
Molecular basis of a high Hb A2/Hb Fβ-thalassemia trait: a retrospective analysis, genotype-phenotype interaction, diagnostic implication, and identification of a novel interaction with α-globin gene triplication
- Published
- Accepted
- Received
- Academic Editor
- Abdul Hafeez Kandhro
- Subject Areas
- Biochemistry, Molecular Biology, Hematology, Medical Genetics
- Keywords
- β-hemoglobinopathies, β-thalassemia trait, 3.4 kb deletion β-thalassemia, High Hb F SNPs, High Hb F β-thalassemia trait
- Copyright
- © 2023 Soontornpanawet et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2023. Molecular basis of a high Hb A2/Hb Fβ-thalassemia trait: a retrospective analysis, genotype-phenotype interaction, diagnostic implication, and identification of a novel interaction with α-globin gene triplication. PeerJ 11:e15308 https://doi.org/10.7717/peerj.15308
Abstract
Background
β0-thalassemia deletion removing 5´β-globin promoter usually presents phenotype with high hemoglobin (Hb) A2 and Hb F levels. We report the molecular characteristics and phenotype-genotype correlation in a large cohort of the β0-thalassemia with 3.4 kb deletion.
Methods
A total of 148 subjects, including 127 heterozygotes, 20 Hb E-β-thalassemia patients, and a double heterozygote with α-globin gene triplication, were recruited. Hb and DNA analysis were performed to identify thalassemia mutations and four high Hb F single nucleotide polymorphisms (SNPs) including four base pair deletion (-AGCA) at Aγ-globin promoter, rs5006884 on OR51B6 gene, −158 Gγ-XmnI, BCL11A binding motifs (TGGTCA) between 3´Aγ-globin gene and 5´δ-globin gene.
Results
It was found that heterozygous β0-thalassemia and Hb E-β0-thalassemia with 3.4 kb deletion had significantly higher Hb, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin and Hb F values as compared with those with other mutations. Co-inheritance of heterozygous β0-thalassemia with 3.4 kb deletion and α-thalassemia was associated with even higher MCV and MCH values. The Hb E-β0-thalassemia patients carried a non-transfusion-dependent thalassemia phenotype with an average Hb of around 10 g/dL without blood transfusion. A hitherto undescribed double heterozygous β0-thalassemia with 3.4 kb deletion and α-globin gene triplication presented as a plain β-thalassemia trait. Most of the subjects had wild-type sequences for the four high Hb F SNPs examined. No significant difference in Hb F was observed between those of subjects with and without these SNPs. Removal of the 5´β-globin promoter may likely be responsible for this unusual phenotype.
Conclusions
The results indicate that β0-thalassemia with 3.4 kb deletion is a mild β-thalassemia allele. This information should be provided at genetic counseling and prenatal thalassemia diagnosis.
Introduction
β-hemoglobinopathies are inherited hemoglobin (Hb) disorders caused by mutations in the β-globin gene, resulting in quantitatively reduced (β+-thalassemia) or absent (β0-thalassemia) β-globin chain synthesis and qualitative β-globin chain defect called structural β-globin chain variant. Although some β-globin gene deletions have been reported, most defects are non-deletional mutants of the β-globin gene (Steinberg et al., 2009). In Thailand, a high prevalence of β-hemoglobinopathies has been described, including 20–30% of Hb E (HBB:c.79G>A) and 3–9% of β-thalassemia (Paiboonsukwong et al., 2022). In northeast Thailand, eight β-thalassemia mutations including CD 41/42 (-TTCT) (HBB:c.126_129delCTTT), CD17 (A-T) (HBB:c.52A>T), NT-28 (A-G) (HBB:c.-78A>G), CD71/72 (+A) (HBB:c.217dupA), IVSI-1 (G-T) (HBB:c.92+1G>T), IVSII-654 (C-T) (HBB:c.316-197C>T), 3.4 kb deletion and IVSI-5 (G-C) (HBB:c.92+5G>C) accounted for 97.4% of the total mutations (Yamsri et al., 2011). Generally, β-thalassemia heterozygote has normal or slightly decreased Hb level and reduced mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) with increased Hb A2 to around 5.0% and Hb F 0–3%. Homozygote or compound heterozygote of β-thalassemia with Hb E led to β-thalassemia major and Hb E-β-thalassemia diseases, with variable thalassemia phenotype. The β+-thalassemia has a milder thalassemia phenotype than the β0-thalassemia (Yamsri et al., 2011; Yamsri et al., 2015).
Interestingly, among these common mutations, it was found that the carrier of β-thalassemia with 3.4 kb deletion was associated with higher Hb A2, Hb F, MCV, and MCH values as compared with those with other mutations (Yamsri et al., 2011). A high Hb F expression may presumably lead to a milder β-thalassemia phenotype in homozygotic or compound heterozygotic forms. This 3.4 kb DNA deletion removes an entire β-globin gene, including its 5′ promoter. In this study, we have examined several genetic single nucleotide polymorphisms (SNPs) modifiers affecting Hb A2 and Hb F expression in a large cohort of β0-thalassemia with 3.4 kb deletion. The molecular basis, and phenotype and genotype interaction observed in both heterozygote and compound heterozygote forms, as well as a hitherto undescribed interaction of the 3.4 kb deletion β0-thalassemia with an α-globin gene triplication, are described.
Materials and Methods
Subjects and hematological analysis
Ethical approval of the study protocol was obtained from the Institutional Review Board (IRB) of Khon Kaen University, Khon Kaen, Thailand (HE632216). A total of 618 leftover specimens of β-thalassemia carriers were selectively recruited from our archival at the Center for Research and Development of Medical Diagnostic Laboratories (CMDL), Faculty of Associated Medical Science, Khon Kaen University and Department of Pathology, Faculty of Medicine, Prince of Songkla University, during January 2008 to December 2021. These included leftover DNA specimens of β-thalassemia carriers with 3.4 kb (n = 148), Filipino (n = 9), and 105 bp (n = 7) deletions. The 454 representative specimens of β+-thalassemia trait (n = 70), β0-thalassemia trait (n = 259), and Hb E- β0-thalassemia disease (n = 125) with non-deletional β-thalassemia mutations were used to compare with the deletional β0-thalassemia mutation. Study was done on leftover DNA specimens; consent was not required. Hematological parameters were recorded on a standard blood cell counter; Sysmex XN-9000 (Sysmex Europe SE, Norderstedt, Germany), and Hb analysis was performed using capillary electrophoresis (Capillary II Flex piercing, Sebia, France).
Routine DNA analysis
Genomic DNA was prepared from peripheral blood leukocytes using the GF-1 Blood DNA extraction kit (Vivantis Technologies Sdn. Bhd, Selangor Darul Ehsan, Malaysia). Identification of α0-thalassemia (SEA and THAI deletions), α+-thalassemia (3.7 kb and 4.2 kb deletions), Hb Constant Spring, Hb Paksé, α-globin gene triplication and common β-thalassemia mutations were performed using polymerase chain reaction (PCR)-based techniques (Yamsri et al., 2010; Singha et al., 2013; Chaibunruang et al., 2018; Charoenwijitkul et al., 2019). Identification of the Filipino deletion and the 105 bp deletion β-thalassemia were performed using gap-PCR assays as described previously (Nopparatana et al., 1999; Yamsri et al., 2012). The Xmn I polymorphism at −158 Gγ (C>T) (rs7482144) was determined by PCR-restriction fragment length polymorphism (PCR-RFLP) as described elsewhere (Phanrahan et al., 2019).
