

Insights on the evolution of Coronavirinae in general, and SARS-CoV-2 in particular, through innovative biocomputational resources
- Published
- Accepted
- Received
- Academic Editor
- Siouxsie Wiles
- Subject Areas
- Biodiversity, Bioinformatics, Virology, Epidemiology, COVID-19
- Keywords
- Coronavirus, Host-virus interaction, Chimerism, Evolutionary constraints, Virology, Zoonotic reservoirs, Viral proteomes, Viral assembly
- Copyright
- © 2022 Dos Santos et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2022. Insights on the evolution of Coronavirinae in general, and SARS-CoV-2 in particular, through innovative biocomputational resources. PeerJ 10:e13700 https://doi.org/10.7717/peerj.13700
Abstract
The structural proteins of coronaviruses portray critical information to address issues of classification, assembly constraints, and evolutionary pathways involving host shifts. We compiled 173 complete protein sequences from isolates belonging to the four genera of the subfamily Coronavirinae. We calculate a single matrix of viral distance as a linear combination of protein distances. The minimum spanning tree (MST) connecting the individuals captures the structure of their similarities. The MST re-capitulates the known phylogeny of Coronovirinae. Hosts were mapped onto the MST and we found a non-trivial concordance between host phylogeny and viral proteomic distance. We also study the chimerism in our dataset through computational simulations. We found evidence that structural units coming from loosely related hosts hardly give rise to feasible chimeras in nature. This work offers a fresh way to analyze features of SARS-CoV-2 and related viruses.
Introduction
Viruses can be considered molecular parasites (Koonin, Dolja & Krupovic, 2015) with an asexual type of reproduction (assisted by cells’ replication mechanisms) in which gene exchanges do not take place. Novel hybrid infectious particles—or chimeras—may be generated when a host cell is infected with at least two viral genomes at the same time (Simon-Loriere & Holmes, 2011). Based on this phenomenon, chimeric viruses, have also been created in laboratories by gain-of-function experiments in order to make, for instance, novel attenuated pan-vaccines (Jochmus et al., 1999; Whitehead et al., 2003; Akhand et al., 2020 among others).
Although viruses cannot be considered as biological entities, they are evolutionary entities; thus, the inquiry about their evolutionary constraints is pertinent. Phenotypic limits in evolutionary entities can be detected by comparing theoretical vs. empirical morphospaces (McGhee, 1999; Eble, 2000). A theoretical morphospace is an n-dimensional space describing and relating phenotypic configurations. It is generated by the systematic variation of the parameters underlying selected construction rules (Raup, 1967; Mitteroecker & Gunz, 2009). An empirical morphospace is the set of entities observed in nature (McGhee, 1999). Viral proteomes can be analyzed under a morphospace framework to delineate a gradient across the domains of existent, plausible, and impossible entities, henceforth the feasibility gradient. Morphospace analytical approaches include tools such as combinatorics, probability, network analysis, among others.
After two years of research and a myriad of publications on SARS-Cov2, the origin of this virus is still a matter not resolved. Moreover, the WHO-led international mission that has begun investigations in China to try to establish the origin of SARS-CoV-2 estimates that could take years to track the zoonotic jumps behind the viral origin (Zarocostas, 2021). The rise of emergent viruses, coming from wild reservoirs can occur as a consequence of increased opportunities for transmission due to perturbations in the ecosystem, independently of changes in their genetic structure (Solé & Elena, 2018). The most closely related virus to SARS-CoV-2 is RaTG13, identified from a Rhinolophus affinis bat. However, the receptor-binding domain (RBD) of SARS-CoV-2 is more similar to its analog to the pangolin-CoV-2020 isolated from Malayan pangolins (Manis javanica) (Segreto & Deigin, 2020). The ambiguous nature of SARS-CoV-2 suggests two main hypotheses regarding its origin: (1) A zoonotic origin with R. affinis as a wild host reservoir, in which mutational changes have occurred in the RBD, conferring the virus the ability to infect humans. This sequence is similar by convergence to the pangolin-CoV-2020; (2) A chimeral origin in which the backbone of the novel virus comes from RaTG13 and the RBD comes from the pangolin-Cov-2020. Interestingly, using an analytic strategy based on shell disorder models, Goh et al. (2020), Goh et al. (2022) offered a renewed view that went against the mainstream on the subject. They measured the percentage of intrinsic disorder of proteins from the viral inner and outer shells, and suggested a silent spreading among humans of a SARS-CoV-2 precursor coming from pangolins. It is, therefore, possible that pangolins play an important role in the ecology and/or evolution of SARS-CoV-2. If this is true, either natural or artificial chimerism becomes a working hypothesis.
Known members of the subfamily Coronavirinae include four genera alpha (α), beta (β), gamma (γ), and delta (δ) (Payne, 2017). Their genome encodes, among others, four main structural proteins essential for viral assembly: envelope (E), membrane (M), nucleocapsid (N), and spike (S) (Yao et al., 2020). E, M, N, and S proteins constitute the basic phenotypic configuration of Coronavirinae that varies between different viral species regarding their specific amino acid sequence. Considering each protein as our building block of the Coronavirinae diversity, we can compose a viral theoretical morphospace as the free combination of those proteins in 4-tuples. Let suppose the next 4-tuple describing the proteinic structure of a given empirical coronavirus x, Ex-Mx-Nx-Sx, and the 4-tuple of another hypothetical empirical coronavirus Ey-My-Ny-Sy from the coronavirus y, and let suppose that hosts in which they were found are Hx and Hy. We hypothesize that any new combination of a theoretical virus (e.g., Ex-My-Ny-Sx) is plausible to the extent that the involved hosts are closely related and viral proteins are similar with regard to their analogs in the observed assembly of empirical viruses (co-occurring proteins).
In this paper, we study the Coronavirinae under an exploration of sequence space. Based on the empirical set of viral protein sequences, we calculate the similarities among them. We simulate a theoretical viral morphospace and propose a scoring system to measure the degree of plausibility of occurrence in nature for every theoretical phenotypical option. We focus on two factors to assess the degree of chimerality for any viral protein assembly: The chemical protein affinities by one side, and the phylogenetic relatedness of the hosts by the other. By chimerality we understand the result of a recombination process as being, for example, the result of a coinfection. Through this approach, (1) we bring the attention to hot topics such as the prediction of unobserved virus lineages, (2) we provide insights to settle debates about the dichotomy artificial-natural emergence of new viruses, and (3) we offer clues to inquire about intermediary hosts in zoonotic jumps.
Materials & Methods
The analysis was based on viral individuals from different vertebrate taxa, belonging to the four genera of Coronavirinae. Therefore, we consulted the NCBI database and utilized a downloaded data table with all the applicable annotations for which complete data of sequences were available for the totality of structural proteins. We end up with a collection of 173 coronaviruses, two of them belonging to unknown genera. Accession numbers in addition to relevant metadata can be found in Table 1. Complete sequences of E, M, N, and S, were aligned using the MUSCLE algorithm (Edgar, 2004) in MEGA X software (Kumar et al., 2018). The number of amino acid substitutions per site was calculated between sequences using the Poisson correction model (Zuckerkandl & Pauling, 1965). The rate variation among sites was modeled with a gamma distribution (shape parameter = 1). These analyses involved 173 amino acid sequences for each class of protein. All positions containing gaps and missing data were eliminated (complete deletion option).
