
The role of NF-kappaB in the inflammatory processes related to dental caries, pulpitis, apical periodontitis, and periodontitis–a narrative review
- Published
- Accepted
- Received
- Academic Editor
- Xin Zhang
- Subject Areas
- Cell Biology, Dentistry
- Keywords
- NF-κB, Caries, Pulpitis, Apical periodontitis, Periodontitis
- Copyright
- © 2024 Chen et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2024. The role of NF-kappaB in the inflammatory processes related to dental caries, pulpitis, apical periodontitis, and periodontitis–a narrative review. PeerJ 12:e17953 https://doi.org/10.7717/peerj.17953
Abstract
Tooth-related inflammatory disorders, including caries, pulpitis, apical periodontitis (AP), and periodontitis (PD), are primarily caused by resident oral microorganisms. Although these dental inflammatory conditions are typically not life-threatening, neglecting them can result in significant complications and greatly reduce an individual’s quality of life. Nuclear factor κB (NF-κB), a family formed by various combinations of Rel proteins, is extensively involved in inflammatory diseases and even cancer. This study reviews recent data on NF-κB signaling and its role in dental pulp stem cells (DPSCs), dental pulp fibroblasts (DPFs), odontoblasts, human periodontal ligament cells (hPDLCs), and various experimental animal models. The findings indicate that NF-κB signaling is abnormally activated in caries, pulpitis, AP, and PD, leading to changes in related cellular differentiation. Under specific conditions, NF-κB signaling occasionally interacts with other signaling pathways, affecting inflammation, bone metabolism, and tissue regeneration processes. In summary, data collected over recent years confirm the central role of NF-κB in dental inflammatory diseases, potentially providing new insights for drug development targeting NF-κB signaling pathways in the treatment of these conditions. Keywords: NF-κB, dental caries, pulpitis, apical periodontitis, periodontitis.
Introduction
In a physiological state, the mouth hosts over 700 species of bacteria, existing in a biofilm to maintain ecological balance with the host. Preserving the stability of this oral microbiome balance is crucial, as its disruption can lead to various oral infectious diseases, including dental caries, periodontitis (PD), and apical periodontitis (AP) (Anne Marie et al., 2023). Despite the rapid economic growth and improvements in living conditions over the past few decades, the public continues to be afflicted by inflammatory dental disorders. Dental caries, a widespread and chronic degenerative condition affecting nearly half of the world’s population (Alawady et al., 2023), is particularly concerning. This bacterial infection-driven disease disproportionately affects individuals in developing nations, where prevalence rates are notably higher. If untreated, bacteria can infiltrate the pulp through damaged tooth tissue, leading to pulpitis and periapical inflammation, eventually resulting in tooth loss. A recent systematic review revealed that approximately 50% of adults worldwide have at least one tooth affected by AP (Tibúrcio-Machado et al., 2021). Meanwhile, PD is a prevalent chronic inflammatory disease, second only to cavities, affecting more than 10% of individuals (Khajavi, Radvar & Moeintaghavi, 2022), with a prevalence rate negatively correlated with the level of economic development (Zhang et al., 2022). Investigating the underlying mechanisms of these inflammatory diseases is essential. Previous research has shown that pro-inflammatory agents can stimulate the activation of NF-κB, which plays a critical role in regulating both inflammatory responses and bone metabolism processes.
NF-κB, a transcription factor first identified in 1986 within the nuclear extracts of B lymphocytes, has the distinctive capability to bind specifically with the enhancer B sequence found in the immunoglobulin kappa light chain gene, thereby stimulating and increasing the expression of this gene (Sen & Baltimore, 1986). NF-κB regulates the expression of genes and proteins, influencing various biological processes such as cell proliferation, apoptosis, inflammation, oxidative stress, and angiogenesis (Govindappa et al., 2020). The NF-κB transcription factor consists of five Rel family members: RelA (p65), RelB, c-Rel, NF-κB1 (p105/p50), and NF-κB2 (p100/p52) (Pai & Sukumar, 2020). Among these, NF-κB1 and NF-κB2 are considered repressors due to the absence of C-terminal transcriptional activation domains (TADs) (Zhang et al., 2019). Consequently, NF-κB dimers composed solely of NF-κB1 or NF-κB2 are not capable of initiating transcription activity both in vivo and in vitro (Barnabei et al., 2021). Conversely, RelA, RelB, and c-Rel, which possess TADs, can form homologous and heterodimers with other family members to act as transcriptional activators. Additionally, RelB can function as both an activator (Ryseck et al., 1992) and an inhibitor (Ruben et al., 1992). Except for RelA, the transcription of genes encoding NF-κB polypeptides is upregulated by NF-κB, creating a positive feedback loop upon cell stimulation (Ghosh et al., 2012). The Both NF-κB1 and NF-κB2 subunits have the Rel homology domain (RHD) structure, a fundamental feature shared among all NF-κB family members. The RHD region contains a nuclear localization sequence (NLS) essential for dimerization, sequence-specific DNA binding, and interaction with inhibitory IκB proteins (Ghosh, May & Kopp, 1998). In most cell types, IκBs, such as IκBα, IκBβ, IκBγ, and IκBε, sequester NF-κB dimers in the cytoplasm, preventing their nuclear translocation and transcriptional activation (Karin et al., 2002). The NF-κB signaling pathway can be activated through two primary routes: the classical and non-classical pathways, as shown in Fig. 1. The classical pathway is mainly triggered by stimuli such as TNF-α, IL-1β, LPS, and RANKL, which activate specific Rel family proteins, including c-Rel, Rel A, and NF-κB1. The phosphorylation of IκBα, IκBβ, or IκBγ by IKKβ results in their degradation in the proteasome (Annunziata et al., 2007). This process permits the activation of c-Rel, Rel A, and NF-κB1, regulating the expression of targeted genes. In contrast, the non-classical pathway, mediated by Rel B and NF-κB2, is regulated by the stimulation of NEMO-independent signaling modules, such as NF-κB-inducible kinase (NIK) and IKKα. This pathway is stimulated by certain TNF family members, including CD40 ligand (CD40L), lymphotoxin-β (LT-β), and RANKL (Ghosh & Karin, 2002). Specifically, the alternative pathway facilitates the cleavage of p100 into NF-κB2 through the activation of IKKα, supporting the nuclear translocation of NF-κB2 without relying on IKKβ and IKKγ (Sun, 2011). The important roles of NF-κB signaling have been reported in various diseases, such as arthritis (Wang et al., 2017), cancer (Yu et al., 2020), colitis (He et al., 2019), and mastitis (He et al., 2015; Liang et al., 2014). NF-κB activation may aid antiviral responses by inducing interferon genes (Boehm et al., 1997; Stark et al., 1998). Recent research indicates that excessive activation of NF-κB signaling in individuals with COVID-19 may worsen the body’s inflammatory response (Amini-Farsani et al., 2021). Notably, the NF-κB signaling pathway has significant potential in regulating dental inflammation, such as caries (Feng et al., 2021), pulpitis (Nam et al., 2024), AP (Guan et al., 2021), and PD (Tian et al., 2022). Studies have demonstrated that inhibiting the NF-κB signaling pathway may promote tooth/bone matrix formation and exert anti-inflammatory effects (Aoki et al., 2023; He et al., 2017; Liu et al., 2013). However, research on NF-κB signaling in dental-related inflammation is still limited, and its specific mechanisms remain unclear. Currently, there are no clinical drugs or materials based on this pathway. Future research should focus on the NF-κB pathway to develop more effective drugs for preventing or treating dental-related inflammatory diseases, such as caries preventive agents, pulp capping agents, root canal disinfectants, or bone regeneration materials.