DNA sequencing
Direct DNA sequencing was used to determine deletion breakpoints of the β0-thalassemia with 3.4 kb deletion generated using primers G9 (5′-TCCCCAGTTAACCTCCTATT-3′) and N1 (5′-ACATATGAGCAAGGTTGTGT-3′) on a specific fragment of 456 base pair (bp). To examine three BCL11A binding motifs (TGGTCA) located between +3 kb 3′of Aγ-globin gene and about 1 kb 5′δ-globin gene, the three amplified fragments using primer pairs F29 (5′-CACGAGTCAGCCTTCAGAAA-3′) & G188 (5′- GCAGAGGAGACCAGCATACA-3′) (1,386 bp), F18 (5′-GACATGGACTATTGTTCAATGA-3′) & G191 (5′-AGACAACGGGCTAATCCCTC-3′) (1,286 bp), and F31 (5′-ACCCACATT GGCATTACAC-3′) & G192 (5′-AGAATGTGTTTGTGAGGGAGGA-3′) (1,111 bp) were generated as shown in Fig. 1 and the fragments were sequenced directly. To analyze the whole γ-globin genes, primer F22 (5′-TACTGCGCTGAAACTGTGGC-3′) & γ35 (5′-AGGTAGTTGTTCCCCTTCAA-3′) and SF5 (5′-TTAACGTCTTCAGCCTACAA-3′) & SF6 (5′-CAATCTGCACACTTGAGGGC-3′) were used to amplify DNA fragments of 1,843 and 993 bp, specific for Aγ-globin gene, and primers γ4 (5′-GGCCTAAAACC ACAGAGA-3′) & γ34 (5′-AGGTAGTTGTTGTTGTTGCA-3′) and SF3 (5′-GTTTGTGTGTG TGTGAGAGC-3′) & SF4 (5′-TCTTTAGGCATGCGTCAACA-3′) were used to amplify DNA fragments of 1,787 and 1,121 bp of Gγ-globin gene, followed by DNA sequencing (Fucharoen, Shimizu & Fukumaki, 1990; Panyasai et al., 2004; Svasti et al., 2007; Singha et al., 2015) [9-12]. DNA sequencing was carried out using Sanger sequencing method on an ABI PRISM™ 3730XL analyzer (Applied Biosystems, Foster City, CA, USA).
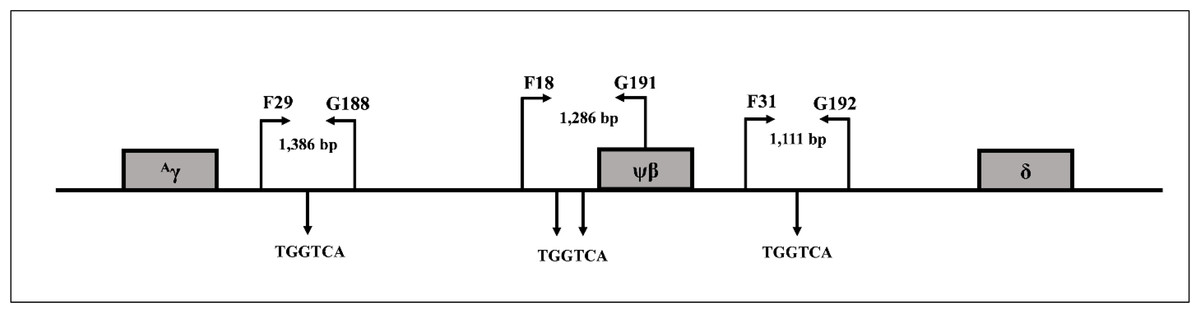
Figure 1: Locations and orientations of amplification primers for BCL11A.
Locations and orientations of amplification primers for identification of BCL11A binding motifs (TGGTCA) by DNA sequencing within DNA sequences between +3 kb 3′ of Aγ-globin gene to about −1 kb 5′ of δ-globin gene.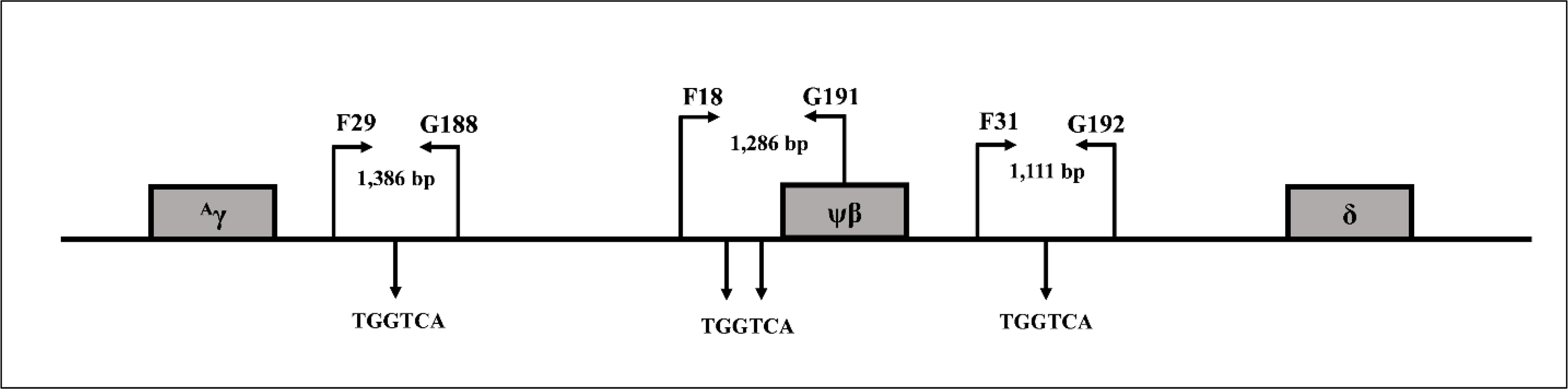{kind=link}
Identification of the four bp deletion (-AGCA) of theAγ-globin gene promoter
The PCR-RFLP assay was developed to detect four bp deletion (-AGCA) at −225 to −222 of the Aγ-globin gene promoter (rs1451276553). Specific amplification of Aγ-globin gene promoter was performed using primers F22 and γ35 to produce a fragment of 1,847 bp wild-type or 1,843 bp specific for the four bp deletion. This was confirmed by DNA sequencing and PCR-RFLP assay. The PCR product was digested with a Mse I restriction enzyme (5′-T▾TAA-3′) (Vivantis Technologies Sdn. Bhd, Selangor Darul Ehsan, Malaysia). After digestion, the 1,021 bp and 167 bp indicate wild type, whereas the 1,184 bp is a specific fragment for the AGCA deletion, as shown in Fig. 2.
