO-1 and inhibiting the production of ROS, thereby reducing the inflammatory response.Red: activating effect. Green: inhibitory effect. (B) Mice that over-express TFEB exhibit reduced leukocyte adhesion, which attenuates plaque formation and slows down the progression of AS. (C) After myocardial ischemia-reperfusion, the expression of AMPKα1 and AMPKα2 was reduced. This inhibition of TFEB occurred through the AMPKα1-mTOR and AMPKα2-skp2-CARM1 pathways, respectively. As a result, lysosomal genesis was reduced, leading to impaired autophagic copper beam and ultimately impaired mitochondrial function. (D) Doxorubicin, a chemotherapeutic drug, inhibits TFEB expression, leading to IKK-β and NFκB activation and subsequent inflammatory response. (E) In endothelial cells, TFEB upregulates IRS1 and IRS2 expression, which activate the Akt signalling pathway, phosphorylate Akt, and facilitate glucose transport into the cytosol. TFEB, transcription factor EB; CVD, cardiovascular disease; HO1, oxygenase 1; SOD2, superoxide dismutase 2; ROS, reactive oxygen species; AS, atherosclerosis; IRS, insulin receptor substrate. Image source credit: Motifolio.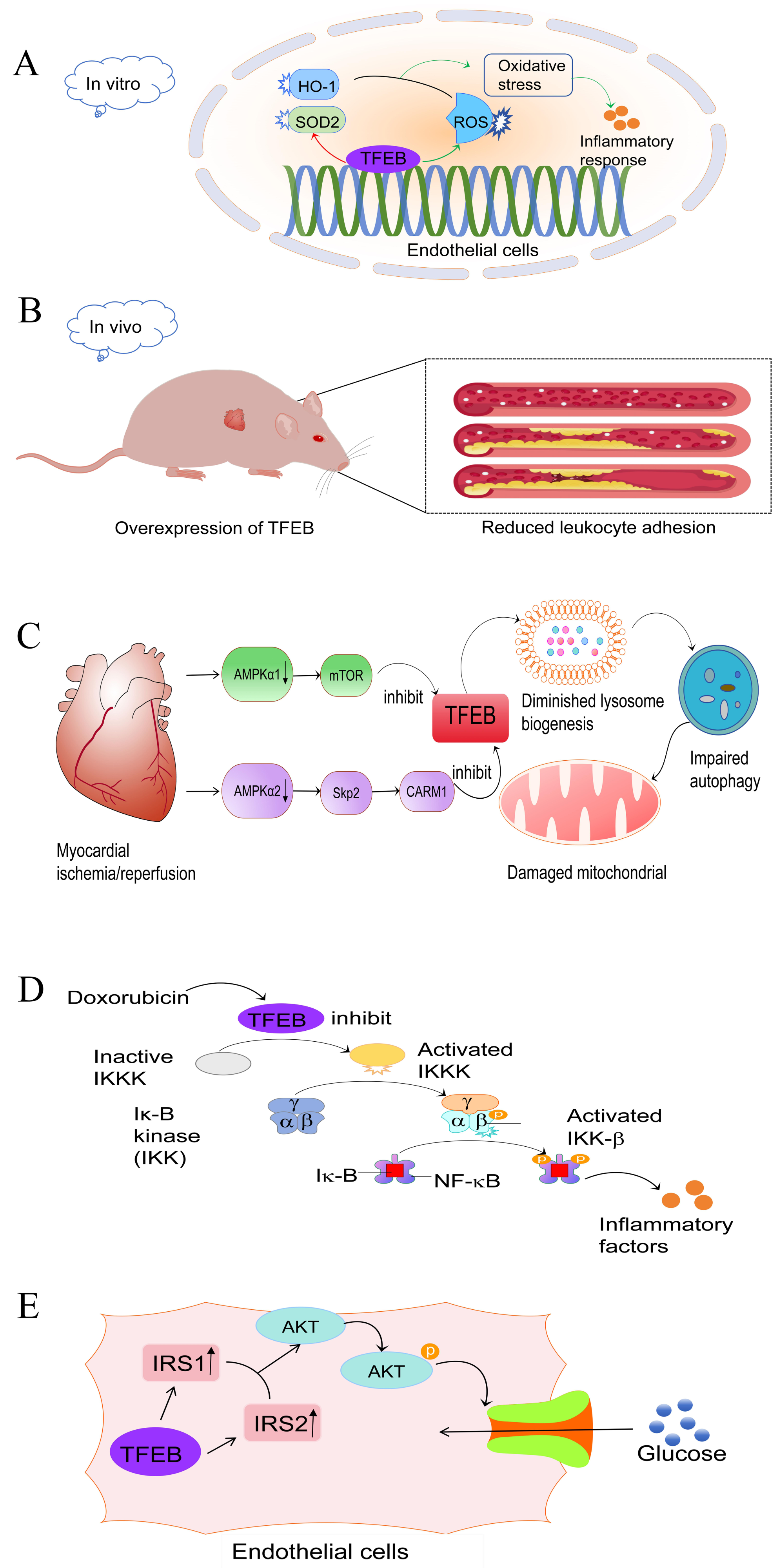{kind=link}
Under in vivo inflammatory conditions, transgenic mice with endothelial cell-specific expression of TFEB exhibited reduced endothelial cell-leukocyte adhesion (Fig. 1B), and atherosclerosis development was reduced (Lu et al., 2017). In addition, EC-TFEB/ApoE-/- mice exhibited a reduction in atherosclerotic lesion formation compared to their littermate ApoE-deficient (ApoE-/-) mice. This suggests that TFEB activation has a protective effect against atherosclerosis in vivo. Chen et al. (2022a) conducted a study demonstrating how bromelain stimulates antioxidant production through the activation of TFEB, thereby slowing the progression of atherosclerosis. These findings highlight the benefits of TFEB in vascular diseases.
Additionally, numerous studies have confirmed that TFEB acts as a master regulator, promoting the expression of autophagic and lysosomal genes (Moore, Sheedy & Fisher, 2013), primarily by targeting intracellular cholesteryl ester-rich lipid droplets (LDs) for degradation to free cholesterol, orchestrating autophagic lysosomes, and promoting lipid degradation. Therefore, TFEB may act as an antioxidant activator and promote autophagy to delay the progression of atherosclerosis. The data indicate that TFEB plays a significant role in the regulation of atherosclerosis, encompassing processes such as angiogenesis, autophagy, inflammation, and oxidative metabolism.
TFEB and myocardial ischemia/reperfusion injury