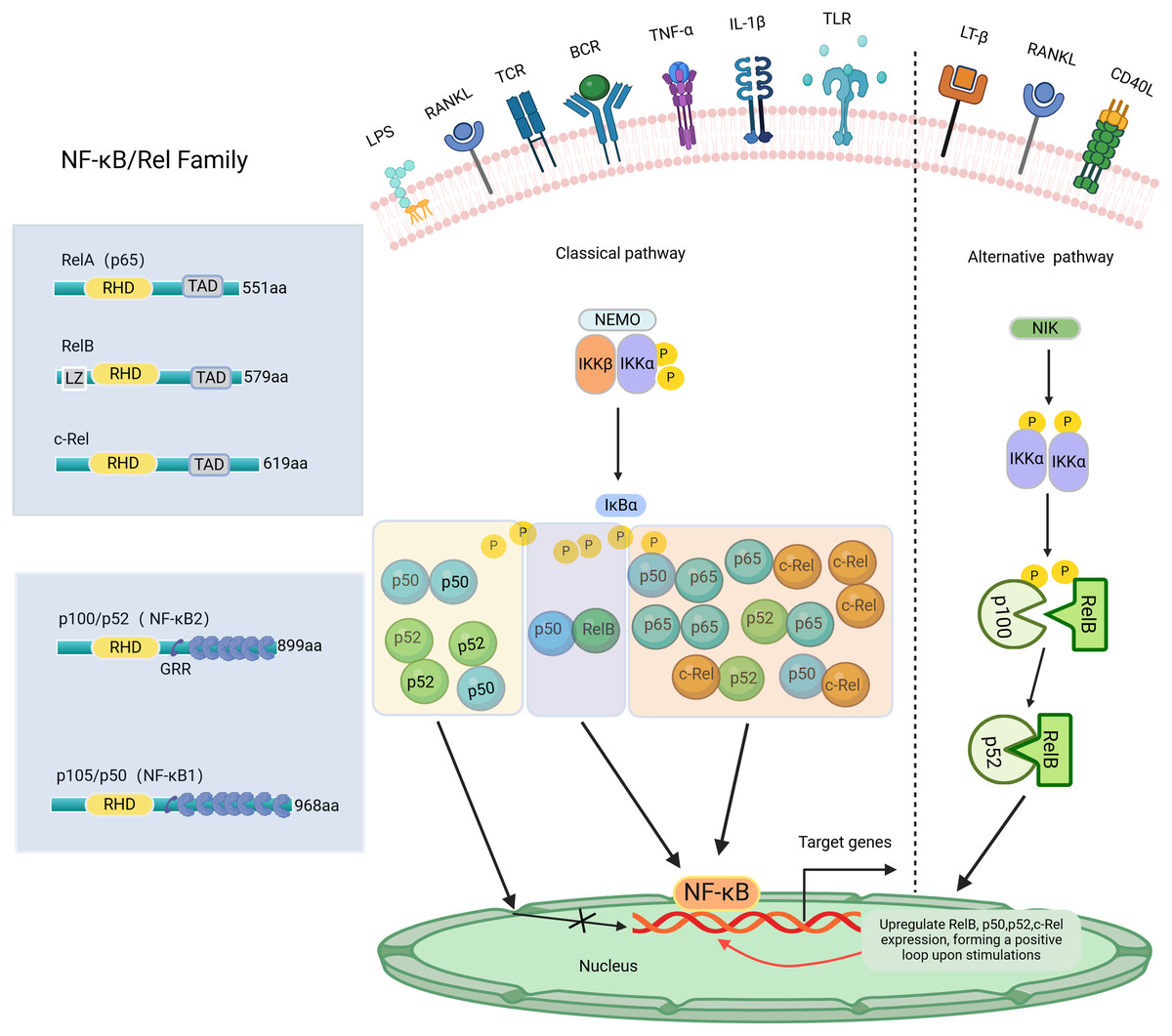
Figure 1: The NF-κB family, member characteristics and signaling pathways involved.