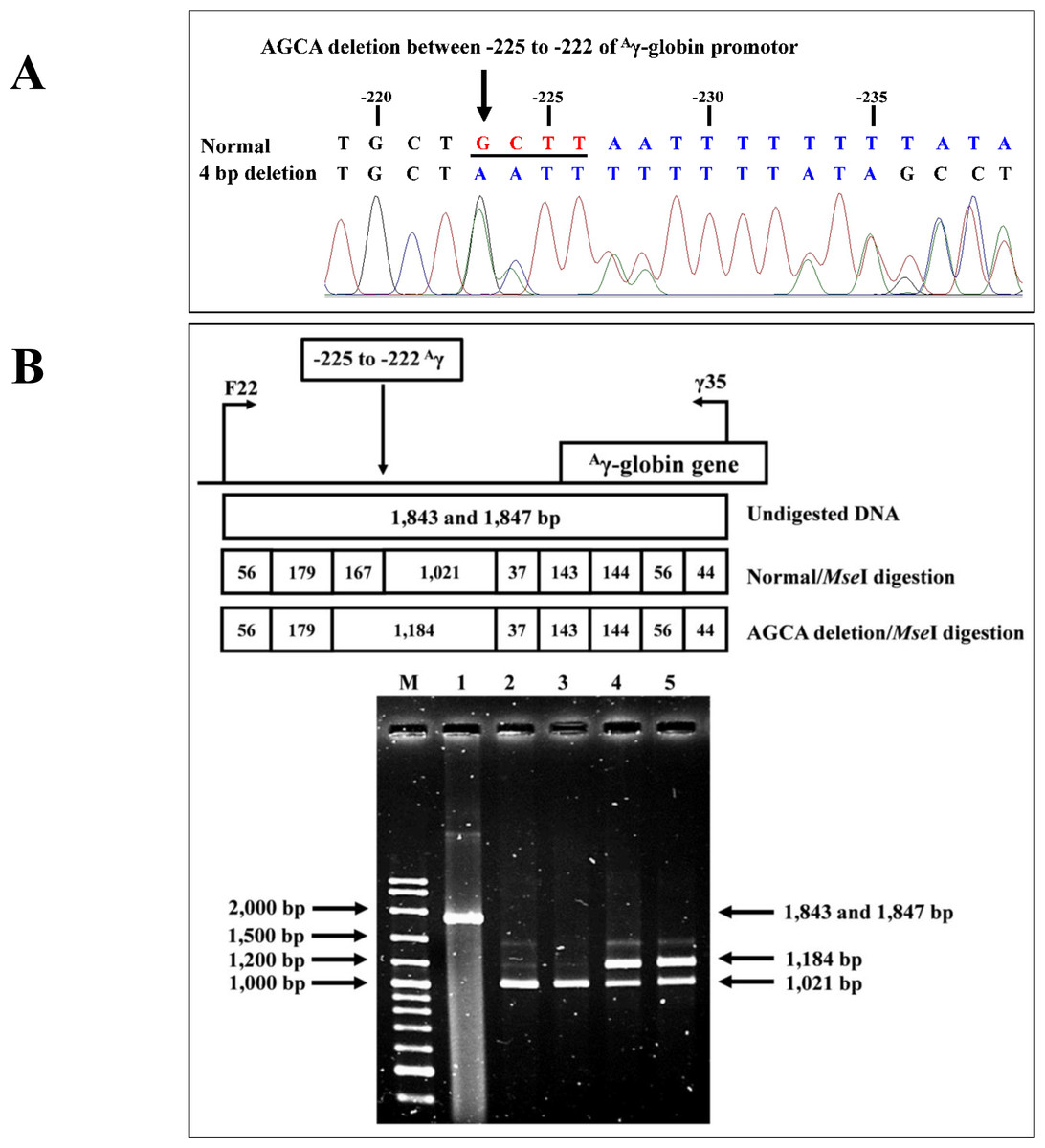
Figure 2: DNA sequencing profile and identification of four bp (-AGCA) deletion in the A-gamma globin gene.
(A) DNA sequencing profile of a representative subject with heterozygosity for the four bp deletion (-AGCA) between −225 to −222 of Aγ-globin gene. (B) Identification of four bp deletion (-AGCA) between −225 to −222 of Aγ-globin gene using PCR-RFLP by Mse I digestion. Locations and orientations of primers F22 and γ35 are indicated to produce PCR fragment of 1,843 bp or 1,847 bp. The Mse I-digested fragments of 1,021 bp and 167 bp for wide-type allele and 1,184 bp for mutant allele are depicted. M represents the GeneRular 100 bp Plus DNA ladder. Lane 1: undigested amplified DNA, lanes 2 & 3: Mse I-digested amplified DNA of homozygous wide-type allele, and lanes 4 & 5: Mse I-digested amplified DNA of heterozygous for the four bp deletion (-AGCA) of Aγ-globin gene.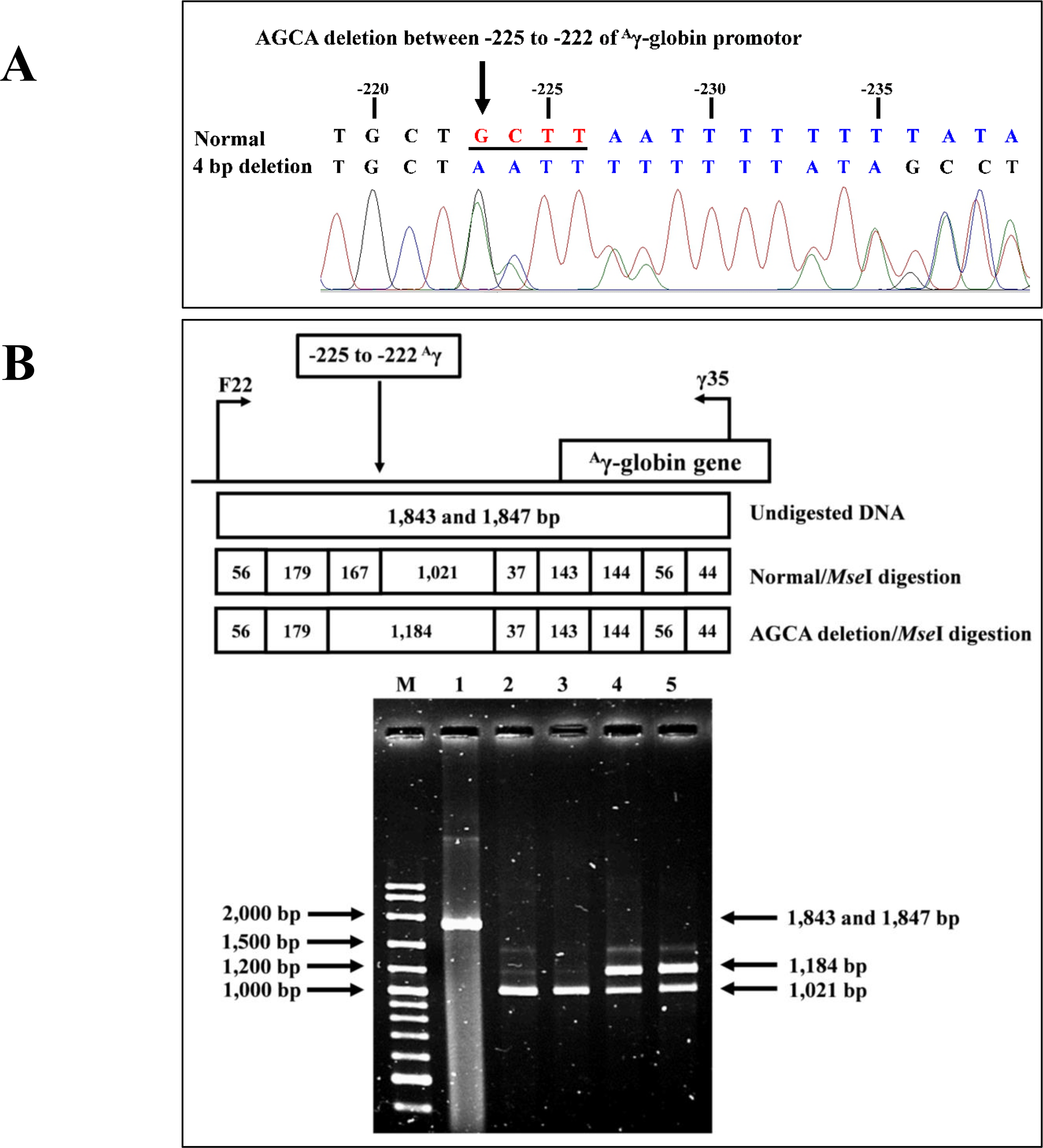{kind=link}
Allele-specific PCR assay for identification of rs5006884 polymorphism in the OR51B6 gene
Allele-specific PCR was developed to identify the rs5006884 (NC_000011.10:g.5352021C>T) in the OR51B6 gene on chromosome 11. In this system, primers G197 (5′-AAGACCAGG ATGCAGCAGAT-3′) and G198 (5′-ACCTCGGAGTGACATTGACC-3′) were used to produce an amplified fragment of 544 bp internal control. Specific amplification of the C allele (252 bp) and T allele (331 bp) were obtained using primers G197 & G199 (5′-CCTACTGTCGATCCCATGTAC-3′), and G200 (5′-AGACAGAAAGCATGGGAGAA-3′) & G198, respectively. The PCR reaction mixture (50 µL) contains 50–200 ng of genomic DNA, 200 µM of dNTPs, 30 pmol of each primer, and 1 unit of Taq DNA polymerase (New England Biolab, Inc., USA) in 10 mM Tris–HCl buffer pH 8.3, 50 mM KCl, 3 mM MgCl2, and 0.01% gelatin. The PCR consists of an initial denaturation at 94 °C for 3 min followed by 30 cycles of amplification at (94 °C for 1 min and 65 °C for 90 s). The PCR product was analyzed on 2% agarose gel electrophoresis and visualized under UV light after ethidium bromide staining. Confirmation of SNP rs5006884 was done using DNA sequencing (Fig. 3).