Although there have been significant advances in understanding ischaemic heart disease, the underlying mechanisms remain incompletely elucidated (Severino et al., 2020). Studies have indicated that autophagy has emerged as a key factor in maintaining cardiac homeostasis and function, as it contributes to the reduction of cardiac damage by facilitating cellular adaptation to misfolded protein accumulation, mitochondrial dysfunction and oxidative stress (Sciarretta et al., 2018a). As previously mentioned, TFEB is a master regulator of autophagy genesis. Therefore, it plays a crucial role in maintaining cardiac homeostasis by mediating autophagy. Studies have reported that in myocardial ischemia/reperfusion injury (IRI), both cytoplasmic AMPKα1 and nuclear α2 subunits are inhibited. This leads to impaired autophagic flux by suppressing TFEB through the AMPKα1-mTOR and AMPKα2-Skp2-CARM1 signaling pathways, respectively (Wang et al., 2019b). Similarly, post-ischemic reperfusion increased the levels of myocardial BECLIN-1 protein, which inhibits the activation of TFEB (Dhingra et al., 2017), resulting in impaired autophagic flux (Oliveira et al., 2021). Autophagy is not an independent process; it is closely linked to mitochondrial and lysosomal functions. BNIP3, a protein interacting with BCL-2 and adenovirus E1B 19kDa, has been reported to play a role in IRI (Diwan et al., 2007). Its up-regulation leads to lysosomal depletion and promotes autophagosome accumulation, impairing mitochondrial autophagy and leading to cardiomyocyte death. On the other hand, TFEB expression stimulates lysosomal biogenesis, restores autophagosome processing and attenuates mitochondrial damage (Fig. 1C) (Ma et al., 2012). During hypoxia/reoxygenation,TFEB knockdown abrogated the clearance of autophagosomes and restoration of cell viability that resulted from LAPTM4B overexpression. Conversely, TFEB overexpression rescued the injury to cardiac tissue and restored the autophagic fluxes that resulted from LAPTM4B knockdown during ischemia and reperfusion (Gu et al., 2020). In addition, Javaheri et al. (2019) discovered that macrophage-specific over-expression of the transcription factor EB (M ϕ-TFEB) enhances ventricular function following IR injury. Additionally, they found that TFEB in macrophages contributes to ventricular remodeling after MI by mediating inflammatory responses. Therefore, it is clear that TFEB may impact IRI through modulation of various biological functions (Javaheri et al., 2019). Several studies have confirmed ways to improve the prognosis of myocardial infarction. For example, Sciarretta et al. (2018b) demonstrated that alginate, a naturally occurring non-reducing disaccharide, improves myocardial remodeling after myocardial infarction (MI). This improvement relies on TFEB-mediated activation of autophagy. Liu et al. (2020) reported that upregulation of TFEB induced by donor mesenchymal stem cell (MSC) apoptotic vesicle release promotes autophagy and angiogenesis, thereby improving post-MI cardiac dysfunction. In summary, TFEB plays a pivotal role in protecting against cardiovascular diseases and more in-depth studies are needed to explore its underlying mechanisms.