| Node ID | Acc. No. | Size | Host | Genus | GenBank Title |
|---|---|---|---|---|---|
| 1 | AY278489 | 29757 | NA | β-coronavirus | SARS coronavirus GD01 |
| 2 | AY390556 | 29760 | NA | β-coronavirus | SARS coronavirus GZ02 |
| 3 | AY572035 | 29518 | Viverridae | β-coronavirus | SARS coronavirus civet010 |
| 4 | AY686864 | 29525 | Paradoxurus hermaphroditus | β-coronavirus | SARS coronavirus B039 |
| 5 | DQ022305 | 29728 | NA | β-coronavirus | Bat SARS coronavirus HKU3-1 |
| 6 | DQ071615 | 29736 | Chiroptera | β-coronavirus | Bat SARS coronavirus Rp3 |
| 7 | DQ084200 | 29711 | NA | β-coronavirus | bat SARS coronavirus HKU3-3 |
| 8 | DQ412042 | 29709 | Rhinolophus ferrumequinum | β-coronavirus | Bat SARS coronavirus Rf1 |
| 9 | DQ412043 | 29749 | Rhinolophus macrotis | β-coronavirus | Bat SARS coronavirus Rm1 |
| 10 | DQ648856 | 29704 | NA | β-coronavirus | Bat coronavirus (BtCoV/273/2005) |
| 11 | DQ648857 | 29741 | NA | β-coronavirus | Bat coronavirus (BtCoV/279/2005) |
| 12 | DQ811787 | 27550 | Sus scrofa | α-coronavirus | PRCV ISU-1 |
| 13 | EF065505 | 30286 | Chiroptera | β-coronavirus | Bat coronavirus HKU4-1 |
| 14 | EF065506 | 30286 | Chiroptera | β-coronavirus | Bat coronavirus HKU4-2 |
| 15 | EF065507 | 30286 | Chiroptera | β-coronavirus | Bat coronavirus HKU4-3 |
| 16 | EF065508 | 30316 | Chiroptera | β-coronavirus | Bat coronavirus HKU4-4 |
| 17 | EF065509 | 30482 | Chiroptera | β-coronavirus | Bat coronavirus HKU5-1 |
| 18 | EF065510 | 30488 | Chiroptera | β-coronavirus | Bat coronavirus HKU5-2 |
| 19 | EF065511 | 30488 | Chiroptera | β-coronavirus | Bat coronavirus HKU5-3 |
| 20 | EF065512 | 30487 | Chiroptera | β-coronavirus | Bat coronavirus HKU5-5 |
| 21 | EF065513 | 29114 | Chiroptera | β-coronavirus | Bat coronavirus HKU9-1 |
| 22 | EF065514 | 29107 | Chiroptera | β-coronavirus | Bat coronavirus HKU9-2 |
| 23 | EF065515 | 29136 | Chiroptera | β-coronavirus | Bat coronavirus HKU9-3 |
| 24 | EF065516 | 29155 | Chiroptera | β-coronavirus | Bat coronavirus HKU9-4 |
| 25 | EF424615 | 31017 | Bos taurus | β-coronavirus | Bovine coronavirus E-AH65 |
| 26 | EF424616 | 30970 | Bos taurus | β-coronavirus | Bovine coronavirus E-AH65-TC |
| 27 | EF424617 | 31016 | Bos taurus | β-coronavirus | Bovine coronavirus R-AH65 |
| 28 | EF424618 | 30995 | Bos taurus | β-coronavirus | Bovine coronavirus R-AH65-TC |
| 29 | EF424619 | 30995 | Bos taurus | β-coronavirus | Bovine coronavirus E-AH187 |
| 30 | EF424620 | 30964 | Bos taurus | β-coronavirus | Bovine coronavirus R-AH187 |
| 31 | EF424621 | 30995 | Bovidae | β-coronavirus | Sable antelope coronavirus US/OH1/2003 |
| 32 | EF424622 | 30979 | Giraffa camelopardalis | β-coronavirus | Giraffe coronavirus US/OH3-TC/2006 |
| 33 | EF424623 | 31002 | Giraffa camelopardalis | β-coronavirus | Giraffe coronavirus US/OH3/2003 |
| 34 | EF424624 | 30979 | Giraffa camelopardalis | β-coronavirus | Calf-giraffe coronavirus US/OH3/2006 |
| 35 | FJ376620 | 26476 | Pycnonotus sinensis | δ -coronavirus | Bulbul coronavirus HKU11-796 |
| 36 | FJ415324 | 31029 | Homo sapiens | β-coronavirus | Human enteric coronavirus 4408 |
| 37 | FJ425184 | 30995 | Kobus ellipsiprymnus | β-coronavirus | Waterbuck coronavirus US/OH-WD358-TC/1994 |
| 38 | FJ425185 | 30940 | Kobus ellipsiprymnus | β-coronavirus | Waterbuck coronavirus US/OH-WD358-GnC/1994 |
| 39 | FJ425186 | 30962 | Kobus ellipsiprymnus | β-coronavirus | Waterbuck coronavirus US/OH-WD358/1994 |
| 40 | FJ425187 | 31020 | Odocoileus virginianus | β-coronavirus | White-tailed deer coronavirus US/OH-WD470/1994 |
| 41 | FJ425188 | 30995 | Rusa unicolor | β-coronavirus | Sambar deer coronavirus US/OH-WD388-TC/1994 |
| 42 | FJ425189 | 30997 | Rusa unicolor | β-coronavirus | Sambar deer coronavirus US/OH-WD388/1994 |
| 43 | FJ647218 | 31285 | Mus musculus | β-coronavirus | Murine coronavirus RA59/R13 |
| 44 | FJ647219 | 31427 | Mus musculus | β-coronavirus | Murine coronavirus RJHM/A |
| 45 | FJ647220 | 31429 | Mus musculus | β-coronavirus | Murine coronavirus RA59/SJHM |
| 46 | FJ647221 | 31456 | Mus musculus | β-coronavirus | Murine coronavirus repA59/RJHM |
| 47 | FJ647222 | 31283 | Mus musculus | β-coronavirus | Murine coronavirus SA59/RJHM |
| 48 | FJ647223 | 31386 | Mus musculus | β-coronavirus | Murine coronavirus MHV-1 |
| 49 | FJ647224 | 31448 | Mus musculus | β-coronavirus | Murine coronavirus MHV-3 |
| 50 | FJ647225 | 31293 | Mus musculus | β-coronavirus | Murine coronavirus inf-MHV-A59 |
| 51 | FJ647226 | 31473 | Mus musculus | β-coronavirus | Murine coronavirus MHV-JHM.IA |
| 52 | FJ647227 | 31250 | Mus musculus | β-coronavirus | Murine coronavirus repJHM/RA59 |
| 53 | FJ882963 | 29682 | Homo sapiens | β-coronavirus | SARS coronavirus P2 |
| 54 | FJ884686 | 31275 | Mus musculus | β-coronavirus | Murine hepatitis virus strain A59 B11 variant |
| 55 | FJ938051 | 29232 | Felis catus | α-coronavirus | Feline coronavirus RM |
| 56 | FJ938052 | 29306 | Felis catus | α-coronavirus | Feline coronavirus UU11 |
| 57 | FJ938053 | 29277 | Felis catus | α-coronavirus | Feline coronavirus UU7 |
| 58 | FJ938054 | 29269 | Felis catus | α-coronavirus | Feline coronavirus UU4 |
| 59 | FJ938055 | 29285 | Felis catus | α-coronavirus | Feline coronavirus UU8 |
| 60 | FJ938056 | 29253 | Felis catus | α-coronavirus | Feline coronavirus UU5 |
| 61 | FJ938057 | 29275 | Felis catus | α-coronavirus | Feline coronavirus UU15 |
| 62 | FJ938058 | 28479 | Felis catus | α-coronavirus | Feline coronavirus UU16 |
| 63 | FJ938059 | 29295 | Felis catus | α-coronavirus | Feline coronavirus UU10 |
| 64 | FJ938060 | 29256 | Felis catus | α-coronavirus | Feline coronavirus UU2 |
| 65 | FJ938061 | 29130 | Felis catus | α-coronavirus | Feline coronavirus UU3 |
| 66 | FJ938062 | 29266 | Felis catus | α-coronavirus | Feline coronavirus UU9 |
| 67 | FJ938063 | 31024 | Bos taurus | β-coronavirus | Bovine coronavirus E-DB2-TC |
| 68 | FJ938064 | 30995 | Bos taurus | β-coronavirus | Bovine coronavirus E-AH187-TC |
| 69 | FJ938065 | 30969 | Bos taurus | β-coronavirus | Bovine respiratory coronavirus AH187 |
| 70 | FJ938066 | 30953 | Bos taurus | β-coronavirus | Bovine respiratory coronavirus bovine/US/OH-440-TC/1996 |
| 71 | FJ938067 | 30953 | Homo sapiens | β-coronavirus | Human enteric coronavirus strain 4408 |
| 72 | GQ153544 | 29695 | NA | β-coronavirus | Bat SARS coronavirus HKU3-9 |
| 73 | GU553361 | 29264 | Feliformia | α-coronavirus | Feline coronavirus UU22 isolate TCVSP-ROTTIER-00022 |
| 74 | GU553362 | 29264 | Feliformia | α-coronavirus | Feline coronavirus UU23 isolate TCVSP-ROTTIER-00023 |
| 75 | HM211098 | 29136 | Chiroptera | β-coronavirus | Bat coronavirus HKU9-5-1 |
| 76 | HM211099 | 29112 | Chiroptera | β-coronavirus | Bat coronavirus HKU9-5-2 |
| 77 | HM211100 | 29136 | Chiroptera | β-coronavirus | Bat coronavirus HKU9-10-1 |
| 78 | HM211101 | 29122 | Chiroptera | β-coronavirus | Bat coronavirus HKU9-10-2 |
| 79 | HM245926 | 28915 | Neovison vison | α-coronavirus | Mink coronavirus strain WD1133 |
| 80 | HQ392469 | 29233 | Felis catus | α-coronavirus | Feline coronavirus UU40 |
| 81 | HQ392470 | 29255 | Feliformia | α-coronavirus | Feline coronavirus UU19 |
| 82 | HQ392471 | 29252 | Feliformia | α-coronavirus | Feline coronavirus UU20 |
| 83 | HQ392472 | 29233 | Feliformia | α-coronavirus | Feline coronavirus UU30 |
| 84 | JF705860 | 27673 | Anatidae | γ-coronavirus | Duck coronavirus isolate DK/CH/HN/ZZ2004 |
| 85 | JF792616 | 31286 | Rattus | β-coronavirus | Rat coronavirus isolate 681 |
| 86 | JN183882 | 29243 | Felis catus | α-coronavirus | Feline coronavirus UU47 |
| 87 | JN183883 | 29222 | Felis catus | α-coronavirus | Feline coronavirus UU54 |