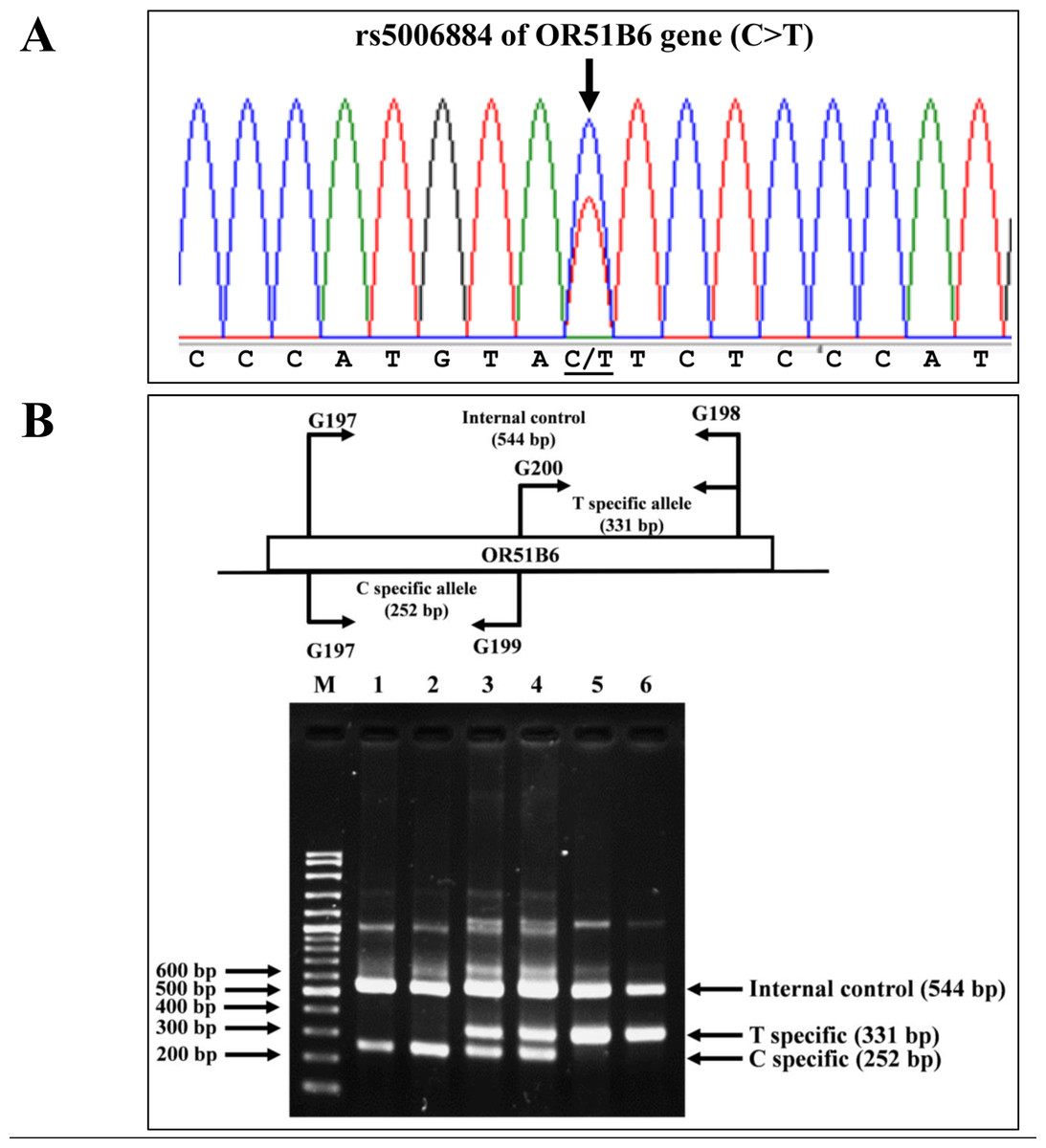
Figure 3: DNA sequencing profile and identification of the rs5006884 on OR51B6.
(A) DNA sequencing profile of a representative subject with heterozygosity for the rs5006884 on OR51B6 gene. (B) Characterization of the rs5006884 on OR51B6 gene by allele-specific PCR. The locations and orientations of primers (G197 & G198), (G200 & G198), and (G197 & G199) are indicated to produce fragments of 544 bp, 331 bp, and 252 bp specific for internal control, T-specific allele (mutant) and C-specific allele (wide-type) of rs5006884, respectively. M represents the GeneRular 100 bp Plus DNA ladder. Lanes 1 & 2: homozygous wide-type allele (C/C), lanes 3 & 4: heterozygous for C/T alleles, and lanes 5 & 6: homozygous for T/T alleles.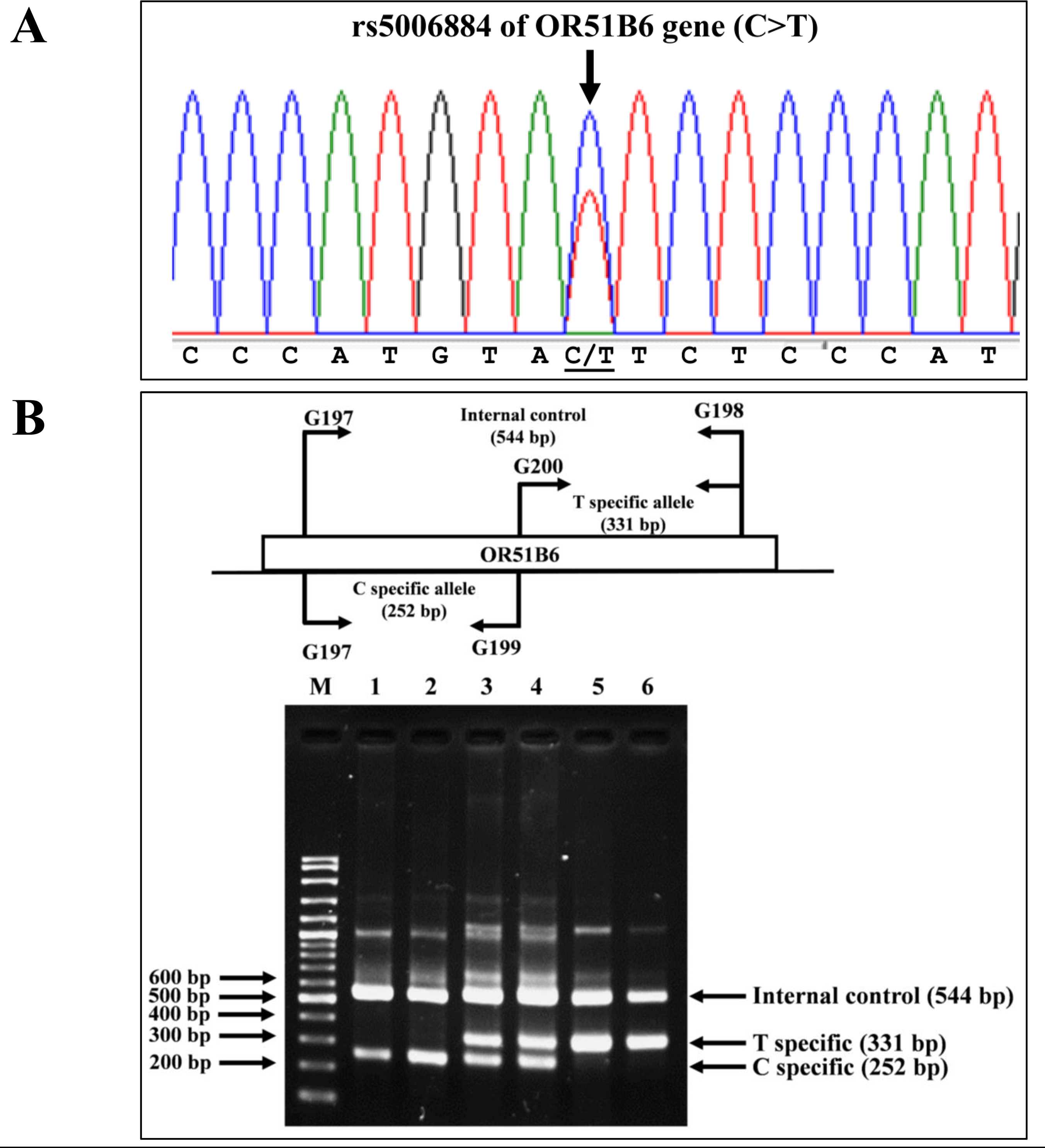{kind=link}
Statistical analysis
Hematological parameters of the subjects were presented as descriptive statistics, mean and standard deviation. The Mann–Whitney U test or Wilcoxon test was performed to compare variables between two dependent groups with non-normal distribution using the R software (version 4.1.0) (The R Foundation, Vienna, Austria). Statistical significance was set at P-value <0.05.
