
Transcriptional and posttranscriptional regulation of Bacillus sp. CDB3 arsenic-resistance operon ars1
- Published
- Accepted
- Received
- Academic Editor
- Abhishek Kumar
- Subject Areas
- Microbiology, Molecular Biology
- Keywords
- Arsenic resistance, Operon expression, RNA degradation, Transcription attenuation
- Copyright
- © 2015 Yu et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2015. Transcriptional and posttranscriptional regulation of Bacillus sp. CDB3 arsenic-resistance operon ars1. PeerJ 3:e1230 https://doi.org/10.7717/peerj.1230
Abstract
Bacillus sp. CDB3 possesses a novel eight-gene ars cluster (ars1, arsRYCDATorf7orf8) with some unusual features in regard to expression regulation. This study demonstrated that the cluster is a single operon but can also produce a short three-gene arsRYC transcript. A hairpin structure formed by internal inverted repeats between arsC and arsD was shown to diminish the expression of the full operon, thereby probably acting as a transcription attenuator. A degradation product of the arsRYC transcript was also identified. Electrophoretic mobility shift analysis demonstrated that ArsR interacts with the ars1 promoter forming a protein-DNA complex that could be impaired by arsenite. However, no interaction was detected between ArsD and the ars1 promoter, suggesting that the CDB3 ArsD protein may not play a regulatory role. Compared to other ars gene clusters, regulation of the Bacillus sp. CDB3 ars1 operon is more complex. It represents another example of specific mRNA degradation in the transporter gene region and possibly the first case of attenuator-mediated regulation of ars operons.
Introduction
Arsenic compounds are widely dispersed in the environment, which poses a threat to all living organisms. Many bacteria have evolved arsenic resistance (ars) operons located on either chromosomes or plasmids, which encode a specific detoxification pathway for arsenic extrusion (Rosen, 2002; Kruger, Bertin & Heipieper, 2013).
The transcription pattern of a common three-gene ars operon (arsRBC) is relatively simple: intracellular arsenite binds to the repressor (ArsR), inducing a conformational change of the protein and dissociation from the promoter, thereby allowing expression to proceed (Shi, Wu & Rosen, 1994). The transcriptional regulation of a common five-gene ars operon (arsRBCDA) has also been well characterised and a model was proposed (Chen & Rosen, 1997). The initiation of operon transcription is the same as arsRBC; however, when the membrane transporter ArsB protein is largely expressed, it may become toxic to host bacterial cells. Therefore, transcription of the operon can be repressed via the second repressor ArsD binding to the regulatory region, to which ArsR binds (Bruhn et al., 1996), even though ArsR and ArsD share no significant sequence similarity (Chen & Rosen, 1997). With the increasing concentration of arsenite, ArsD tends to dissociate from operator region, re-activating transcription of ars operon. This mode employs a secondary repressor to guard the upper level of gene expression, but still only involves single transcript. Apart from these two well characterised regulatory mechanisms, other transcriptional modes have also emerged along with the discoveries of more ars operons. For example, the basic repressor ArsR is not always operon born. The arsBHC operon of Synechocystis sp. Strain PCC6803 missing transcriptional repressor gene has been proved to be mediated by an ArsR protein that is encoded away from the operon (López-Maury, Florencio & Reyes, 2003). Similarly, the Ochrobactrum tritici SCII24 arsR1C1YC2H operon is also regulated by another ArsR, which is constitutively expressed and resides outside the operon in opposite direction (Branco, Chung & Morais, 2008). Moreover, some bidirectional ars operons have been found mediated by single ArsR but expression levels of two divergent transcripts appear different in response to arsenic stress changes, such as the ars operons from Acidithiobacillus ferrooxidans (Butcher & Rawlings, 2002), Corynebacterium glutamicum ATCC13032 (Ordóñez et al., 2005), Microbacterium sp. Strain A33 (Achour-Rokbani et al., 2010) and pR478 of Serratia marsescens (Ryan & Colleran, 2002).