The amino-terminal half of these proteins contains a highly conserved 300-amino-acid Rel homology domain (RHD) shared by all members. RelA, c-Rel, and RelB possess transactivation domains (TADs) involved in transcription. In contrast, p50 and p52 lack transactivation domains, thus inhibiting rather than stimulating gene transcription. The length of each protein is indicated on the right side. NF-κB signaling pathway: Classical and Alternative Arms. The classical NF-κB pathway is triggered by TNF-α, IL-1β, LPS, RANKL, etc. The initiation of this pathway heavily relies on the IκB kinase (IKK) complex known as IKKEMO, leading to the rapid degradation of IκBα through phosphorylation. The alternative pathways are activated by LT-β, CD40L, RANKL, etc. This pathway involves the activation of NIK and IKKα, resulting in the gradual processing of p100 and the formation of p52. The p52/RelB heterodimer is then activated by dimerization. P, phosphorylation; LZ, leucine zipper domain on IKK/and Rel-B, and GRR, glycine-rich region. Created with BioRender.com.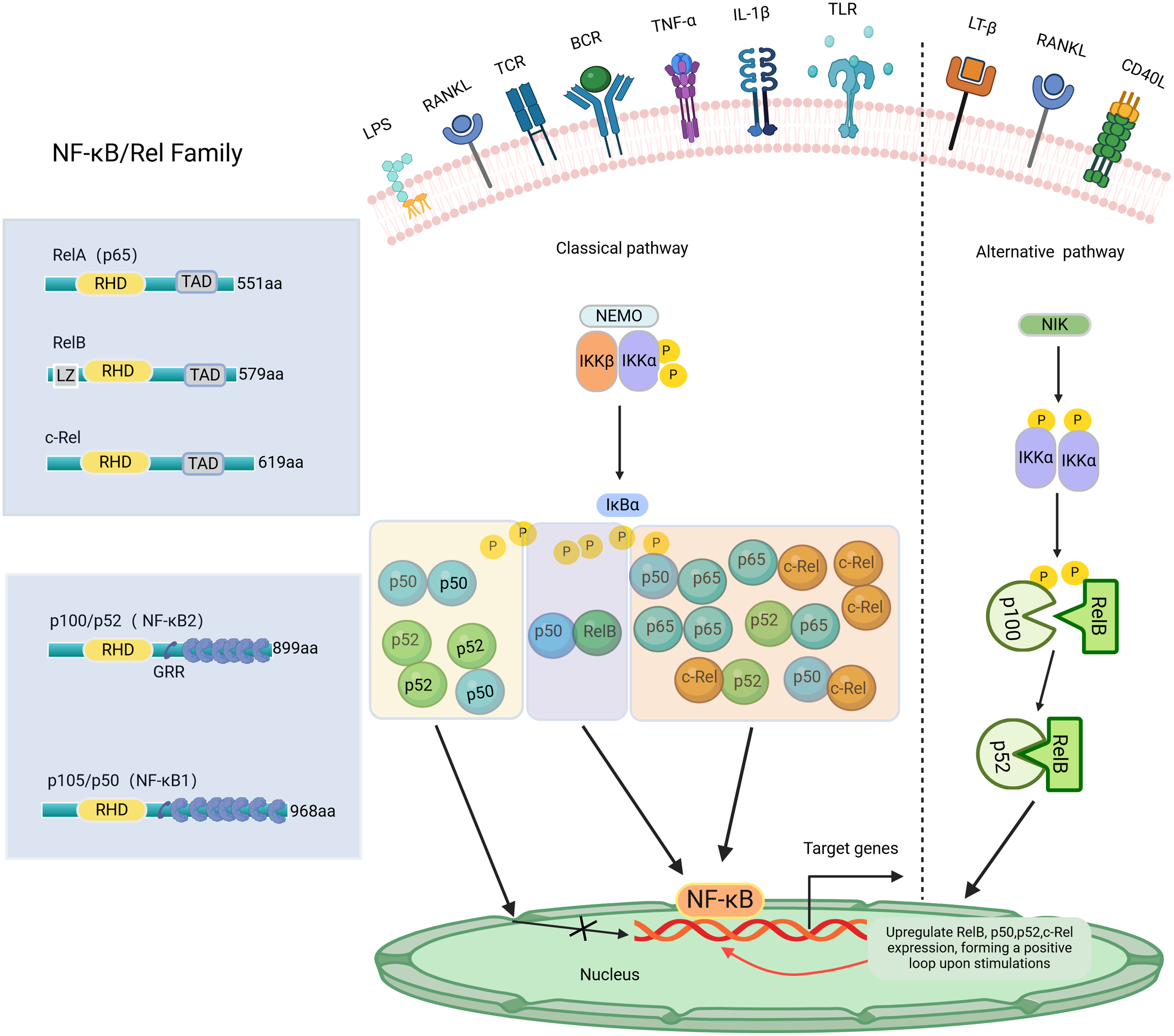{kind=link}
Methods
This study conducted comprehensive literature searches in the PubMed and Web of Science databases, focusing on studies published before 2024. Searches were performed using the terms “NF-κB (Title)”, “caries (Title)”, “pulpitis (Title)”, and “periodontitis (Title)”. Duplicate articles and those not indexed in SCI were excluded, as well as those without full text availability. The articles were then ranked based on relevance, prioritizing timeliness, explanatory value, and comprehensiveness when reading the full texts, with a focus on citing 42 articles for professional background. The key search terms “NF-κB and dental caries” (n = 45), “NF-κB and pulpitis” (n = 73), “NF-κB and apical periodontitis” (n = 69), and “NF-κB and periodontitis” (n = 1,531) guided the search process. Articles were further excluded based on duplication, non-SCI indexing, lack of relevance to the target diseases and/or NF-κB, unavailability of full-text, articles with comorbidities, and less common etiologies. Titles and abstracts were reviewed, and original articles were selected for full-text reading, adhering to the SANRA quality criteria, resulting in the selection of 74 articles. A total of 116 references were cited, as shown in Fig. 2.
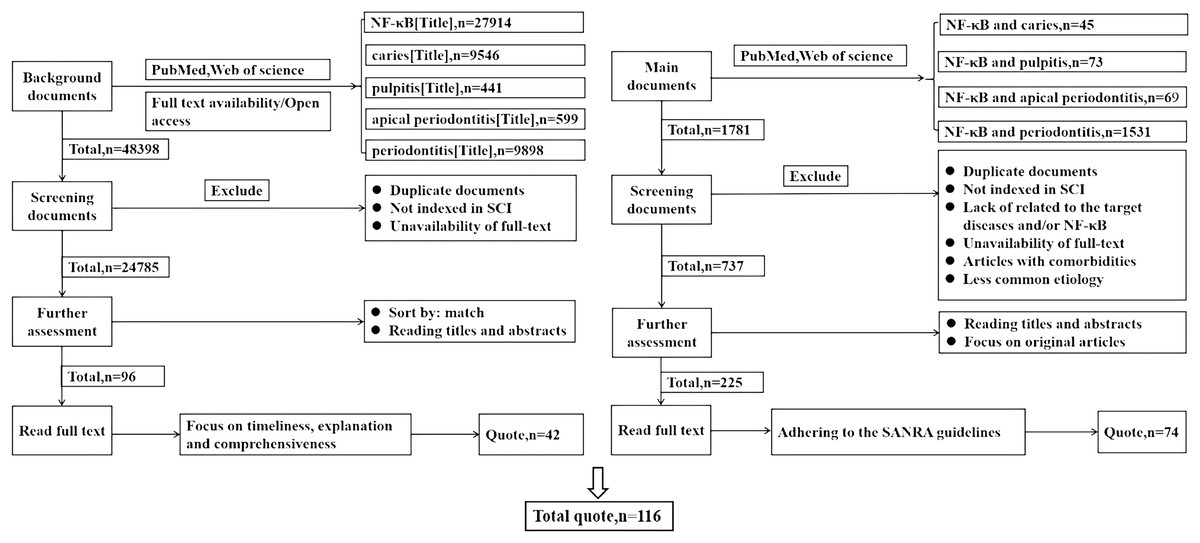
Figure 2: This review literature search flowchart.