| 88 | JQ410000 | 27374 | Vicugna pacos | α-coronavirus | Alpaca respiratory coronavirus isolate CA08-1/2008 |
| 89 | JQ989272 | 28483 | Chiroptera | α-coronavirus | Hipposideros bat coronavirus HKU10 isolate TLC1343A |
| 90 | JX860640 | 31028 | Canis lupus familiaris | β-coronavirus | Canine respiratory coronavirus strain K37 |
| 91 | JX869059 | 30119 | Homo sapiens | β-coronavirus | Human β-coronavirus 2c EMC/2012 |
| 92 | JX993987 | 29484 | Rhinolophus pusillus | β-coronavirus | Bat coronavirus Rp/Shaanxi2011 |
| 93 | JX993988 | 29452 | Chaerephon plicatus | β-coronavirus | Bat coronavirus Cp/Yunnan2011 |
| 94 | KC667074 | 30112 | Homo sapiens | β-coronavirus | Human β-coronavirus 2c England-Qatar/2012 |
| 95 | KC776174 | 30030 | Homo sapiens | β-coronavirus | Human β-coronavirus 2c Jordan-N3/2012 |
| 96 | KC881005 | 29787 | Rhinolophus sinicus | β-coronavirus | Bat SARS-like coronavirus RsSHC014 |
| 97 | KC881006 | 29792 | Rhinolophus sinicus | β-coronavirus | Bat SARS-like coronavirus Rs3367 |
| 98 | KF294457 | 29676 | Rhinolophus monoceros | β-coronavirus | SARS-related bat coronavirus |
| 99 | KF367457 | 30309 | Rhinolophus sinicus | β-coronavirus | Bat SARS-like coronavirus WIV1 |
| 100 | KF569996 | 29805 | Rhinolophus affinis | β-coronavirus | Rhinolophus affinis coronavirus isolate LYRa11 |
| 101 | KF906249 | 31052 | Camelus bactrianus | β-coronavirus | Dromedary camel coronavirus HKU23 strain HKU23-265F |
| 102 | KJ473811 | 29037 | Rhinolophus ferrumequinum | β-coronavirus | BtRf-BetaCoV/JL2012 |
| 103 | KJ473812 | 29443 | Rhinolophus ferrumequinum | β-coronavirus | BtRf-BetaCoV/HeB2013 |
| 104 | KJ473813 | 29461 | Rhinolophus ferrumequinum | β-coronavirus | BtRf-BetaCoV/SX2013 |
| 105 | KJ473814 | 29658 | Rhinolophus sinicus | β-coronavirus | BtRs-BetaCoV/HuB2013 |
| 106 | KJ473815 | 29161 | Rhinolophus sinicus | β-coronavirus | BtRs-BetaCoV/GX2013 |
| 107 | KJ473816 | 29142 | Rhinolophus sinicus | β-coronavirus | BtRs-BetaCoV/YN2013 |
| 108 | KJ473821 | 30423 | Vespertilio sinensis | β-coronavirus | BtVs-BetaCoV/SC2013 |
| 109 | KJ481931 | 25406 | Sus scrofa | δ -coronavirus | δ -coronavirus PDCoV/USA/Illinois121/2014 from USA |
| 110 | KJ567050 | 25422 | Sus scrofa | δ -coronavirus | Porcine δ -coronavirus 8734/USA-IA/2014 |
| 111 | KJ601777 | 25408 | Sus scrofa | δ -coronavirus | δ -coronavirus PDCoV/USA/Illinois133/2014 from USA |
| 112 | KJ601778 | 25404 | Sus scrofa | δ -coronavirus | δ -coronavirus PDCoV/USA/Illinois134/2014 from USA |
| 113 | KJ601779 | 25404 | Sus scrofa | δ -coronavirus | δ -coronavirus PDCoV/USA/Illinois136/2014 from USA |
| 114 | KJ601780 | 25404 | Sus scrofa | δ -coronavirus | δ -coronavirus PDCoV/USA/Ohio137/2014 from USA |
| 115 | KJ769231 | 25433 | Sus scrofa | δ -coronavirus | Swine δ -coronavirus OhioCVM1/2014 |
| 116 | KM820765 | 25422 | Sus scrofa | δ -coronavirus | Porcine δ -coronavirus KNU14-04 |
| 117 | KP886808 | 29723 | Rhinolophus ferrumequinum | β-coronavirus | Bat SARS-like coronavirus YNLF_31C |
| 118 | KT444582 | 30290 | Rhinolophus sinicus | β-coronavirus | SARS-like coronavirus WIV16 |
| 119 | KU973692 | 29722 | Chiroptera | β-coronavirus | UNVERIFIED: SARS-related coronavirus isolate F46 |
| 120 | KY352407 | 29274 | Rhinolophus | β-coronavirus | Severe acute respiratory syndrome-related coronavirus strain BtKY72 |
| 121 | KY417142 | 29725 | Aselliscus stoliczkanus | β-coronavirus | Bat SARS-like coronavirus isolate As6526 |
| 122 | KY417143 | 29741 | Rhinolophus sinicus | β-coronavirus | Bat SARS-like coronavirus isolate Rs4081 |
| 123 | KY417150 | 30311 | Rhinolophus sinicus | β-coronavirus | Bat SARS-like coronavirus isolate Rs4874 |
| 124 | KY417151 | 30307 | Rhinolophus sinicus | β-coronavirus | Bat SARS-like coronavirus isolate Rs7327 |
| 125 | MG772933 | 29802 | Rhinolophus sinicus | β-coronavirus | Bat SARS-like coronavirus isolate bat-SL-CoVZC45 |
| 126 | MG772934 | 29732 | Rhinolophus sinicus | β-coronavirus | Bat SARS-like coronavirus isolate bat-SL-CoVZXC21 |
| 127 | MK211374 | 29648 | Rhinolophus | β-coronavirus | Coronavirus BtRl-BetaCoV/SC2018 |
| 128 | MK211375 | 29698 | Rhinolophus affinis | β-coronavirus | Coronavirus BtRs-BetaCoV/YN2018A |
| 129 | MK211376 | 30256 | Rhinolophus affinis | β-coronavirus | Coronavirus BtRs-BetaCoV/YN2018B |
| 130 | MK211377 | 29689 | Rhinolophus affinis | β-coronavirus | Coronavirus BtRs-BetaCoV/YN2018C |
| 131 | MK211378 | 30213 | Rhinolophus affinis | β-coronavirus | Coronavirus BtRs-BetaCoV/YN2018D |
| 132 | MN908947 | 29903 | Homo sapiens | β-coronavirus | Severe acute respiratory syndrome coronavirus 2 (Wuhan) |
| 133 | MN996532 | 29855 | Rhinolophus affinis | β-coronavirus | Bat coronavirus RaTG13 |
| 134 | MT121216 | 29521 | Manis javanica | β-coronavirus | Pangolin coronavirus isolate MP789 |
| 135 | NC_006577 | 29926 | Homo sapiens | β-coronavirus | Human coronavirus HKU1 |
| 136 | NC_009019 | 30286 | Chiroptera | β-coronavirus | Bat coronavirus HKU4-1 |
| 137 | NC_009020 | 30482 | Chiroptera | β-coronavirus | Bat coronavirus HKU5-1 |
| 138 | NC_009021 | 29114 | Chiroptera | β-coronavirus | Bat coronavirus HKU9-1 |
| 139 | NC_010646 | 31686 | Delphinapterus leucas | γ-coronavirus | Beluga Whale coronavirus SW1 |
| 140 | NC_010800 | 27657 | Meleagris gallopavo | γ-coronavirus | Turkey coronavirus |
| 141 | NC_011547 | 26487 | Pycnonotus jocosus | δ -coronavirus | Bulbul coronavirus HKU11-934 |
| 142 | NC_011549 | 26396 | Turdus hortulorum | δ -coronavirus | Thrush coronavirus HKU12-600 |
| 143 | NC_011550 | 26552 | Lonchura striata | δ -coronavirus | Munia coronavirus HKU13-3514 |
| 144 | NC_012936 | 31250 | Rattus | β-coronavirus | Rat coronavirus Parker |
| 145 | NC_014470 | 29276 | Rhinolophus blasii | Unknown | Bat coronavirus BM48-31/BGR/2008 |
| 146 | NC_016992 | 26083 | Passeridae | δ -coronavirus | Sparrow coronavirus HKU17 |
| 147 | NC_016993 | 26689 | Muscicapidae | δ -coronavirus | Magpie-robin coronavirus HKU18 |
| 148 | NC_016994 | 26077 | Ardeidae | δ -coronavirus | Night-heron coronavirus HKU19 |
| 149 | NC_016995 | 26227 | Mareca | δ -coronavirus | Wigeon coronavirus HKU20 |
| 150 | NC_016996 | 26223 | Gallinula chloropus | δ -coronavirus | Common-moorhen coronavirus HKU21 |
| 151 | NC_017083 | 31100 | Oryctolagus cuniculus | β-coronavirus | Rabbit coronavirus HKU14 |
| 152 | NC_018871 | 28494 | Chiroptera | α-coronavirus | Rousettus bat coronavirus HKU10 |
| 153 | NC_019843 | 30119 | Homo sapiens | β-coronavirus | Middle East respiratory syndrome coronavirus |
| 154 | NC_023760 | 28941 | Neovison vison | α-coronavirus | Mink coronavirus strain WD1127 |
| 155 | NC_025217 | 31491 | Hipposideros pratti | β-coronavirus | Bat Hp- β-coronavirus/Zhejiang2013 |
| 156 | NC_026011 | 31249 | Rattus norvegicus | β-coronavirus | β-coronavirus HKU24 strain HKU24-R05005I |
| 157 | NC_028752 | 27395 | Camelus | α-coronavirus | Camel α-coronavirus isolate camel/Riyadh/Ry141/2015 |
| 158 | NC_028806 | 28111 | Sus scrofa | α-coronavirus | Swine enteric coronavirus strain Italy/213306/2009 |
| 159 | NC_028811 | 27935 | Myotis ricketti | α-coronavirus | BtMr-AlphaCoV/SAX2011 |
| 160 | NC_028814 | 27608 | Rhinolophus ferrumequinum | α-coronavirus | BtRf-AlphaCoV/HuB2013 |
| 161 | NC_028824 | 26975 | Rhinolophus ferrumequinum | α-coronavirus | BtRf-AlphaCoV/YN2012 |
| 162 | NC_028833 | 27783 | Nyctalus velutinus | α-coronavirus | BtNv-AlphaCoV/SC2013 |
| 163 | NC_030292 | 28434 | Mustela putorius | α-coronavirus | Ferret coronavirus isolate FRCoV-NL-2010 |
| 164 | NC_030886 | 30161 | Rousettus leschenaultii | β-coronavirus | Rousettus bat coronavirus isolate GCCDC1 356 |