TFEB and chemotherapy-related cardiac toxicity
Chemotherapeutic agents are essential in the treatment of tumors, but their clinical use is severely hampered by their unexpected cardiotoxicity. Clinicians and scientists have long been aware of doxorubicin (DOX)-induced cardiotoxicity (DIC), and its molecular mechanisms are still being discovered. The known mechanisms involved in DIC include oxidative stress, Ca2+ overload, DNA damage, mitochondrial dysfunction, and autophagic flux impairment (Rawat et al., 2021). One study found that human cardiac tissues from doxorubicin-induced heart failure exhibited an increase in nuclear TFEB protein (Bartlett et al., 2016), suggesting that there may be some association between TFEB and DIC, and in vitro experiments, cardiomyocyte-specific TFEB over-expression induced cardiac remodeling, whereas TFEB knockdown attenuated DIC. Bartlett et al. (2016) have reported that DOX inhibited TFEB expression in a time- and dose-dependent manner, leading to disruption of autophagic flux and deterioration of cardiac function. However, TFEB activation prevented DIC by ameliorating lysosomal dysfunction and autophagy inhibition, reducing ROS overload and increasing cell viability (Bartlett et al., 2016; Wang et al., 2019a). A significant decrease in TFEB mRNA levels was observed in DOX-treated H9C2 cardiac fibroblasts, but not in DOX-treated Sprague-Dawley rat hearts. This suggests that the effect of DOX on TFEB transcriptional repression is cell-type and/or tissue-specific (Bartlett et al., 2016). In contrast, another study on chemotherapeutic agents found that TFEB exacerbated DOX-induced cardiotoxicity after the addition of alternate day fasting (ADF) treatment, resulting in increased nuclear TFEB, impaired cardiac function, and increased mortality in mice, which may be mediated by TFEB/MuRF1 or TFEB/GDF15 leading to a decrease in left ventricular (LV) mass decreased (Ozcan et al., 2023). Recently, it has been demonstrated that TFEB plays important and multiple roles. The study discovered that doxorubicin treatment reduced TFEB expression in the nucleus and increased IKKα/β and NF-κB phosphorylation (Wang et al., 2020). This suggests a possible connection between TFEB activation and NF- κB, a well-known inflammation-associated factor (Fig. 1D). Therefore, DIC may be achieved by inhibiting the anti-inflammatory activity of TFEB through the activation of the NF- κB signaling pathway. Taken together, in most in vitro model studies, TFEB exerts a protective effect against cardiotoxicity, whereas in the in vivo model, an increase in TEFB may exacerbate myocardial fibrosis and cardiomyocyte death, ultimately leading to HF.