How the literature cited in this review is selected. Left: the screening of literature cited for professional background. Right: the screening of literature cited in the main text.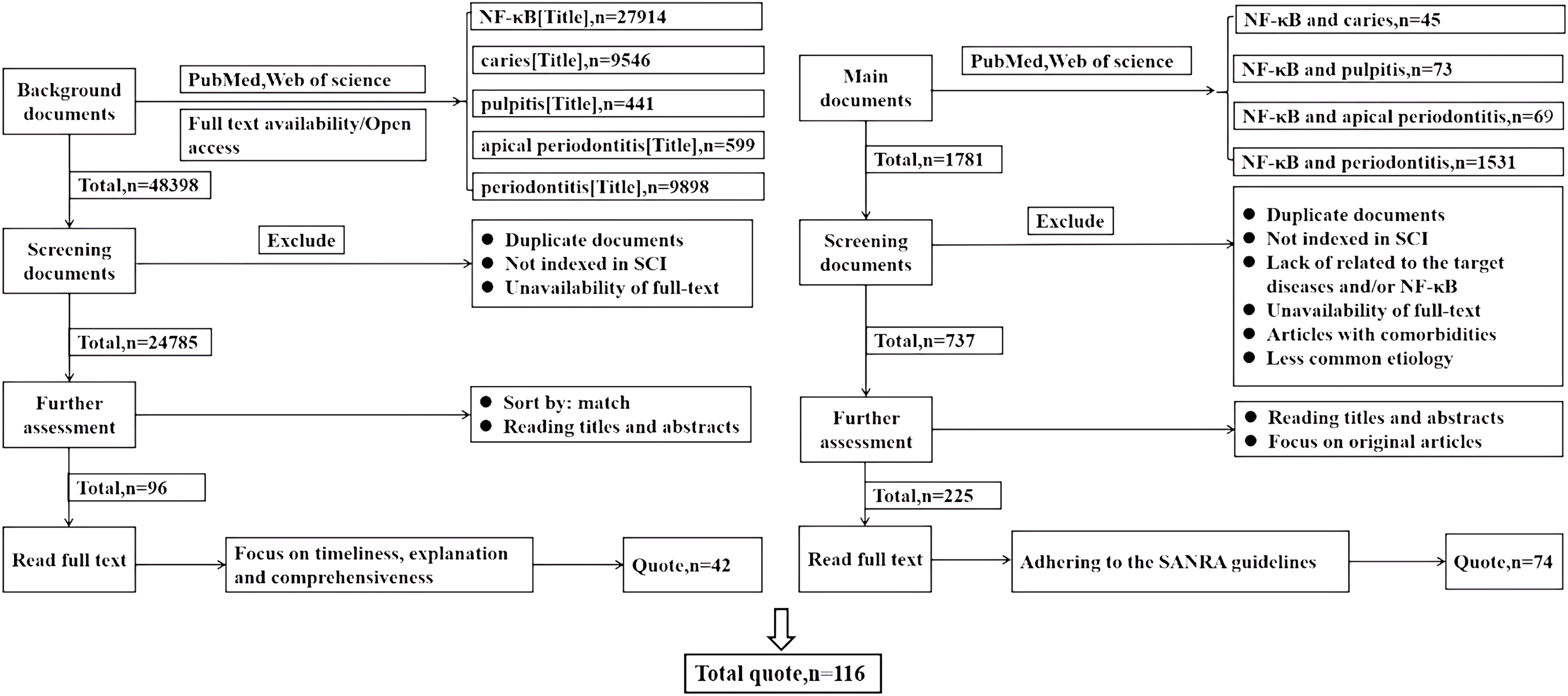{kind=link}
NF-κB in dental caries
Dental caries results from the complex interaction between the resident commensal microbiota in dental biofilms, host susceptibility traits, and environmental factors, with diet being particularly significant (Keller et al., 2017). The pathological changes typically involve the demineralization and destruction of hard tissues in teeth, primarily affecting the enamel, dentin, and cementum (Kang, Njimbouom & Kim, 2023). The onset of dental caries begins with the formation of a dental plaque biofilm on the tooth’s surface, as oral microorganisms adhere to the enamel or cementum. These bacteria metabolize nutrients, producing acids that lead to the demineralization of tooth structures. Over time, this process results in cavities, causing dental caries (Gao et al., 2016). The presence of Streptococcus mutans (S. mutans) in the oral cavity is widely acknowledged as a key factor in the formation of dental plaque biofilm and the onset of dental caries (Li et al., 2015; Zhang et al., 2021b). It has been recognized that inhibiting or breaking down oral biofilms could serve as an effective approach to halt the development of dental caries (Kawada-Matsuo & Komatsuzawa, 2017). This concept has gained traction in recent research and is increasingly considered a potential strategy for preventing oral health issues. Previous studies found that S. mutans can activate NF-κB in a mouse macrophage cell line (Kim et al., 2012). One study demonstrated that the antimicrobial peptide LL-37 derivatives IG-13-1 and IG-13-2 not only inhibit S. mutans but also reduce the levels of the transcription factor NF-κB, suggesting a potential role of NF-κB in their antimicrobial activity (Chen et al., 2019b). Currently, the main focus of NF-κB in dental caries is on regulating the differentiation of odontoblasts and dental pulp stem cells (DPSCs) in an inflammatory environment. In the tooth, odontoblasts are considered the initial defense for the host and are involved in initiating, developing, and maintaining the pulp’s immune and inflammatory responses to dentine invaders (Farges et al., 2015; Goldberg & Lasfargues, 1995). Studies have shown a link between the formation of reactive dentine and the NF-κB pathway. Specifically, when NF-κB signaling is blocked by NF-κB inhibitors MG132 or p65 siRNA in DPSC, odontoblastic differentiation and collagen matrix formation significantly improve, suggesting that inhibiting the NF-κB signaling pathway promotes dentin formation (Hozhabri et al., 2015). Other research indicates that blocking NF-κB promotes odontoblast differentiation by promoting lipopolysaccharide (LPS)-induced autophagy (Pei et al., 2016). Additionally, the phosphorylation of NF-κB signaling proteins such as IKKα/β, IκBα, and p65 increased with the deletion of DNA methyltransferase (DNMT)-1 in the lipoteichoic acid (LTA)-induced inflammatory environment of human odontoblasts (Meng et al., 2019). Another study showed that NF-κB signaling might play a role in upregulating adhesion molecules and chemotactic factors in the LPS-induced inflammatory environment of hDPSCs (Li & Wang, 2014). In the LPS-induced DPCs model, inhibiting the NF-κB signaling pathway can reduce inflammation (Long et al., 2023) and increase the odontogenic differentiation potential of DPCs (Zhu et al., 2019). NF-κB not only regulates odontoblasts but also controls DPSCs during dental caries development. DPSCs are known for their ability to self-renew and differentiate into multiple lineages. Research has shown that increased nuclear translocation of phosphorylated NF-κB p-p65 negatively impacts the differentiation of DPSCs into odontoblasts. This hindrance occurs when muramyl dipeptide (MDP), a component influencing DPSC differentiation under inflammatory conditions, is present. MDP exposure leads to a substantial upregulation of nucleotide-binding oligomerisation domain 2 (NOD2), a protein that regulates inflammatory homeostasis (Xiao et al., 2022). Existing studies show that activating the NF-κB signaling pathway can exacerbate the inflammatory response and inhibit the differentiation of DPSCs into odontoblasts (Song et al., 2018). In conclusion, NF-κB signaling is involved in the anti-caries process by reducing inflammation and promoting the differentiation of odontoblasts and DPSC/DPCs in inflammatory conditions.