| 165 | NC_032107 | 28363 | Triaenops afer | α-coronavirus | NL63-related bat coronavirus strain BtKYNL63-9a |
| 166 | NC_032730 | 28763 | Rattus norvegicus | α-coronavirus | Lucheng Rn rat coronavirus |
| 167 | NC_034440 | 29642 | Pipistrellus | Unknown | Bat coronavirus isolate PREDICT/PDF-2180 |
| 168 | NC_034972 | 27682 | Apodemus chevrieri | α-coronavirus | Coronavirus AcCoV-JC34 |
| 169 | NC_035191 | 25995 | Suncus murinus | α-coronavirus | Wencheng Sm shrew coronavirus |
| 170 | NC_038294 | 30111 | Homo sapiens | β-coronavirus | β-coronavirus England 1 |
| 171 | NC_038861 | 28586 | Sus scrofa | α-coronavirus | Transmissible gastroenteritis virus genomic RNA |
| 172 | NC_039207 | 30148 | Erinaceus europaeus | β-coronavirus | β-coronavirus Erinaceus/VMC/DEU/2012 |
| 173 | NC_039208 | 25425 | Sus scrofa | δ -coronavirus | Porcine coronavirus HKU15 strain HKU15-155 |
For each type of structural protein, a matrix of distance between amino acid sequences was normalized to the maximum from the combined perspective of rows and columns. The normalized score of each cell depends on the respective row and column maxima simultaneously (mean fraction). So, final values fall symmetrically within the unit interval [0, 1]. The correlation between matrices of distances was studied via Mantel’s test. A single matrix of distances between viral units was obtained as a linear combination of the normalized distances between involved proteins. We construct a proximity network from such a unified matrix of viral distance. This proximity network refers to the minimum spanning tree that connects all sampled viruses at the minimum cost (i.e., the sum of distances across edges is minimized). The proximity network encodes the backbone of the similarity relationships between studied items.
After identifying the set of hosts defined to the highest possible degree of taxonomic resolution, we focused on the phylogenetic tree subtended by them. The phylogeny for the vertebrate hosts was recovered from the VertLife dataset at http://vertlife.org (mammals: Upham, Esselstyn & Jetz, 2019; birds: Jetz et al., 2012). Branch lengths of the tree were computed using the method of Grafen (1989). The distance to root is set to 1. The patristic distance was adopted as a measure of phylogenetic distance between terminals, namely the sum of the lengths of the branches that link two taxa at the leaves of the tree. The phylogenetic diversity associated with a set of taxa was derived from the average value of pairwise patristic distances. Then, we match any available host with the pertinent phylogenetic node, either a terminal or an inner node, depending on the taxonomic resolution of the item. This step is necessary for calculating later the phylogenetic distance between hosts. Computational null experiments were run to assess the coupled information between the phylogeny of hosts and the configuration of similarities between viral entities.
Chimerism was theoretically studied. Structural proteins of the same kind are here called homotopic (e.g., orthologous pairs Sx − Sy, Ex-Ey, Nx-Ny, Mx-My coming from viral sources x and y), otherwise they are called heterotopic (non-orthologous pairs Sx-Ey; Sx-Ny; Sy-Mx and all possible cross combinations). We can establish the distances among homotopic proteins, but this cannot be done among heterotopic ones. Therefore, we used the distances among homotopic proteins to infer the associations between the heterotopic ones, based on the crossed distances they have with the respective homotopic proteins. The inferred associations between the heterotopic proteins are defined as heterotopic disaffinity.
Whenever high correlation among matrices of distances is detected, the next assumption would gain support: Similar homotopic proteins (low distance between them) are likely exchangeable in the assembly they occur. So, for two heterotopic proteins recorded in different virions, their feasibility of being combined into a new theoretical structure can be estimated from the similarity between homotopic elements of the virions where they are actually embedded into. We will refer to this as the interchangeability property of structural proteins (IPSP). The lower the heterotopic disaffinity between a pair of proteins, the larger the chance of being integrated into a common viral assemblage. As a corollary of this statement, chimerism understood as a mosaic of proteins already recognized in distant viruses, is hardly expected to occur in nature. The average of pairwise disaffinities in a tetrameric assembly estimates its degree of chimerality. Using combinatorial simulations, we study the behavior of the coefficient of chimerality in mixtures of proteins randomly drawn from the empirical sets already compiled by us.
Finally, we assess the co-structure between phylogeny and chimerism in our dataset. We run computational experiments of chimeras (combinatorial urn models) that represent our theoretical morphospace and we study their association with the content of phylogenetic information. We draw 100,000 proteins of classes E, M, N, and S from the respective urns. Then, we assemble them in tetrads. In parallel, we calculate the phylogenetic diversity associated with the pool of hosts in which the sampled proteins were observed to occur. All statistical tests, analyses, and graphics were carried out with the R software (R Core Team, 2020, version 4.0.0). See supplementary material: https://github.com/GFontanarrosa/Viral-Morphospace-Dos-Santos-et-al/blob/main/Coronavirinae_complete_analysis.R.
Results
Basic information about amino acid sequences of the major structural proteins (E, M, N, and S) is reported. Proteins can be strictly ordered by length, i.e., E < M < N < S, across the entire sample of sequences (Table 2). A significant correlation was detected among the four matrices of distances (P < 0.01, Mantel’s test), suggesting that they are congruent regarding the configuration of pairwise similarities between data points. Correlation scores are consistently higher than 0.7 (Table 3). This finding suggests the average distance between proteins serves as a proxy to assess the dissimilarity between virions as a whole.
| E protein | M protein | N protein | S protein | |
|---|---|---|---|---|
| Tukey Five-Number Summaries | [65, 76, 82, 83, 109] | [185, 221, 222, 230, 268] | [342, 379, 421, 448, 470] | [1126, 1241, 1324, 1363, 1472] |
| Mean (SD) | 80.7 (4.8) | 229.9 (15.8) | 413.5 (35.5) | 1308.7 (91.2) |
| E | M | N | S | |
|---|---|---|---|---|
| E | – | 0.83 | 0.82 | 0.74 |
| M | – | – | 0.87 | 0.80 |
| N | – | – | – | 0.79 |
| S | – | – | – | – |
Figure 1 depicts the minimum spanning tree (proximity network) of Coronavirinae isolates. Topologically, the four genera of Coronavirinae could be segregated into connected components after removing the only inter-genera links. The α-CoVs lie always at the intermediacy along the shortest paths connecting β-CoVs with the remaining coronaviruses genera. The two unknown items (nodes 167 and 145) are located within the β-coronavirus set. SARS-CoV-2 is located at just one-step distance from the network periphery and lies between the RaTG13 (the peripheral node 133) and the pangolin-CoV-2020 (the inner node 134) (Fig. 1). In terms of protein distances, SARS-CoV-2 is consistently closer to RaTG13 than any other sequenced element. Additional information about nodes is displayed in Table 1.
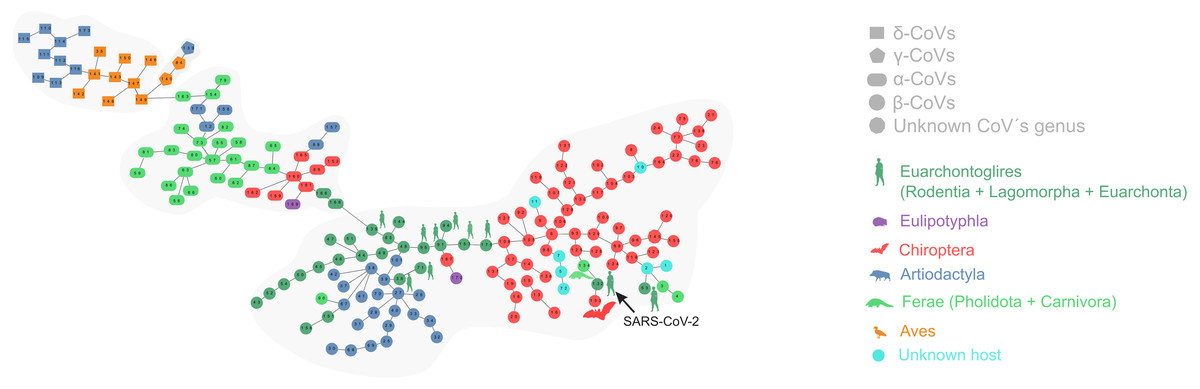
Figure 1: Proximity network spanning over 173 samples from the group Coronavirinae of viruses.