Results
DNA sequencing of the deletion breakpoint of the β0-thalassemia with 3.4 kb deletion encountered in Thai patients identified the deletion as NG_000007.3:g.69825_73314del3488 or NC_000011.10:g.5224302_5227791del3490 (GenBank Accession no. OQ161123), exactly the same with that reported previously in a Chinese patient (He et al., 2018). Table 1 and Fig. 4 demonstrated the hematological parameters associated with the β0-thalassemia trait with 3.4 kb deletion (n = 103) compared to those of the β0-thalassemia trait with other mutations (n = 259) encountered in our series. As shown in the table, significant higher Hb (11.6 ± 1.7 vs. 11.2 ± 1.6 g/dL), hematocrit (Hct) (36.1 ± 5.2 vs. 34.8 ± 5.2%), MCV (66.3 ± 4.3 vs. 63.2 ± 3.8 fL), MCH (21.3 ± 1.3 vs. 20.2 ± 1.4 pg), Hb A2 (6.9 ± 0.8 vs. 5.7 ± 0.6%) and Hb F (6.5 ± 3.4 vs. 1.3 ± 0.9%) values were observed for the former group of subjects. These are also the cases for the patients with compound heterozygous β0-thalassemia and Hb E. We observed significant higher Hb, Hct, MCV, MCH, and Hb F (48.1 ± 8.3 vs. 32.2 ± 15.6%) but lower Hb A2 (3.9 ± 1.1 vs. 6.5 ± 1.9%) values for the patients with 3.4 kb deletion (n = 20) as compared to those with other β0-thalassemia mutations (n = 125). These results indicate that the 3.4 kb deletion β0-thalassemia is associated with high Hb A2 and Hb F in a heterozygote state and a mild hematological phenotype when found in association with Hb E in a Hb E- β0-thalassemia disease. The effect of co-inheritance of α-hemoglobinopathy on the hematological phenotype of heterozygous β-thalassemia with 3.4 kb deletion was summarized in Table 2. Significant elevation of MCV and MCH values and a reduction of Hb F (3.0 ± 3.3%) were observed in heterozygous β-thalassemia with 3.4 kb deletion with heterozygous α+-thalassemia (– α/αα, n = 13). Lower Hb F but not Hb A2 were also observed in double heterozygotes for 3.4 kb deletion β-thalassemia and other deletional forms of α-thalassemia including homozygous α+-thalassemia (– α/– α, n = 1), αo-thalassemia (--/ αα, n = 2) and Hb H disease (--/- α, n = 1). In contrast, we observed that co-inheritance of a non-deletional α-thalassemia, the Hb Constant Spring (αCSα/αα, n = 7), did not lead to a reduction in Hb F expression, although it did have some hematological improvement. However, no statistical analysis was performed due to the small sample sizes of these α-hemoglobinopathies.
| Group | β-genotype | N | Sex (M/F) |
RBC (10 12/L) |
Hb (g/dL) |
Hct (%) |
MCV (fL) |
MCH (pg) |
MCHC (g/dL) |
RDW (%) |
Hbs E+A2 (%) |
Hb A2 (%) |
Hb F (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pure β0-thalassemia trait a | β0/βA | 259 | 119/140 | 5.5 ± 0.9 | 11.2 ± 1.6 | 34.8 ± 5.2 | 63.2 ± 3.8 | 20.2 ± 1.4 | 32.2 ± 1.8 | 17.1 ± 1.8 | – | 5.7 ± 0.6 | 1.3 ± 0.9 |
| Pure β3.4-thalassemia trait | β3.4/βA | 103 | 55/48 | 5.5 ± 0.8 | 11.6 ± 1.7 | 36.1 ± 5.2 | 66.3 ± 4.3 | 21.3 ± 1.3 | 32.2 ± 1.3 | 19.2 ± 2.0 | – | 6.9 ± 0.8 | 6.5 ± 3.4 |
| P-value | 0.50 | 0.030* | 0.052 | <0.001* | <0.001* | 0.099 | <0.001* | – | <0.001* | <0.001* | |||
| β0-thalassemia/Hb E b | β0/βE | 125 | 60/65 | 4.1 ± 1.1 | 7.8 ± 1.5 | 24.8 ± 4.9 | 60.4 ± 8.6 | 19.0 ± 2.9 | 31.7 ± 1.9 | 29.5 ± 3.9 | 63.2 ± 16.2 | 6.5 ± 1.9 | 32.2 ± 15.6 |
| β3.4-thalassemia/Hb E | β3.4/βE | 20 | 8/12 | 4.4 ± 0.7 | 10.2 ± 1.4 | 30.5 ± 4.6 | 68.3 ± 5.7 | 23.0 ± 2.1 | 33.4 ± 1.8 | 22.4 ± 1.7 | 48.4 ± 6.3 | 3.9 ± 1.1 | 48.1 ± 8.3 |
| P-value | 0.150 | <0.001* | <0.001* | <0.001* | <0.001* | 0.009* | <0.001* | <0.001* | <0.001* | <0.001* |
Interestingly, an adult male patient was encountered with a double heterozygote for β-thalassemia with 3.4 kb deletion and an α-globin gene triplication (αααanti−3.7/ αα), a hitherto undescribed condition. He had relatively normal Hb and Hct values with reduced MCV & MCH characteristics of a plain β-thalassemia carrier. Elevated Hb A2 (7.2%) and Hb F (6.8%) were observed. The result indicates that, unlike other β0-thalassemia alleles, the interaction of α-globin gene triplication (αααanti−3.7/ αα) and a β0-thalassemia with 3.4 kb deletion does not lead to a β-thalassemia disease in heterozygotic form.
SNPs were examined in representative subjects to determine if the existence of known Hb F associated SNPs are responsible for the high Hb A2 and Hb F phenotype in our patients. As shown in Table 3, the majority of the subjects had wild-type sequences for the three SNPs, including deletion of four nucleotides (-AGCA) between −225 to −222 on Aγ-globin promotor, rs5006884 (C>T) of the OR51B6 gene, and Gγ-Xmn I of Gγ-globin gene promoter. In addition, no significant difference in Hb F and other hematological parameters was observed between those of subjects with and without these SNPs. DNA sequencing of the three BCL11A binding motifs (TGGTCA) between the 3′ Aγ-globin gene and the 5′ δ-globin gene (Fig. 1) identified no alteration of the nucleotide at the binding motifs and their vicinities. Further sequencing of the whole Aγ- and Gγ-globin genes in representative subjects identified no mutation.