TFEB and metabolism-related cardiotoxicity
Both hyperglycemia and fatty acid overload contribute to a condition known as ‘glycolipotoxicity’, which leads to the accumulation of toxic metabolites in the cardiovascular system and is increasingly recognized as a major driver of cardiac pathology and a contributor to the progression of end-stage heart failure (Sanbe et al., 2004; Weekes et al., 2003; Su & Wang, 2020). Numerous studies have demonstrated that glycolipotoxic effects on cardiomyocytes primarily originate or terminate in the mitochondria and endoplasmic reticulum (ER) (Boudina & Abel, 2006; Cai et al., 2002; González-Rodríguez et al., 2014; Karunakaran et al., 2012; Yang et al., 2015). Transcriptomic data from ventricular tissue of constitutive cardiomyocyte-specific TFEB-/- mice suggest that TFEB regulates a network of genes involved in lipid and carbohydrate metabolism. Modulation of cardiomyocyte lipid metabolism by TFEB is achieved through modulation of prominent lipid targets such as peroxisome proliferator-activated receptor alpha (PPARα) (Trivedi et al., 2020). In the liver, TFEB acts in an autophagy-dependent manner to reduce lipid accumulation (Settembre et al., 2013). Lack of TFEB action resulted in significant LD accumulation, whereas over-expression of TFEB reduced LD size and accumulation. This demonstrates an unusual function of TFEB in regulating substrate metabolic pathways in cardiomyocytes, rather than its usual role in regulating lysosomal signaling and function. In endothelial cells, TFEB up-regulates Insulin Receptor