The four distinct CoV genera can be easily segregated after removing the unique between-genera links, and are highlighted through a gray halo. Nodes have been colored by clade membership of host in which virus was isolated. SARS-CoV-2 and adjacent nodes have been tagged with the respective host icon. Human silhouette was also added to all those viruses infecting humans. Note the overall co-structure between viral proteome distance and phylogenetic distance of respective hosts, leading to a broad agreement between connected clusters of CoV genera and host clades. Additional information about nodes of the network are available in Table 1. Silhouette images were freely obtained from http://phylopic.org/.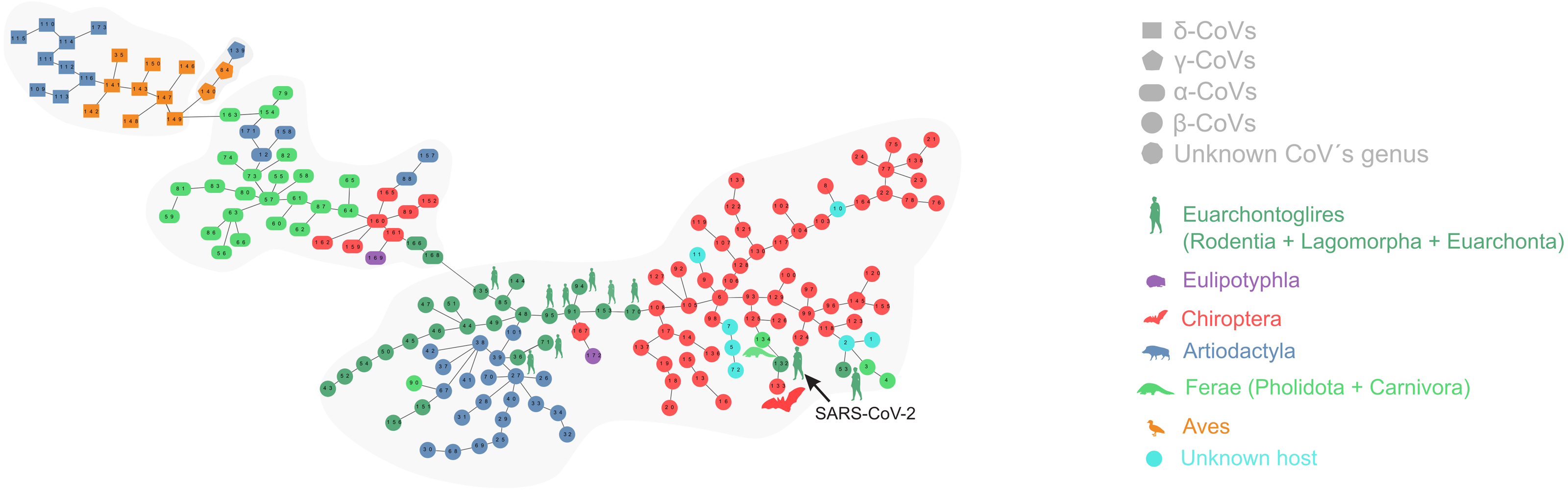{kind=link}
The minimal set of links of MST grasps the skeleton of relationships between viral samples. It synthesizes the structure of similarities held by data. Focusing on it, the main patterns are easily recognized and hypothesis generation becomes facilitated. In observing the network links of Fig. 1 with nodes tagged with the respective host, we track the phylogenetic relatedness between pairs of hosts across the total set of links (Fig. 2). In Fig. 2, each link is represented like a parabolic arc between hosts at the terminals in the phylogeny. The height of the parabola is dictated by the distance between nodes of Fig.1. A minor proportion of links (14%) bridge less similar viruses (distance > 0.1) and are frequently associated with weakly related hosts (0.65 ± 0.13, mean ± standard error of patristic distance). The staircase pattern of parabolic arcs shown by birds is eloquent in this regard (Fig. 2). We test this claim through randomization. We run 10,000 random experiments of host allocation in the same network or backbone of proximity relationships. Phylogenetic distance between neighboring hosts increases steeply in the random scenarios (Fig. 3). The observed distribution of hosts across the proximity network is compact in phylogenetic terms. In general, hosts of very similar viruses are also close phylogenetically.
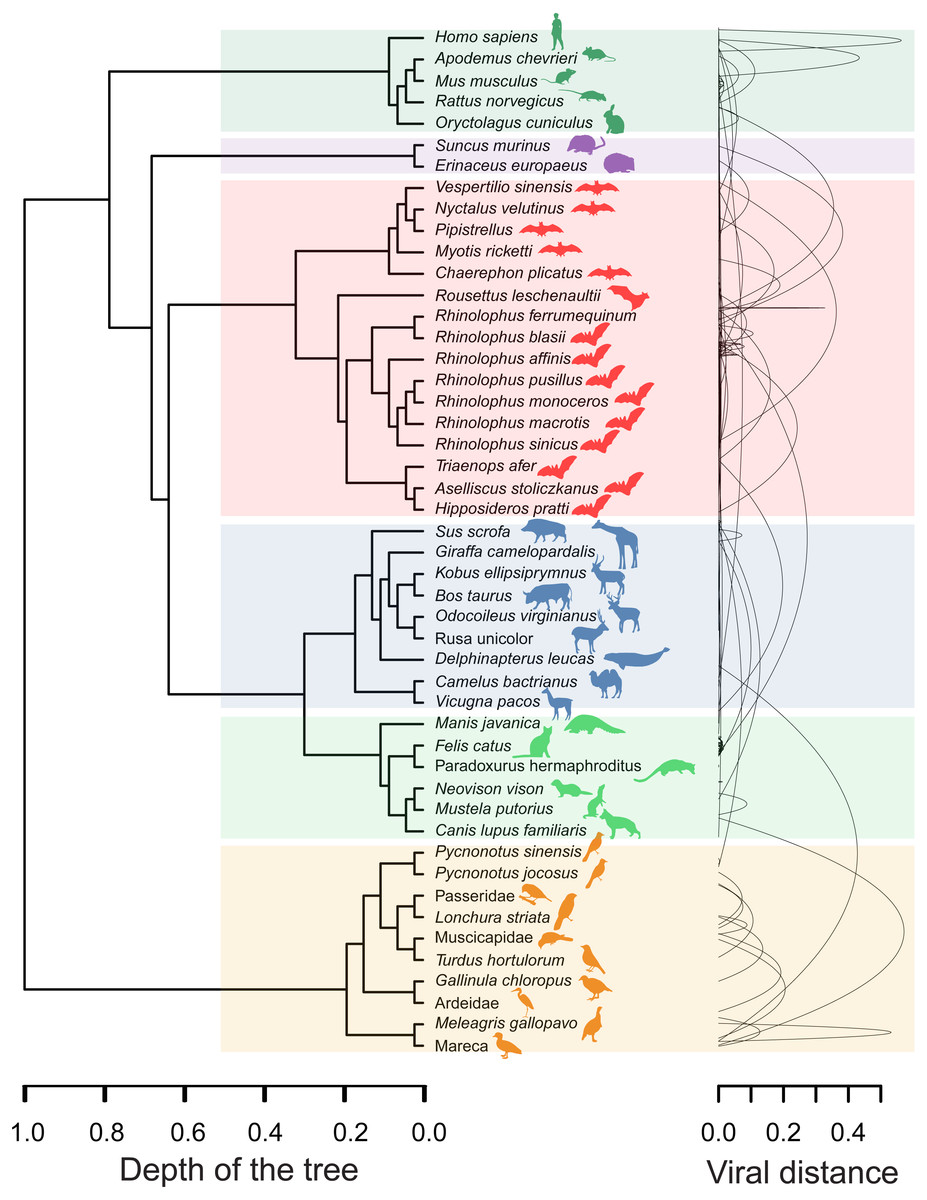
Figure 2: Graphical representation of hosts associated with the endpoints of links in the proximity network of Fig. 1.