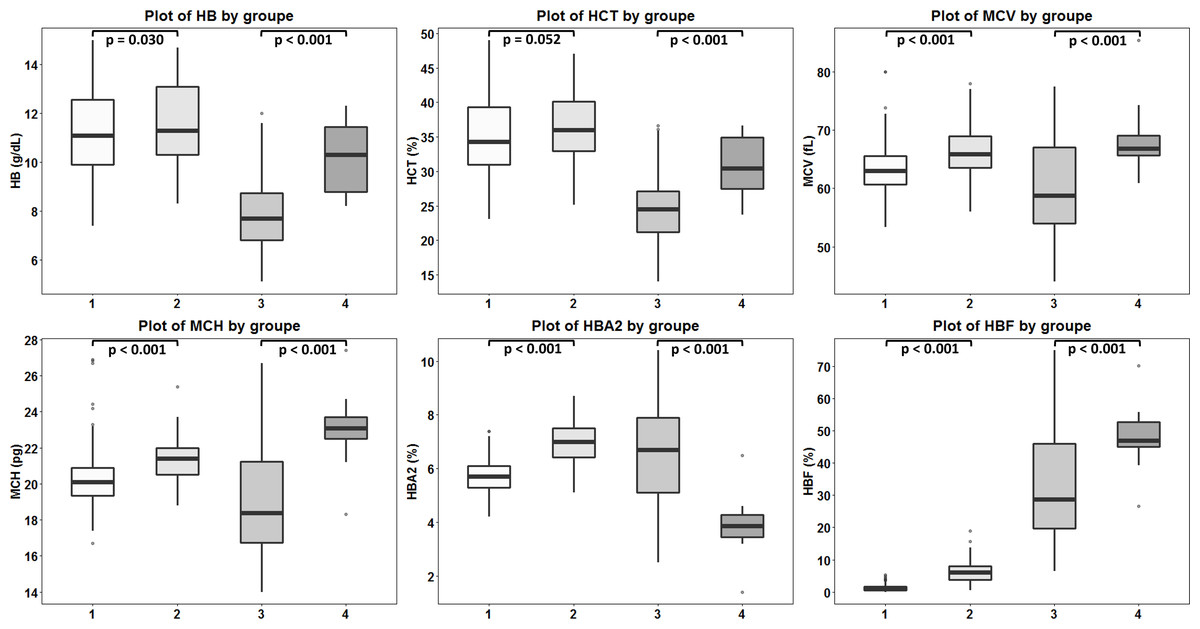
Figure 4: Comparison of hematological values of the four groups of β -thalassemia.
Box plots demonstrating medians and interquartile ranges in comparison of Hb, Hct, MCV, MCH, Hb A2 and Hb F values of four β-thalassemia genotypes including (1): β0-thalassemia trait with point mutations (n = 259), (2): β0-thalassemia trait with 3.4 kb deletion (n = 103), (3): Hb E- β0-thalassemia with point mutations (n = 125) and (4): Hb E- β0-thalassemia with 3.4 deletion (n = 20).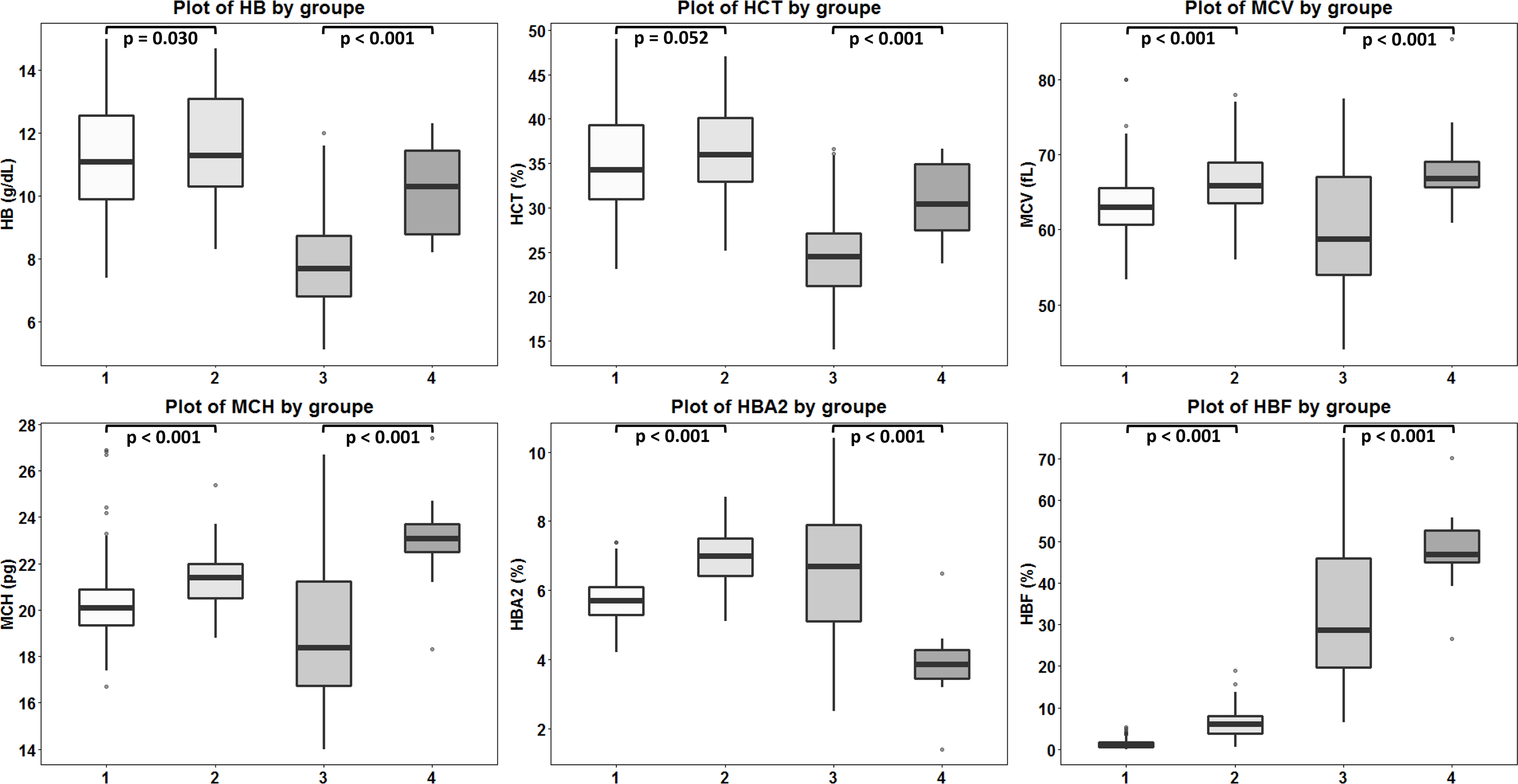{kind=link}
Discussion
The carrier of β-thalassemia usually accompanies elevated Hb A2 and normal Hb F levels. Hb A2 isa product of tetrameric assembly of two α- and two δ-globin chains (α2δ2), which is synthesized at a low level (2.5−3.5%) in normal adult individuals. Several genetic and non-genetic conditions are associated with Hb A2 expression (Singha et al., 2021). Among many different β-thalassemia genes in Thailand, we observed that only the 3.4 kb deletion β0-thalassemia is always associated in heterozygotic form with high Hb A2 (6.9 ± 0.8%) and Hb F (6.5 ± 3.4%) levels. Heterozygosity for other β-thalassemia alleles is usually found with elevated Hb A2 (5.7 ± 0.6%) but not Hb F (1.3 ± 0.9%) (Yamsri et al., 2010; Yamsri et al., 2011; Yamsri et al., 2015; Yamsri et al., 2022). Hematological characteristics in a large cohort of β-thalassemia carriers with this mutation listed in Table 1 confirmed this phenotype characteristic. This 3.4 kb deletion β0-thalassemia has been frequently described among Southeast Asian and Chinese patients. It has rarely been encountered on population of India, Pakistan and Bangladesh (Nadkarni et al., 2019; Khaliq, 2022; Chowdhury, Sultana & Das, 2022). This 3,488 bp deletion β-thalassemia allele removes DNA between positions −125 to +78 of the β-globin gene, eliminating the CACCC, CCAAT, and TATA elements in the β-globin gene promoter. The high Hb F characteristic of this β0-thalassemia allele could likely ameliorate the patient’s clinical phenotype. As shown in Table 1, when found in combination with Hb E, it could lead to non-transfusion-dependent thalassemia (NTDT). As compared to other β0-thalassemia/Hb E, which is generally associated with severe transfusion-dependent thalassemia (TDT), the patients with β3.4-thalassemia/Hb E had milder NTDT phenotype with significantly higher Hb, Hct, MCV, MCH, MCHC, and Hb F but lower RDW values. We have also documented recently that the patients with homozygous β0-thalassemia of 3.4 kb deletion were presented with mild NTDT phenotype with Hb ranging from 7–9 g/dL and did not require regular blood transfusion (Soontornpanawet, Singha & Fucharoen, 2022). It is noteworthy for a compound Hb E- β-thalassemia with 3.4 kb deletion that the patient had significant higher Hb, Hct, MCV, MCH, and Hb F (48.1 ± 8.3 vs. 32.2 ± 15.6%) but lower Hb A2 (3.9 ± 1.1 vs. 6.5 ± 1.9%) as compared to those of other β0-thalassemia mutations (Table 1). This lower Hb A2 in Hb E-β0-thalassemia with 3.4 kb deletion as compared with Hb E-β0-thalassemia with non-deletional mutations is most likely due to the higher Hb F expression. There is an inverse relationship between Hb A2 and Hb F expression in β-thalassemia. Red blood cells that produce relatively large amount of Hb F will produce less Hb A2, and vice versa (Singha et al., 2018). It is conceivable that although the 3.4 kb deletion results in β0-thalassemia, the increase in Hb F may compensate for the complete absence of Hb A and ameliorate the severity of the disease as seen in our patients.