NF-κB in pulpitis
Pulp infection and necrosis can occur due to microbial invasion of teeth through dentinal tubules, deep cavities, dental trauma, or tooth fractures (Chang et al., 2022). In cases of pulpitis, there is typically activation and infiltration of inflammatory cells, including T lymphocytes, monocytes, macrophages, and eosinophils. Previous reports have indicated that reducing the nuclear translocation of NF-κB is beneficial for maintaining dental pulp health (Galler et al., 2021). Blocking NF-κB signaling can decrease inflammation and cytokine release while promoting the differentiation of DPSC and collagen matrix formation (Hozhabri et al., 2015). Wang et al. (2024) found that inhibiting NF-κB may partially restore the mineralization function of DPSCs in an inflammatory environment. Conversely, activation of the NF-κB signaling pathway can lead to inflammatory cell infiltration and exacerbate pulpitis (Liu et al., 2020b). The LPS of Gram-negative bacteria is a major pathogenic factor known to cause severe inflammation in dental pulp tissues (Qiao et al., 2019). LPS stimulation leads to the translocation of NF-κB p65 from the cytoplasm to the nucleus (Pei et al., 2016). When LPS binds to its specific receptor toll-like receptor 4 (TLR4), it activates the MyD88-dependent signaling cascade, triggering a transcriptional response involving NF-κB p65 and inducing the secretion of proinflammatory cytokines by epithelial cells, macrophages, and dendritic cells (Ciesielska, Matyjek & Kwiatkowska, 2021) in dental pulp inflammation (Wang et al., 2019). In a rat model of acute pulpitis, the addition of TLR4 antagonists significantly down-regulated MyD88 and NF-κB to combat pulp inflammation (Lin et al., 2015). However, an in vitro study found that LPS stimulation of human dental pulp cells (hDPCs) caused a protective inflammatory response. Concretely speaking, immunofluorescence revealed that nuclear translocation of NF-κB promotes the differentiation of hDPCs (Huang et al., 2015). This may be because LPS from different bacteria cannot mimic the same receptor, resulting in varying effects on other receptors. Subsequent cellular trials showed that downregulation of nuclear translocation of NF-κB p65 can maintain pulp viability by knockdown of astrocyte elevated gene-1 (AEG-1) (Wu et al., 2018). Current research in pulpitis is heavily focused on pulp tissue regeneration, a complex and challenging area that represents both a persistent challenge and a promising trend within the field. Revascularization is a crucial component for achieving successful pulp regeneration (Siddiqui et al., 2022). Inhibition of NF-κB p65 has been shown to have anti-inflammatory and pro-angiogenic effects when the angiogenic factor G-patch and FHA domain 1 (AGGF1) is regulated (Shen et al., 2021). Rescue experiments have demonstrated that breaking the low AGGF1 levels promotes p65 nuclear transfer and suppresses DPCs angiogenesis. Furthermore, overexpression of Pannexin3 in hDPCs suppresses TNF-α-induced NF-κB transcriptional activity by attenuating IκBα degradation and inhibiting p65 nuclear translocation, which significantly reduces the expression of inflammatory cytokines IL-6 and IL-1β (Song et al., 2017). Additionally, targeting regulation in human dental pulp fibroblasts (HDPF) shows potential as an effective anti-inflammatory approach in pulpitis treatment. In the HDPF inflammation model, p65 and IKK expression significantly decreased, and the inflammatory response in LPS-stimulated HDPF was suppressed with the addition of berberine (BBR), leading to a significant reduction in RelA mRNA expression and the ratio of n/c-p65 (Song et al., 2020). NF-κB activation was effectively suppressed by oxidoreductase E1 (RvE1) and lipoxygenase A4 (LXA4), which decreased the production of pro-inflammatory molecules and promoted the resolution of pulpitis inflammation (Liu et al., 2022). In summary, NF-κB signaling regulates the expression of pro-inflammatory molecules and influences the differentiation of DPCs and HDPFs, helping to counteract inflammation in the tooth pulp. This regulation contributes to preventing excessive dental pulp inflammation and preserving the viability of dental pulp tissues, especially in the early stages of inflammation.