To the left, phylogenetic tree of involved hosts. To the right, links/edges of proximity network represented as parabolic arcs bridging the hosts associated with endpoints of such links. The height of arcs correspond to the distance between nodes/virus connected by the respective link, so that flat arcs represent links between similar viruses whereas bumpy arcs join dissimilar ones. All taxa from the main clades (highlighted through transparent rectangles) retrieve always a between-clade patristic distance larger than unity (>1.0). Silhouette images were freely obtained from http://phylopic.org/.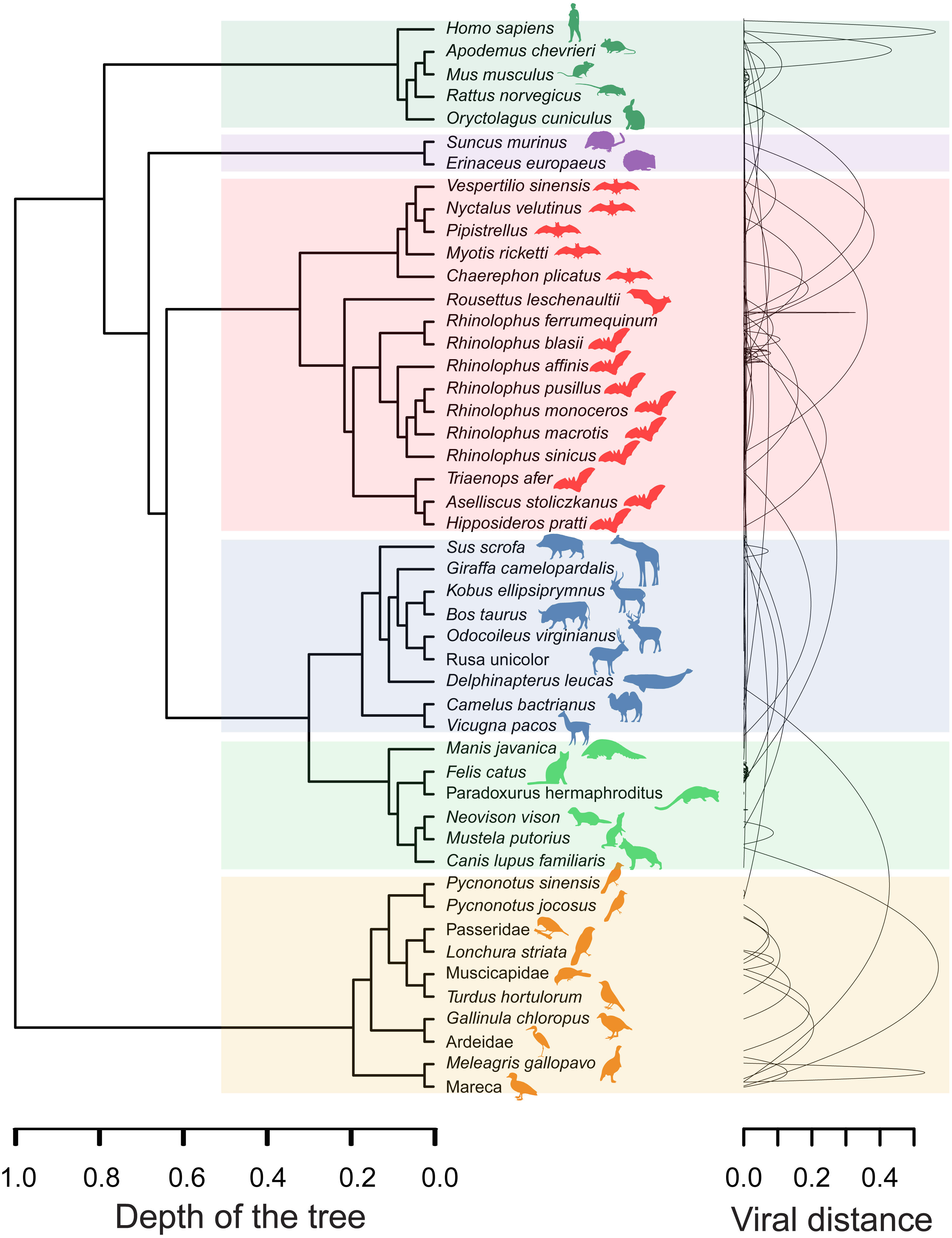{kind=link}
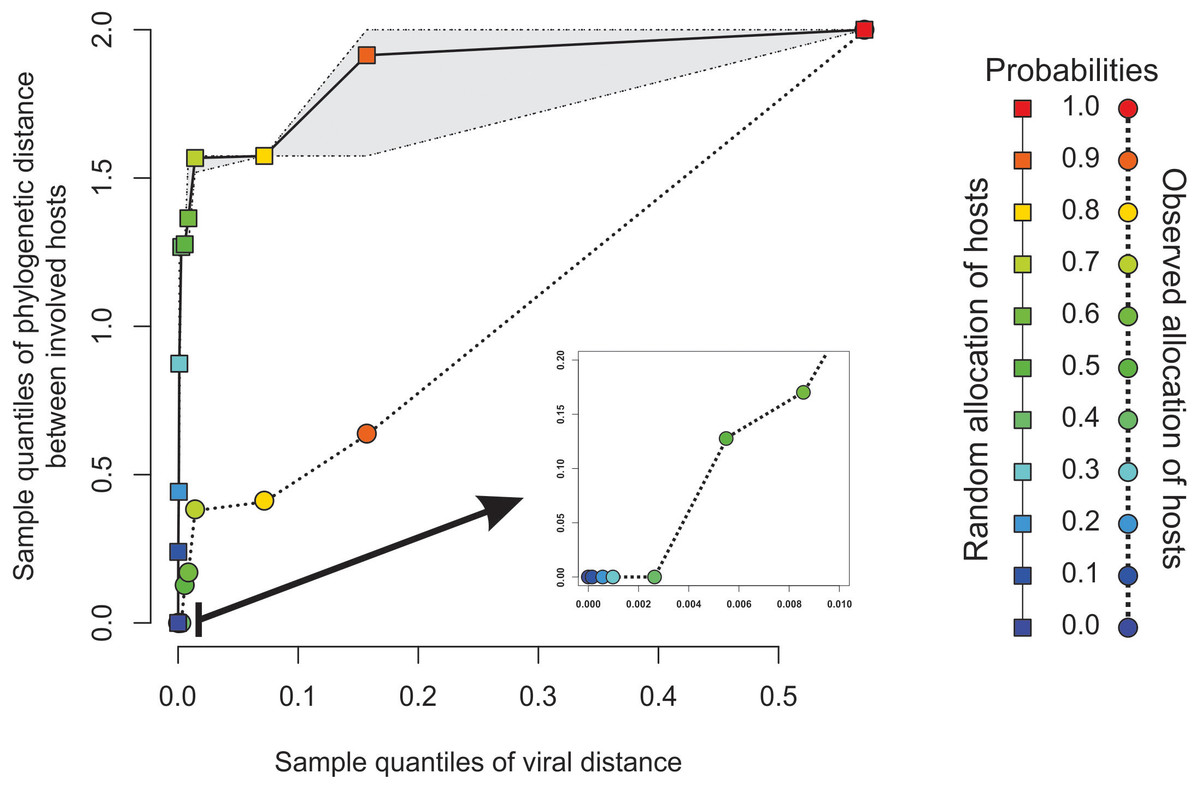
Figure 3: Expected phylogenetic distance between hosts under random scenarios of host allocation on the same proximity network represented in Fig. 1.
Quantiles of viral distance are plotted against quantiles of phylogenetic distance between hosts. Dotted polyline, the observed distribution of values. Solid polyline, values obtained after randomization. The 95% confidence interval is drawn around this last line. Departure of observed values from randomness indicates that hosts of viruses directly connected in the proximity network tends to be closely related.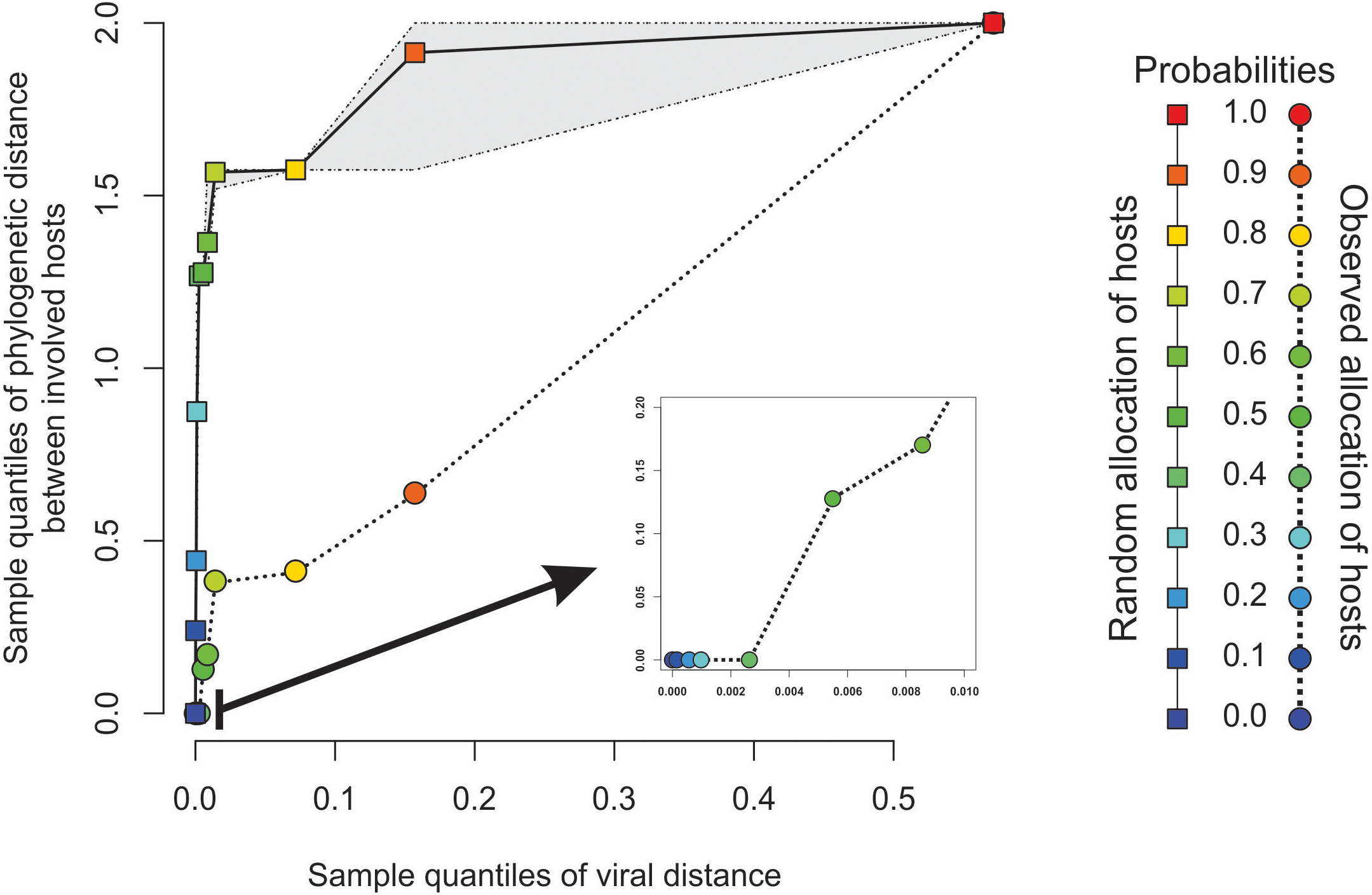{kind=link}
Figures 4A–4B depict, in a didactic way, the flow work leading to the calculation of chimerality. Figure 4A shows three hypothetical viral configurations. The heterotopic disaffinity is calculated from the distance between involved homotopic proteins. Thus, for instance, the heterotopic disaffinity between protein M from the leftmost viral configuration and the s protein from the rightmost one comes from the mean distance between the respective homotopic partners (i.e., distances M-m and S-s). Figure 4B shows the 81 possible tetrads (hypothetical theoretical morphospace) obtained by a free combination of protein precursors. This figure highlights both the heterotopic disaffinity between pairs of proteins of each configuration and the chimerality coefficient of the whole configuration.