Substrate (IRS1) 1 and 2 through different mechanisms to activate Akt signaling and increase glucose uptake (Fig. 1E) (Sun et al., 2021). In a mouse model, TFEB overexpression reduces diet-induced weight gain and obesity and improves glucose intolerance and insulin sensitivity (Evans et al., 2019). On the other hand, mtorc2-Akt-mediated inactivation of GSK3β under glucose deprivation conditions leads to nuclear retention of TFEB in the human colorectal adenocarcinoma cell line HT2951 (Li et al., 2018). Thus, there may be an interaction between TFEB and Akt to maintain internal homeostasis. Altogether, all these data suggest that TFEB is an important regulator of glucose homeostasis and lipid homeostasis.
Conclusion
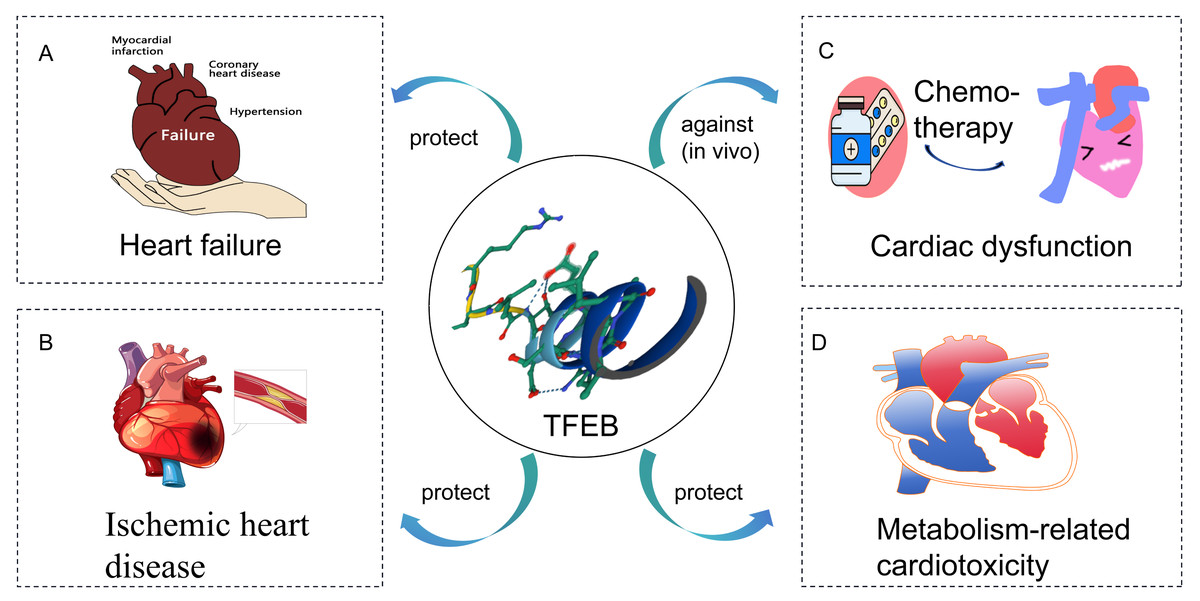
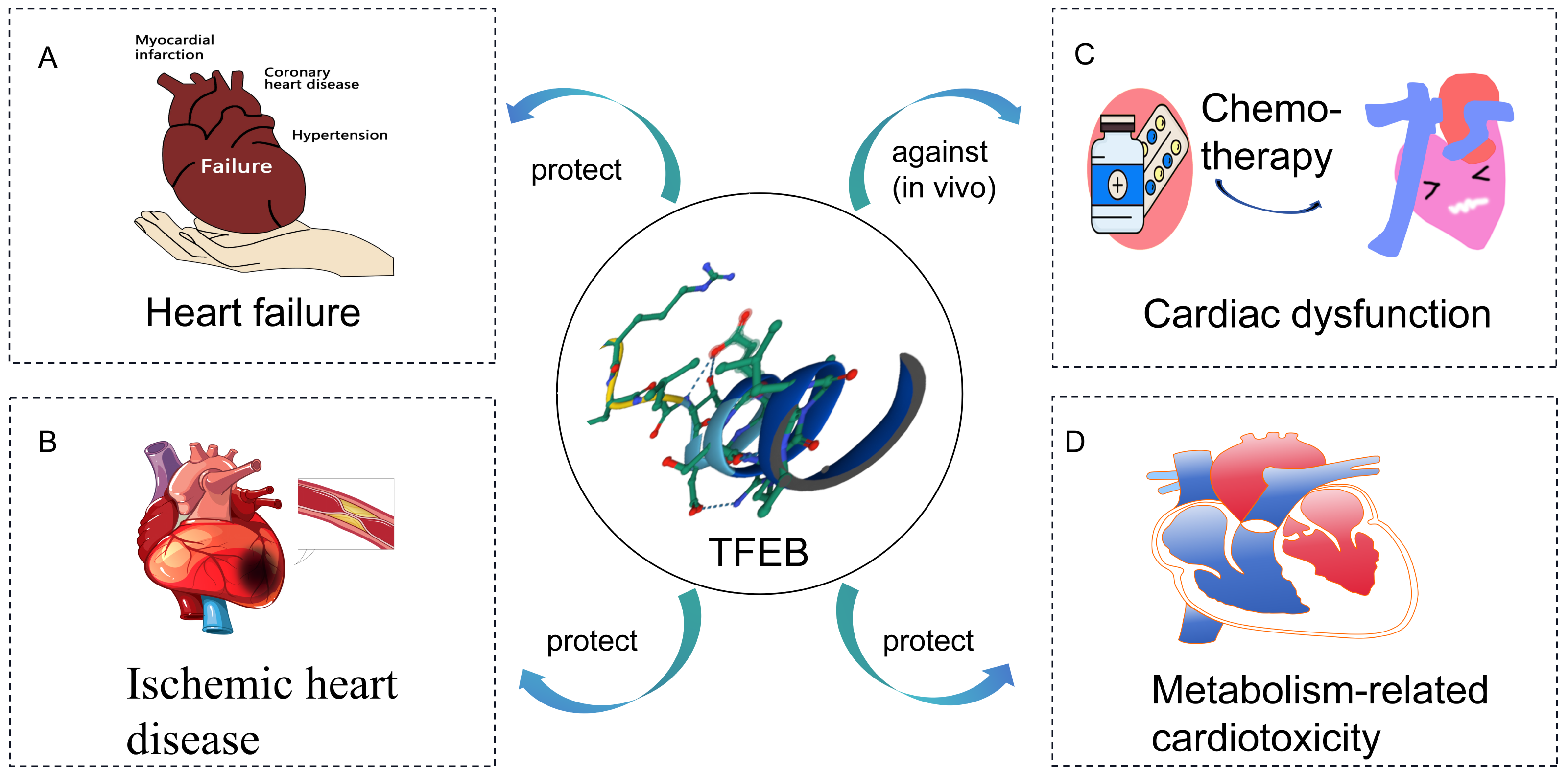
In this review, the role of TFEB in CVD is discussed (Fig. 2). It is found that stimulation of TFEB is an effective strategy to ameliorate cardiac dysfunction, mainly associated with improved lysosomal and mitochondrial dysfunction and reduced inflammation. Increased TFEB helps clear damaged mitochondria and inflammatory factors, thus improving oxidative stress in the heart. Additionally, TFEB has non-classical roles in metabolic pathways, besides regulating lysosomal biogenesis and autophagy. However, the mechanisms underlying TFEB’s role in CVD have not been fully elucidated. Understanding TFEB’s role in CVD and its associated molecular mechanisms is important. Manipulating TFEB activity may provide a promising target for treating CVD.
{kind=link}