NF-κB in apical periodontitis
Apical periodontitis (AP) is an inflammatory and immune response caused by anaerobic polymicrobial infection of the dental pulp and root canals (Siqueira & Rôças, 2009; Sjögren et al., 1997). It is characterized by the formation of granulation tissue and the destruction of the alveolar bone around the root apex of the tooth, eventually leading to bone resorption or tooth loss (Luo et al., 2022). In immunology, NF-κB signaling pathways are increasingly recognized for their crucial role in regulating immune responses in AP (Silva-García, Valdez-Alarcón & Baizabal-Aguirre, 2019). Immunohistochemical staining and Western blotting have shown that NF-κB (p65) expression is relatively low in healthy individuals but significantly upregulated in the thyroid tissues of human AP patients. A rat model of AP confirmed the positive correlation between the NF-κB signaling pathway and AP progression (Guan et al., 2021). In cell experiments, exposure to LTA led to the translocation of NF-κB p65 into the nucleus, worsening periapical inflammation (Wang et al., 2016). Additionally, research by Dong et al. (2018) found that osteopontin, through the activation of NF-κB (p65) signaling, contributes to bone destruction in patients with periapical inflammation at the tooth root. Further studies have shown that activation of hypoxia-inducible factor 1 (HIF-1) can reduce periapical inflammation and bone loss by down-regulating NF-κB, pro-inflammatory cytokines, M1 macrophages, and osteoclastogenesis (Hirai et al., 2018). Other research indicates that NF-κB activation exacerbates periapical inflammation in mice, attributed to increased TLR4 signaling due to TLR2 deficiency (Rider et al., 2016). It has been previously demonstrated that upregulation of NF-κB exacerbates periapical bone destruction (Wei et al., 2022). This is primarily due to the significantly increased expression of NF-κB ligand-receptor activator (RANKL) mRNA in patients with AP compared to normal subjects, which activates osteoclasts and ultimately worsens periapical bone destruction (Sabeti et al., 2005). The impact of RANKL on osteoclasts in AP was further confirmed through experiments on mice (Ikeda et al., 2022) and rats (Zhang & Peng, 2005). In one study, exposure of periodontal ligament cells (PDLC) to LPS induced activation of the NF-κB pathway via translocation and phosphorylation, followed by IκB degradation. This process resulted in an upregulation of IL-23 expression, promoting osteoclast generation and leading to apical bone destruction (Ma et al., 2017). Additionally, bacterial chaperonin 60 molecules, also known as GroEL, can overexpress and induce acetylation of NF-κB p65 in AP. This acetylated NF-κB p65 transcription factor binds to the promoter regions of matrix metalloproteinase 2 (MMP2) and MMP9 genes, activating their expression (Zhang et al., 2021a). Interestingly, another MMP family member, active MMP-8, has been reported to identify collagenolytic tissue destruction for early diagnosis of peri-implantitis, serving as a convenient and reliable adjunct tool for chairside assay (Guarnieri et al., 2022). Overall, NF-κB influences the progression of periapical inflammation by regulating both the inflammatory response and bone resorption processes.
NF-κB in periodontitis
Periodontitis (PD) is a multifactorial inflammatory disease characterized by the irreversible and progressive degradation of periodontal tissues, including the gingiva, periodontal ligament, and alveolar bone, often leading to tooth loss. The primary cause of PD is a microbial biofilm (He et al., 2022). PD is a significant global public health concern, affecting approximately 60% of the world’s adult population (Wang et al., 2023). Severe PD, recognized as the sixth most common human disease, affects about 11% of the global population (Ebadi et al., 2023). The NF-κB signaling pathway is a major mediator of inflammation and plays a crucial role in the pathogenesis of periodontal disease by activating cytokine genes (Lepetsos, Papavassiliou & Papavassiliou, 2019). Fusobacterium nucleatum (F. nucleatum) is an early pathogenic colonizer in PD, but the mechanism of the host’s immune response to this pathogen remains unclear (Wang et al., 2023). In vitro studies have shown that NF-κB1 and NF-κB2 are upregulated three hours after F. nucleatum infection, with the expression levels of inflammatory-related genes in the NF-κB signaling pathway increasing over time (Wang et al., 2023). Specifically, NF-κB (p65, p-p65, IKK, p-IKK) is activated following stimulation with F. nucleatum, as detected by Western blot analysis. Drug treatment has been shown to reduce NF-κB activity. Previous studies using Western blot analysis have shown increased expression of NF-κB transcription factors (p50/p65) and decreased levels of IκB in gingival tissues of PD patients compared to healthy individuals (Ambili et al., 2005). NF-κB activation rates were much higher (75–90%) in tissues affected by periodontal disease compared to normal tissues (5–30%). Conversely, the expression of IκB (5%) in periodontal disease tissues was significantly lower than in healthy tissue (50%), indicating the vital role of NF-κB in PD. Recent evidence on the role of NF-κB in regulating periodontal inflammation shows that inhibition of this signaling pathway reduces the production of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α (Shi et al., 2021). Further research has shown that inhibition of the NF-κB (p-IκBα, N-p65) signaling pathway significantly downregulates the expression of IL-6 and IL-8 in hPDLCs through low-intensity pulsed ultrasound (LIPUS) stimulation (Liu et al., 2020a). In a separate study, activation of the NF-κB pathway was indicated by increased phosphorylation of p65 in visfatin-overexpressing hPDLCs, which subsequently upregulated the production of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) (Yao et al., 2021). Additionally, NF-κB signaling plays a role in Long non-coding RNA (lncRNA)-regulated periodontal inflammation. Mechanistically, the NF-κB p65 subunit can be directly bound by lncRNA SNHG5, which suppresses its translocation, thereby reducing pathological changes such as inflammasome formation (Han et al., 2022). Another study found that when hPDLCs were exposed to LPS, the lncRNA FGD5 AS1 was overexpressed, leading to a significant decrease in p/t-p65 levels. This effectively inhibited NF-κB signaling and reduced PD severity (Chen et al., 2019a). NF-κB signaling in PD pathogenesis is regulated by both lncRNA and microRNA (miRNA). For example, the loss of microRNA-21 (miR-21) worsens PD by increasing the protein expression of phosphorylated (p)-NF-κB and reducing the total IκB levels both in vivo and in vitro (Zhou et al., 2018). NF-κB also regulates bone metabolism and affects alveolar bone loss in periodontal diseases. Previous research has shown the significant role of NF-κB in bone remodeling. Studies on mice with a double knockout of NF-κB1 and NF-κB2 genes have demonstrated that the absence of