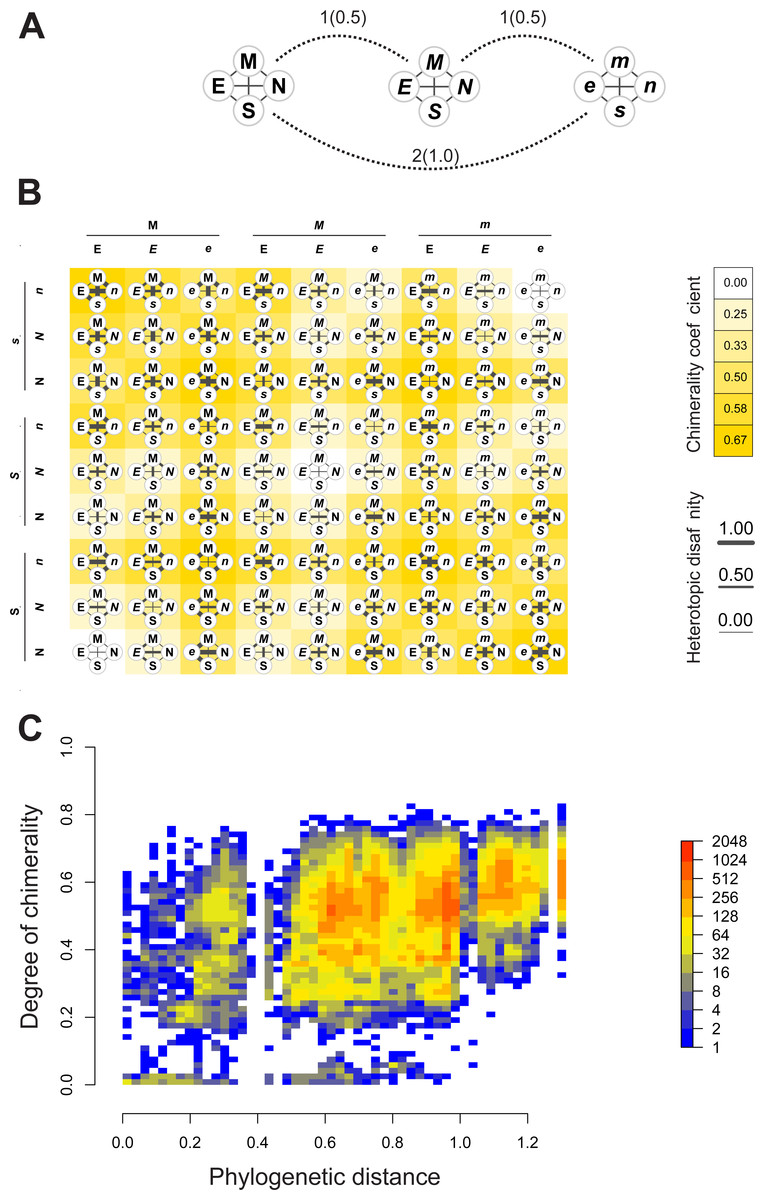
Figure 4: Computational experiments of chimera compositions.
Didactic introduction to concepts (A–B) in addition to results from such experiments (C) applied on our real data. (A) Three hypothetical tetrads of structural proteins coming from three different viruses. The distance between them are indicated (normalized values to the maximum between brackets). Here, the distance denotes the amount of differences in the attributes of letters used to label the protein (upper/lowercase; normal/italics). (B) Showing all the possible combinations of proteins from the above hypothetical viral sources. Heterotopic disaffinity between pairs of distinct proteins is inferred from the distance between proteins of the same kind of the viral precursors. For any assembly, the degree of chimerality is the average heterotopic disaffinity.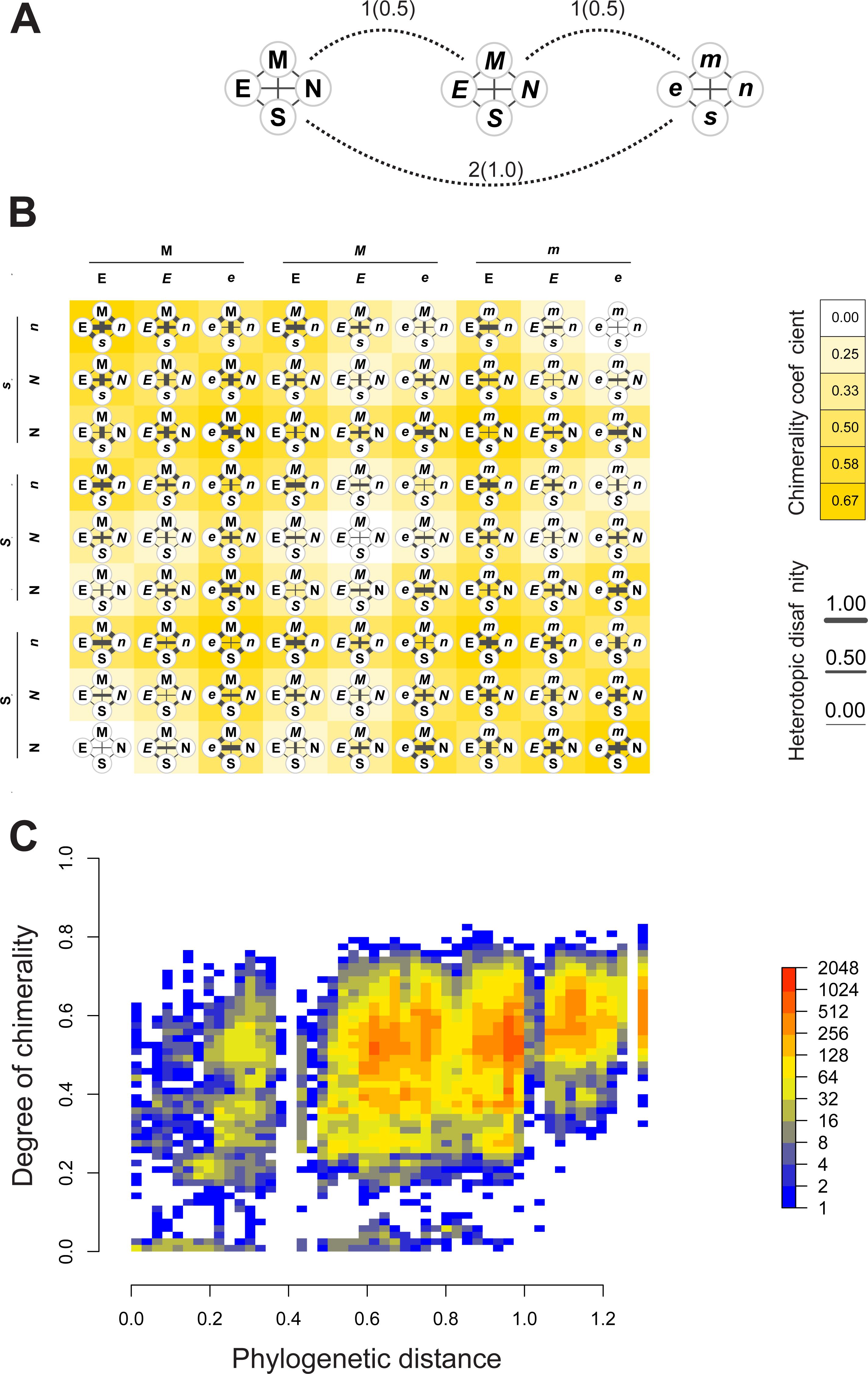{kind=link}
Results of computational experiments of chimeras are plotted in Fig. 4C. It shows the dispersion of both chimerality and phylogenetic diversity of hosts in the random set of tetrads. The frequency of observations is represented through a heatmap. Noticeably, the rarest event is to find simulated viruses that jointly exhibit high intrinsic phylogenetic diversity and low coefficient of chimerality. Since different viruses can be recognized in closely related hosts, it is possible to achieve tetrads of high chimerism (low phylogenetic diversity, high coefficient of chimerality). On the contrary, it is rather difficult to find similar viruses in loosely related organisms (high phylogenetic diversity, low coefficient of chimerality).