| α-genotype | No | Sex (M/F) |
RBC (1012/L) |
Hb (g/dL) |
Hct (%) |
MCV (fL) |
MCH (pg) |
MCHC (g/dL) |
RDW (%) |
Hb A2 (%) |
Hb F (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| αα/αα | 103 | 55/48 | 5.5 ± 0.8 | 11.6 ± 1.7 | 36.1 ± 5.2 | 66.3 ± 4.3 | 21.3 ± 1.3 | 32.2 ± 1.3 | 19.2 ± 2.0 | 6.9 ± 0.8 | 6.5 ± 3.4 |
| - α/αα | 13 | 10/3 | 5.2 ± 1.1 | 12.5 ± 1.9 | 38.7 ± 5.2 | 72.3 ± 6.5a | 22.9 ± 1.7b | 31.6 ± 1.7 | 15.3 ± 1.9c | 6.9 ± 0.7 | 3.0 ± 3.3d |
| αCSα/αα | 7 | 2/5 | 5.4 ± 0.8 | 12.0 ± 1.4 | 36.6 ± 4.6 | 68.3 ± 3.9 | 22.3 ± 1.6 | 32.7 ± 0.8 | 17.9 ± 2.5 | 6.8 ± 0.8 | 9.1 ± 5.6 |
| - α/- α | 1 | 1/0 | na | na | na | 71.4 | na | na | na | 7.2 | na |
| -- /αα | 2 | 0/2 | 4.5 | 10.3 | 30.7 | 67.8 | 22.7 | 33.6 | 18.1 | 6.3, 7.0 | 3.9, 3.6 |
| -- /- α | 1 | 0/1 | 5.3 | 9.2 | 29.7 | 51.0 | 15.8 | 31.2 | Na | 5.0 | 2.5 |
| αααanti−3.7/αα | 1 | 1/0 | 6.4 | 13.0 | 40.0 | 62.0 | 20.3 | 32.5 | 13.7 | 7.2 | 6.8 |
Notes:
- na
-
not available
- CS
-
Hb Constant Spring
| Genotype | N |
Sex (M/F) |
RBC (1012/L) |
Hb (g/dL) |
Hct (%) |
MCV (fL) |
MCH (pg) |
MCHC (g/dL) |
RDW (%) |
Hb A2 (%) |
Hb F (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Four bp deletion (-AGCA) inAγ-globin promoter * | |||||||||||
| WT/WT | 83 | 43/40 | 5.5 ± 0.8 | 11.7 ± 1.7 | 36.1 ± 5.3 | 65.8 ± 4.0 | 21.2 ± 1.3 | 32.3 ± 1.2 | 19.3 ± 2.0 | 7.0 ± 0.7 | 6.3 ± 3.2 |
| WT/-4 bp | 1 | 1/0 | 6.2 | 13.2 | 39.7 | 64.0 | 21.4 | 33.2 | 21.7 | 7.0 | 6.8 |
| rs5006884 (C>T) /OR51B6 gene | |||||||||||
| C/C | 68 | 36/32 | 5.5 ± 0.8 | 11.6 ± 1.7 | 35.8 ± 5.1 | 65.8 ± 4.1 | 21.2 ± 1.3 | 32.3 ± 1.2 | 19.3 ± 1.9 | 6.9 ± 0.7 | 6.4 ± 3.7 |
| C/T | 13 | 5/8 | 5.2 ± 0.8 | 11.0 ± 1.6 | 34.1 ± 5.0 | 66.0 ± 4.0 | 21.4 ± 1.1 | 32.4 ± 1.3 | 19.6 ± 1.3 | 7.3 ± 0.9 | 7.3 ± 2.1 |
| P-value | 0.280 | 0.346 | 0.315 | 0.823 | 0.813 | 0.993 | 0.987 | 0.126 | 0.238 | ||
| Gγ-XmnI in Gγ-globin promoter* | |||||||||||
| C/C | 52 | 26/26 | 5.5 ± 0.8 | 11.8 ± 1.7 | 37.0 ± 5.5 | 67.4 ± 4.2 | 21.5 ± 1.2 | 31.5 ± 2.5 | 18.4 ± 2.1 | 6.9 ± 0.9 | 6.6 ± 3.9 |
| C/T | 3 | 2/1 | 5.3, na | 10.3, na | 40.2, 34.2 | 69.0, 64.4 | 19.3, na | 30.0, na | 22.6, na | 6.7 ± 0.7 | 5.0 ± 5.7 |
As shown in Table 2, co-inheritance of the 3.4 kb deletion β0-thalassemia and α-thalassemia (-α/αα, αCSα/αα, -α/-α, --/αα) could further improve the hematological phenotype of the patients due to the balance between α- and non-α-globin chains. Fortunately, in all cases of these double heterozygotes, the Hb A2 levels are still within the diagnostic range for a β-thalassemia carrier. Interestingly, we have encountered a subject with a hitherto undescribed condition of double heterozygote for the β3.4kb deletion and α-globin gene triplication (αααanti−3.7/αα). This subject presented with β-thalassemia trait phenotype without anemia with Hb 13.0 g/dL and Hct 40.0%, Hb A2 7.2%, and Hb F 6.8% (Table 2). The heterozygous β-thalassemia with α-globin gene triplication generally presents with thalassemia intermedia phenotype due to more excess α-globin chain and imbalance in α- and β-globin chain synthesis (Steinberg et al., 2009). This might be explained by the fact that, unlike other point mutations, the β3.4kb deletion is associated with increased expression of both δ- and γ-globin genes and could therefore improve the balance between α- and non- α-globin chains. This information indirectly supports that the β0-thalassemia with 3.4 kb deletion is a mild β0-thalassemia allele. We observed that this high Hb A2 and Hb F β-thalassemia trait is also the case with the Filipino β-thalassemia deletion occasionally encountered in our setting, although with different extent of Hb F expression. This Filipino β-thalassemia deletion [NG_000007.3:g.66258_184734del118477] was described originally as approximately 45 kb long starting from position -4,279 bp relative to the mRNA cap site of the β-globin gene but with an uncertain 3′ breakpoint. It was later described using gap-PCR and DNA sequencing as 118 kb in length, with the 5′ breakpoint at position −4,279 of mRNA cap site, and the 3′ breakpoint extending to the downstream olfactory receptor (OR) region where it deletes four functional OR genes and two OR pseudogenes including the OR52A1 that contains a γ-globin gene enhancer (Yamsri et al., 2012; Teh et al., 2018; Yasin et al., 2022). Table 4 compared the hematological parameters of β0-thalassemia carriers with three different β-thalassemia deletions in our series, including the 3.4 kb deletion (n = 103), the Filipino β0-thalassemia (n = 9), and 105 bp β0-thalassemia deletion (n = 7), all without α-thalassemia. The 3.4 kb deletion and the Filipino β0-thalassemia are associated with high Hb A2 and high Hb F β-thalassemia trait but not the 105 bp β0-thalassemia. In fact, this is not unexpected. The 105 β0-thalassemia is caused by a DNA deletion of −24 or −25 to +80 or +81 relative to the cap site of β-globin mRNA. Unlike the 3.4 kb deletion and the Filipino β0-thalassemia with 118 kb deletion, this 105 bp deletion does not remove the β-globin gene promoter TATA, CCAAT, and CACCC motifs (Nopparatana et al., 1999).