these proteins leads to osteopetrosis, a condition characterized by abnormally dense bones due to an accumulation of poorly developed osteoclasts (Franzoso et al., 1997; Iotsova et al., 1997). In an experiment using a mouse model infected with Porphyromonas gingivalis (P. gingivalis) to simulate PD, the phosphorylation rate of NF-κB p50 and p65, and TNF-α levels in the gingival epithelium were significantly elevated compared to the control group, which experienced substantial alveolar bone loss (Li et al., 2017). However, mangiferin treatment notably decreased TNF-α expression and the phosphorylation levels of p50 and p65, substantially inhibiting alveolar bone loss. Previous studies have observed that inhibiting IKK/p65/NF-κB promotes osteoblast differentiation and bone formation, with improvements such as increased trabecular numbers and enhanced bone mineral density (Alles et al., 2010; Chang et al., 2009). In addition, the mRNA and protein expression levels of the NF-κB ligand (RANKL) were significantly higher in diseased periodontal tissues compared to healthy ones, leading to increased osteoclast activity and consequent bone damage (Crotti et al., 2003). Research has shown that the internalization of RANK, triggered by its ligand RANKL, is crucial for activating downstream signaling pathways like NF-κB. This activation is essential for initiating and sustaining osteoclast formation and function (Wang et al., 2021). For instance, pirfenidone (PFD) suppresses the NF-κB signaling pathway by inhibiting RANKL-induced osteoclast differentiation and LPS-induced expression of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α), thereby slowing the progression of PD (Zhang et al., 2023). Additionally, when downstream signaling is activated by RANKL, the phosphorylation of IκB activates NF-κB, leading to increased expression of osteoclast-specific genes and increased osteoclast formation and function (Sakamoto et al., 2019). Recent findings suggest that the non-canonical NF-κB signaling pathway plays a significant role in regulating PD. Experimental studies have shown that topical application of the NIK inhibitor Cpd33 suppresses osteoclast formation, reducing alveolar bone resorption and demonstrating the potential therapeutic effects of targeting this pathway in a mouse model of PD (Aoki et al., 2023). Similar results were observed in another study using a NIK inhibitor both in vitro and in vivo (Wang et al., 2020). Furthermore, several studies have shown that lncRNA interacts with proteins in the NF-κB signaling pathway, influencing bone resorption in PD. For example, Li et al. (2022) found that lncRNA Nron accelerates the nuclear transport of the NF-κB inhibitor, significantly reducing osteoclast formation and alveolar bone loss in PD. In another study, the activity of NF-κB (p-p65, p-IκBα) was partially inhibited by lncRNA Dancr expression in MC3T3-E1 cells, promoting osteogenic differentiation (Lu et al., 2020). Overall, these studies provide new insights into treating periodontal bone destruction by targeting NF-κB signaling.
Cementum regeneration is considered the gold standard for periodontal tissue regeneration (Yang, Rossi & Putnins, 2010). Recent research has indicated that NF-κB signaling is activated during the osteoblast differentiation of PDLCs. For instance, one study showed that NF-κB (p-p65) activation by transcription factor 7-like 2 (Tcf7l2) accelerates cementum formation and aids in guiding periodontal tissue regeneration (Zhao et al., 2019). The NF-κB signaling pathway is triggered during the osteogenic differentiation process in PDLCs (Lee et al., 2014, 2016). However, contradictory results have been observed, potentially due to differences in experimental procedures, cell types, or the source and intensity of stimuli. In the inflammation of human periodontal ligament fibroblasts (HPDLFs), significant inhibition of IκBα and P65 phosphorylation can promote osteogenic differentiation and mineralization of HPDLFs treated with proanthocyanidins (PA) (Huang et al., 2020). Furthermore, the NF-κB non-canonical pathway also contributes to periodontal tissue regeneration. In periodontal inflammatory conditions, inhibiting the non-classical NF-κB2 signaling pathway significantly downregulates pro-inflammatory factors while promoting skin healing in mice via FGF-2-inhibited CD40 signaling (Fujihara et al., 2019). Moreover, NF-κB interacts with other signaling pathways to encourage periodontal tissue regeneration. For example, LL37, a robust antimicrobial peptide, increases NF-κB p65 and ERK1/2 phosphorylation levels in hPDLC, and this cascade induces the production of vascular endothelial growth factor-A (VEGF-A), leading to angiogenesis and promoting periodontal regeneration (Kittaka et al., 2013). In summary, NF-κB is involved not only in regulating inflammatory cytokines and bone metabolism in PD but also in periodontal tissue regeneration.
Conclusions
Despite increased awareness and a growing focus on oral health, inflammatory dental diseases remain a significant concern, with their pathogenesis still not fully understood. Research has provided substantial evidence for the role of NF-κB activation in dental inflammation, significantly affecting both the progression of inflammatory responses and bone destruction in the oral cavity (Table 1). Traditional caries treatments, such as topical fluoride, dental sealants, and surgical procedures like fillings (He et al., 2024), often exhibit cytotoxicity to the pulp and lack long-term antimicrobial properties, leading to issues like microleakage and pulpitis. Developing NF-κB-related materials or drugs could provide sustained antimicrobial effects and favorable biological properties. The most common treatment for pulpitis and AP is root canal therapy. For a sucessful canal therapy, the most critical step is the control of intracanal infection which is the main cause of periradicular lesions (Jang et al., 2024). However, current root canal disinfectants often cause significant irritation to periradicular tissues. Therefore, we aim to develop mild and long-lasting anti-infectives targeting the NF-κB signaling pathway to achieve effective antimicrobial properties and prevent apical bone destruction. Management of PD, including initial therapy, drug therapy, and periodontal surgery, focuses on regulating inflammation and bone resorption. Inhibiting NF-κB can provide anti-inflammatory effects (Kim et al., 2023), suppress osteoclast activity (Xie et al., 2024), and enhance angiogenesis (Yang et al., 2024), making it a promising target for clinical drug development. However, few studies focus on how NF-κB signaling works in caries, pulpitis, and periapical inflammation. While targeting the NF-κB signaling pathway holds great promise for treating dental inflammatory diseases, a more thorough understanding of its precise role and therapeutic potential requires further investigation through extensive cell experiments, preclinical, and clinical studies to fully reveal the underlying mechanisms.