Discussion
Our analysis of structural proteins recovered both the four viral genera (α, β, γ, and δ) and SARS-CoV-2 affinities with viruses isolated from bats and pangolins. The approach is useful to address issues of taxonomic classification such as positioning of unknown items. Rapid classification of new viruses is a topic of great concern since it contributes for strategic planning, containment, and treatment (Randhawa et al., 2020). In the proximity network, the β-coronavirus and α-coronavirus sets are neighbors. The β-coronavirus set is structured into two subgroups that are bridged by a sequence of the nodes 95-91-153-170, all isolated from Homo sapiens. Almost all the viruses that infect human hosts in our sample are distributed in the left subgroup regarding node 170, with the exception of SARS-CoV-2 (node 132) and the SarsCovP2 (node 53) which are located in the right subgroup. Considering the remarkable proteomic closeness among most viruses infecting humans (Fig. 1), it could be inferred that viruses located in the vicinity of SARS-CoV-2 are also potentially dangerous to humans. The fact that SARS-CoV-2 rather than their neighbors in the proximity network has emerged recently in the human population could be due to the degree of biogeographic and ecological isolation of its hosts or lack of opportunity (Segreto & Deigin, 2020; Solé & Elena, 2018). The limit between α-coronaviruses and β-coronaviruses is depicted by node 135, also a human parasite. Viruses historically infecting a wide range of vertebrate hosts seem to be converging to infect humans. Human explosive demography jointly with human-driven changes as bringing in close contact farm animals and crops with wild animals and plants are the triggers of viral evolution and spillovers (Woolhouse, Taylor & Haydon, 2001). Notwithstanding, considerations about the bridging role of humans in diversification of β-coronavirus should be taken with caution because of biasing in datasets (e.g., NCBI Virus) towards viral sequence from isolates infecting humans.
The RaTG13 (node 133 isolated from Rhinolophus affinis), previously identified as the closest known relative of SARS-CoV-2 based on genome similarity (Cyranoski, 2020; Zhang & Holmes, 2020; Zhang et al., 2020), is located peripherally in the β-coronavirus set and is the immediate neighbor of the SARS-CoV-2. The two unknown items presumably belong to the genus β-coronavirus based on the membership of their local neighborhood in the network (Fig. 1). Node 134 (isolated from Manis javanica) is also connected with SARS-CoV-2 but showing an inner location within the network. The subset composed of RaTG13, SARS-CoV-2, and pangolin-CoV-2020 is in turn located peripherally in the main network. The peripheral position of RatG13 may be related to its isolated evolution in the Yunnan’s caves (Southern China) where R. affinis inhabits. Accessibility to these caves for researchers did not occur until recently (Segreto & Deigin, 2020).
Coronaviruses infect a range of mammalian and avian species (Latinne, Hu & Olival, 2020). Within them, α-coronaviruses are able to switch hosts more frequently and between more distantly related taxa than β-coronaviruses. These last are specialist strategists infecting mainly bats and also other mammalian species such as humans, camelids, and leporids (Figs. 1 and 2). Nevertheless, the emergence of SARS-CoV-2 suggests a jump between phylogenetically distant hosts, allowed by modifications in the RBD that make it more virulent and host-specific for humans. This modification enables a new range of potential hosts for SARS-CoV-2 (hosts phylogenetically related to humans and domestic and farm animals that co-inhabit with humans).
Our results showed that more similar viruses tend to infect the most phylogenetically related hosts displaying a specialist strategy (Fig. 2). This result is reinforced by the randomized simulations here performed (Fig. 3). Longdon et al. (2011) found evidence that most host shifts occur between closely related hosts, and that the host phylogeny could explain most of the variation in viral replication and persistence. Viruses that co-evolved with a certain species of vertebrates have developed host-specific mechanisms to infect it. This adaptation will be more likely co-opted as an exaptation to jump into a host species closely related to the host in which the virus evolved (Latinne, Hu & Olival, 2020). This specialist strategy is held by the majority of viruses (Solé & Elena, 2018) and represents an ecological constraint on the virus-host available set. However, there are also viruses separated by long distances infecting closely related hosts such as Mareca sp. and Meleagris sp. In this case, the distance is of 0.53 between viruses and belong to δ- and γ- genera, respectively. On the contrary, there are a few viruses with shorter distances infecting distantly related hosts, as in the case of Homo sapiens and Rhinolophus affinis. Succinctly, results show: (1) The minimum spanning network recapitulates the known phylogeny of Coronovirinae, and (2) some concordance is found between host phylogeny and viral genetic distance. With a few exceptions, this result suggests that the overall pattern is not one of frequent host shifts.
Since bats are natural reservoirs for several coronaviruses that can potentially infect humans (Woo et al., 2012), their viruses have been deeply studied and even researchers have been using them to generate chimera coronaviruses for the last 20 years (Segreto & Deigin, 2020). Laboratory chimeras were meant to simulate recombination events that might occur in nature (Menachery et al., 2015). Thus, even when a chimera virus is detected, the distinction between natural and artificial chimeras represent another challenging step. We use the IPSP to obtain the different possibilities of theoretical viruses and relate them to the degree of chimerality. The larger the amount of interactions between proteins coming from dissimilar virus sources, the larger the chimerality of that particular assemblage in the sense of decreasing chance for observing it in nature (lower feasibility). Our results on chimeral virus simulations (Fig. 4C) showed a non-trivial fill of the theoretical morphospace. Whenever protein precursors come from phylogenetically distant hosts, chimerality is expected to achieve high values in the sense of a global entity composed of dissimilar, heterogeneous parts. The relevance of this approach is that it gives us clues to assess the chimeral origin of coronaviruses. To inquire about a potential chimera origin of a certain sampled virus, we can compare it with the viruses belonging to the empirical morphospace and the theoretical morphospace. If our focal virus turns out to be more similar to a theoretical chimera virus (belonging to the theoretical morphospace) than to an observed one (empirical), then the suspicion about a chimera origin increases.
Based on the aforementioned insights, our analysis cannot support hypothesis number 2, since SARS-CoV-2 does not have all the features deemed to be chimeric using just information about amino acid sequences. Even though the chimerism origin theory is consistent with the remarkable proteomic closeness between RaTG13, SARS-CoV-2 and pangolin-CoV-2020, also found in this work, we also observe that when extracting the SARS-CoV-2 from Fig. 1, pangolin-CoV-2020 and RaTG13 do not undergo modifications in their network locations. Andersen et al. (2020) also state that the genetic data irrefutably show that SARS-CoV-2 is not derived from any previously backbone used in chimeras assemblage. Our approach represents a tool that could guide researchers to detect chimerality.
After phylogenetic genome-wide analysis, most studies indicated that Rhinolophus bats may be the natural host of the novel coronavirus (Ma et al., 2021). However, these results should be critically appraised since classic dichotomic phylogenetic tools do not handle recombination well and results could be misleading if recombination occurs (Goh et al., 2022; Posada, 2000). In order to deal with these constraints, Goh et al. (2022) suggest narrowing the phylogenetic analysis to conserved proteins such as the M protein. In doing so, a different tale emerges and pangolin is no longer so easily dismissed as ancestor. We also consider that network and combinatorial approaches can be useful to address issues of recombination. Thus, the intermediary position of SARS-CoV-2 between pangolin and bat calls for a care consideration. New lineages as a result of blending of loosely related predecessors, for instance symbionts or hybrids, pose a challenge for classic phylogenetic reconstruction. Alternatively, phylogenetic networks allow investigation of complex evolutionary histories that involve cross-species gene transfer (Albrecht et al., 2012). On the other hand, combinatorics under a morphospace research program shed light on how likely an entity can occur in nature. Our proposal then expands the repertoire of biocomputational resources to gain a deeper understanding of evolution of items through events other than cladogenesis or speciation.
Conclusions
Since WHO declared the COVID-19 outbreak a pandemic on March 11, 2020, an unprecedented multidisciplinary interest in the responsible coronavirus exploded from all around the world at the same time. One approach to gain a better comprehension of it is by zooming in their structural details. Another approach consists of zooming out and achieving the big picture of Coronavirinae as a whole. We provide a general framework to address issues of viral classification, assembly constraints, degree of chimerism, evolutionary paths, and putative chains of zoonotic jumps. We constructed a proximity network based on the four major structural proteins in coronavirus. Through this, we explored the relationship between host-phylogeny and viral proteomic distance. We also investigated the potential of generating feasible chimeras in nature from loosely related hosts through simulation. Finally, we brought attention to both the molecular and phylogenetic constraints behind the evolution of coronaviruses.