| β-genotype | α-genotype | No. | Sex (M/F) |
RBC (106/ µL) |
Hb (g/dL) |
HCT (%) |
MCV (fL) |
MCH (pg) |
MCHC (g/dL) |
RDW (%) |
Hb A2 (%) |
Hb F (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β0/βA | αα/αα | 259 | 119/140 | 5.5 ± 0.9 | 11.2 ± 1.6 | 34.8 ± 5.2 | 63.2 ± 3.8 | 20.2 ± 1.4 | 32.2 ± 1.8 | 17.1 ± 1.8 | 5.7 ± 0.6 | 1.3 ± 0.9 |
| β+/βA | αα/αα | 70 | 40/30 | 5.5 ± 0.8 | 12.6 ± 1.8 | 38.6 ± 5.2 | 70.5 ± 4.4 | 23.3 ± 2.1 | 32.6 ± 1.8 | 16.7 ± 1.9 | 5.9 ± 0.6a | 1.4 ± 0.8 |
| β3.4/βA | αα/αα | 103 | 55/48 | 5.5 ± 0.8 | 11.6 ± 1.7 | 36.1 ± 5.2 | 66.3 ± 4.3 | 21.3 ± 1.3 | 32.2 ± 1.3 | 19.2 ± 2.0 | 6.9 ± 0.8b,c | 6.5 ± 3.4b,c |
| βFil/βA | αα/αα | 9 | 5/4 | 5.1 ± 0.6 | 11.7 ± 1.3 | 37.1 ± 3.9 | 69.2 ± 5.4 | 23.1 ± 4.0 | 32.0 ± 0.7 | 16.8 ± 1.3 | 6.4 ± 0.6d,e | 4.0 ± 3.2f,g |
| β105/βA | αα/αα | 7 | 2/5 | 5.1 ± 0.5 | 10.6 ± 0.9 | 32.9 ± 2.7 | 63.6 ± 3.4 | 20.6 ± 1.3 | 32.1 ± 0.4 | 16.6 ± 1.0 | 5.3 ± 0.7h | 1.4 ± 1.1 |
Notes:
It has been noted that variation preliminarily in the two SNPs, rs4895441 and rs9399137 in HBS1L-MYB intergenic region, may have a small effect on Hb F expression in this β-thalassemia allele (Tepakhan, Kanjanaopas & Srewaradachpisal, 2020). We have also documented that the G allele of rs4895441 and C allele of rs9399137, alone or in combination with other SNPs, could explain the mild phenotypic expression of NTDT associated with Hb E-β-thalassemia disease (Phanrahan et al., 2019). To understand the high Hb F characteristic of the 3.4 kb deletion β0-thalassemia allele, we have examined other SNPs associated with high Hb A2 & Hb F expression. These included the -AGCA deletion at −225 to −222 of Aγ-globin gene promoter, rs5006884 (C>T) on OR51B6 gene, −158Gγ-Xmn I, BCL11A binding motifs between Aγ- and δ-globin genes as well as the whole γ-globin gene sequencing. It has been shown that the four bp deletion (-AGCA) at −225 to −222 of the Aγ-globin gene was associated with reduced Aγ-globin chain and elevation of Gγ chain with slightly increased Hb F level in β-thalassemia subjects (Manca et al., 1991; Ugrin et al., 2016). The rs5006884 (C>T) on OR51B6 gene located upstream of β-globin gene cluster might intervene in the tetramer formation of α-globin chain with β- and δ-globin chains and regulated Hb A2 level in β-thalassemia carriers (Cyrus et al., 2019). The −158Gγ-Xmn I polymorphism is associated with high Hb F expression in β-hemoglobinopathies during erythropoietic stress (Thein, 2017). This Gγ-Xmn I is almost linked to the +25 (G>A)Aγ-globin promoter (rs368698783). The polymorphism leads to a decreased Ly-1 antibody reactive clone (LYAR) binding efficiency, which is the repressor of γ-globin genes (Bianchi et al., 2016; Chen et al., 2017). BCL11A maintains silencing of γ-globin expression in adult erythroid cells and direct promoter repression to control the fetal to adult Hb switch in humans. The BCL11A binding motifs (TGGTCA or TGACCA) have been identified on the β-globin gene cluster at the third hypersensitive site (HS3) of the β-LCR, downstream of Aγ-globin gene and upstream of δ-globin gene (Sankaran et al., 2008; Sankaran, Xu & Orkin, 2010; Liu et al., 2018). Mutation in these binding motifs may therefore result in increased Hb F expression. As shown in Table 3, we did not observe the association of these SNPs with high Hb F and higher hematological values for the β0-thalassemia with 3.4 kb deletion. In fact, most of the cases had wild-type SNPs sequences, and no mutation was found in the BCL11A binding motifs between 3′Aγ-globin gene and 5′ δ-globin gene and within Aγ- and Gγ-globin genes of the patients examined. It is, therefore, unlikely that the unusually high Hb A2 and Hb F β-thalassemia trait of the β0-thalassemia with 3.4 kb deletion is caused by the effect of these high Hb F SNPs. However, this might be explained by the fact that removal of the 5′ β-globin promoter in the 3.4 kb deletion may release competition of the β-globin gene for the upstream locus control region (β-LCR), leading to its increased interaction with the γ- and δ-globin genes in cis, increasing their expression resulting in elevation of Hb F and Hb A2 (Thein, 2018). Recently, it has been documented that disrupting the adult β-globin promoter reactivates γ-globin gene expression by promoter competition model in the Hb switching (Topfer et al., 2022). The 3.4 kb deletion removing β-globin promoter and the entire β-globin gene is therefore associated with elevated Hb F in addition to elevated Hb A2 in heterozygotic form.
Conclusions
Nonetheless, the high Hb, Hct, MCV, MCH, Hb A2, and Hb F values in the heterozygote state and a mild hematological phenotype observed in Hb E-β3.4kb deletion disease and double heterozygosity for α-globin gene triplication (αααanti−3.7) and β3.4kb deletion support that the β3.4kb deletion is mild β0-thalassemia allele and might ameliorate severity when occurring in homozygote or compound heterozygote state. This high Hb A2 and high Hb F β-thalassemia characteristic could also be a useful marker for the β3.4kb deletion before being further confirmed by PCR analysis. This information related to the 3.4 kb deletion β-thalassemia should be provided to the patient’s family for the appropriate management and genetic counseling. This is especially important information for routine prenatal thalassemia screening in a prevention and control program in the regions.