| Diseases | Main factors | Experiment strategy | NF-κB change | Effect |
|---|---|---|---|---|
 Dental caries |
IG-13-1 and IG-13-2 (Chen et al., 2019b) | Macrophages | NF-κB↓ | Anti-infla |
| NF-κB inhibitor (Hozhabri et al., 2015) | hDPSCs | NF-κB gene↓, N-p6↓ | ||
| Connexin43 (Long et al., 2023) | Rat, hDPCs | NF-κB↓ | ||
| Ag-BG/CS (Zhu et al., 2019) | DPCs | p-p65↓ | ||
| DNMT1 (Meng et al., 2019) | hOB | p-ΙKKα/β↑, p-IκBα↑, p-p65↑ | Pro-infla | |
| AZA (Feng et al., 2021) | hOBs | p-IKKα/β↑, p-IκBα↑, p-p65↑ | ||
| Autophagy (Pei et al., 2016) | mDPC6T (cell line) | p-NF-κB↓ | Pro-differentiation | |
| MDP/NOD2 (Xiao et al., 2022) | hDPSC | p-p65↑, p-IκBα↑ | Inhibition differentiation | |
| Neuropilin-1-FYN (Song et al., 2018) | hDPSC | N-p65↑ | Pro-infla and inhibition differentiation | |
 Pulpitis |
microRNA-506 (Wang et al., 2019) | DPSCs | p-p65↓ | Anti-infla |
| AEG-1 (knockout) (Wu et al., 2018) | Rat, hDPCs | p-p65↓ | ||
| Pannexin3 (Song et al., 2017) | hDPCs | p-p65↓, IκBα↑ | ||
| Berberine (Song et al., 2020) | hDPF | p-p65↓, p-ΙKK↓, IκBα↑ | ||
| Resolvin and lipoxinA4 (Liu et al., 2022) | hDPF, rat pulpitis | RelA mRNA↓, N/C-p65↓ | ||
| G-Rb1 (Nam et al., 2024) | hDPCs | p-p65↓, pIκBα↓ | ||
| TLR4/MyD88 (Lin et al., 2015) | Rat acute pulpitis | p65↑, N-p50↑ | Pro-infla | |
| PECAM1 and CXCR4 (Liu et al., 2020b) | HDPFs | p-p65↑ | ||
| NF-κB inhibitor (Hozhabri et al., 2015) | hDPSCs | NF-κB gene↓, N-p65↓ | Pro-differentiation | |
| Autophagy (Pei et al., 2016) | mDPC6T (cell line) | p-NF-κB↓ | ||
| LPS (Huang et al., 2015) | hDPCs | NF-κB↑ | ||
| TSG-6 (Wang et al., 2024) | DPSC | p-p65↓, pIκBα↓ | ||
| AGGF1 (knockout) (Shen et al., 2021) | hDPCs | p-p65↑ | Pro-infla and inhibition angiogenesis | |
 Apical periodontitis |
LTA/NLRP3 (Wang et al., 2016) | Rat, RAW 264.7 cells | N-p65↑ | Pro-infla |
| LPS (Guan et al., 2021) | Subjects, hDPCs, rat | p-p65↑ | Pro-infla and bone destruction | |
| CD14 and TLR4, TLR2 (Rider et al., 2016) | Mice, L929 cells | NF-κB↑ | ||
| RGS10 (inhibition) (Wei et al., 2022) | Mice | NF-κB↑ | ||
| RANKL (Ikeda et al., 2022) | Subjects | NF-κB↑ | Bone destruction | |
| IL-23 (Ma et al., 2017) | Monocyte/Macrophage cell | p-IκB↑ | ||
| Osteopontin (Dong et al., 2018) | Subjects, RAW264.7 and MC3T3-E1 cells, mice | NF-κB↑, p-p65↑ | ||
| GroEL (Zhang et al., 2021a) | Subjects, MC3T3-E1 cell line, mice primary osteo-blasts | Acetyl-p65↑ | ||
| HIF1 (Hirai et al., 2018) | Mice, macrophages | p-p65↓, p-ΙKK↓ | Anti-infla and bone protection | |
 Periodontitis |
LIPUS (Liu et al., 2020a) | hPDLSCs | p-IκBα↓, N-p65↓ | Anti-infla |
| LncRNA SNHG5 (Han et al., 2022) | Mice PD, subjects | p-p65↓ | ||
| LncRNA FGD5 AS1 (Chen et al., 2019a) | Subjects, hPDLSCs mice, PDLCs | p/t-p65↓, p/t-IκBα↑ | ||
| FGF-2 (Fujihara et al., 2019) | Mice monocyte and fibroblast cells,mice, | P52↓ | ||
| Resveratro liposomes (Shi et al., 2021) | Human vascular endothelial cells | p-p65↓, p-IκBα↓ | ||
| Inflammation (Ambili et al., 2005) | Subjects | p50/p65↑ | Pro-infla | |
| F. nucleatum (Wang et al., 2023) | hPDLSCs | p-ΙKK↑, p-p65↑ | ||
| Visfatin (Yao et al., 2021) | hPDLCs, hGFs | p-p65↑ | ||
| Mangiferin (Li et al., 2017) | Mice | pp50/p50↓, | Anti-infla and bone protection | |
| Prifenidone (Zhang et al., 2023) | Mice, macrophages, mesenchymal stem cells | pp65/p65↓ | ||
| p-p65↓, p-IκBα↓ | ||||
| Non-canonical NF-κB (Aoki et al., 2023) | Mice PD, Primary osteoblasts, | non-canonical NF-κB↓ | ||
| PA (Huang et al., 2020) | Bone marrow cells hPDLFs | p-IκBα↓, p-p65↓ | ||
| microRNA-21 (knockout) (Zhou et al., 2018) | miR-21 deficient mice, subjects, macrophages | p-NF-κB↑, T-IκB↓ | Pro-infla and, bone destruction | |
| NF-κB1, NF-κB2 (Franzoso et al., 1997; Iotsova et al., 1997) | p50 and p52 knockouted mice | NF-κB1↓, NF-κB2↓ | Osteopetrosis | |
| ΙKK/NF-κB (Chang et al., 2009) | Primary calvarial cells, generation of transgenic mice | p-p65↓, NF-κB↓, p-IκBα↓ | Bone protection | |
| NF-κB (Alles et al., 2010) | Mice, osteoblasts, | NF-κB↓ (OD p65↓, p50↓) | ||
| LncRNA Dancr (Lu et al., 2020) | Osteoclasts | p-p65↓, p-IκBα↓ | ||
| LncRNA Nron (Li et al., 2022) | Osteoblastic cell line,Nron knockouted mice | NF-κB↓(NKRF↑) | ||
| rh-BMP-2 and rh-DSP (Lee et al., 2014) | Human cementoblasts and PDLCs | p-p65↓, p-IκBα↓ | ||
| Tcf712 (Zhao et al., 2019) | Mice, cementoblast cell line | p-p65↑ | ||
| Vibration (Sakamoto et al., 2019) | Osteocyte and pre-osteoclast cell line, rat | p-IκB↑, NF-κB↑ | Bone destruction | |
| NIK (Wang et al., 2020) | Mice, osteoclasts | p-IκBα↑, p-p105↑ | ||
| Caveolin-1 (inhibition) (Lee et al., 2016) | Mice, hPDLCs, human cementoblasts | p-IκB↑, N-p65↑ | ||
| LL-37 (Kittaka et al., 2013) | hPL cells | p-p65↑ | Pro-angiogenesis |