Bacillus sp. CDB3 is a highly arsenic-resistant bacterium, containing two ars clusters (Bhat et al., 2011). The ars2 is similar to the four-gene operon arsRorf2YC (Y represents yqcL) from the skin element of B. subtilis (Sato & Kobayashi, 1998), while ars1 is one of the longest ars clusters characterised to date, bearing eight genes (arsRYCDATorf7orf8) of which two are novel (Bhat et al., 2011). The orf7 gene encodes a putative HesB-like protein and orf8 a putative dual-specificity protein phosphatase (DSP). Assessed in E. coli host, orf8 exhibited a significant resistance to arsenate but it could not complement the function-loss of arsC gene, indicating its role not as an arsenate reductase (Zheng et al., 2013). In addition to these new genes, ars1 also displays features consistent with a novel regulation mechanism (Bhat et al., 2011). (1) Arrangement of the five genes (arsRYCDA) differs from the common five-gene ars operon (arsRDABC) in E. coli; (2) two inverted repeats exist between arsC and arsD potentially forming a hairpin structure, which may play a role in transcriptional regulation; and (3) the ArsD protein sequence lacks the C-terminal vicinal cysteine pairs which E. coli ArsD bears, bringing into question its role as a repressor regulator. In this study, we investigated the transcriptional pattern of CDB3 ars1 under varying arsenic stresses and assessed the function of potential regulatory factors.
Materials and Methods
Bacterial strains, plasmids and DNA primers
The bacterial strains, plasmids and primers used in this study are listed in Table 1. LB media with appropriate antibiotics or arsenic compounds were used to culture Bacillus sp. CDB3 at 30 °C or E. coli strains at 37 °C, respectively.
| Strains, plasmids and primers | Description | Source |
|---|---|---|
| Strains | ||
| Escherichia coli | ||
| JM109 | recA1, endA1, gyrA96, thi, hsdR17 (rk−, mk+), supE44, relA1, Δ(lac-proAB), [F’, traD36, proAB, lacIqZΔ M15] | Promega (USA) |
| AW3110 | K-12 F-IN (rrnD-rrnE) ars::cam (Cmr, the chromosomal arsRBC deleted) | Gift of Dr BP Rosen (Carlin et al., 1995) |
| M15 (pREP4) | naIs, strs, rifs, thi−, lac−, ara+, gal+, mtl−, F−, recA−, uvr+, lon+ | Promega (USA) |
| Bacillus sp. CDB3 | Isolated from cattle dip sites | Chopra et al. (2007) |
| Plasmids | ||
| pAR27 | pRYCDATorf7orf8, CDB3 ars1cloned in pGEM7Zf(+) | Bhat et al. (2011) |
| pQE30 | N-His6 expression vector, PT5/Olac, ColEI ori, Apr | Qiagen (Germany) |
| pQENR | pQE30/ArsR expression plasmid | This study |
| pQEND | pQE30/ArsD expression plasmid | This study |
| pAR27HPΔ | Mutant of pAR27 in the inverted repeat region between C and D | This study |
| Primers (5’-3’) | ||
| Probe 1F | CCATAACTTGCACCCAC | |
| Probe 1R | TTAACAAGAGTGTCACAG | |
| Probe 2F | GTCTGGAGCTTGCACAG | |
| Probe 2R | GGACTTCAGATTGTGATCGTC | |
| Probe 3F | TCTGCAATGGAAGAAGC | |
| Probe 3R | GTCTAGACTGTTTCTGTGACATC | |
| ProbeR-F | TTAACAAGAGTGTCACAG | |
| ProbeR-R | CTCTGTTTGGTCTGGTAC | |
| ProbeC-F | TTACCATTGACCGTGAC | |
| ProbeC-R | TCTCCTGTTTCAGCGAAT | |
| RT-DF | CCGAGTGTTGACCCAGAGTT | |
| RT-DR | CATCTGGTTCACTTGCCAAA | |
| pQENR-Fa | (BamHI)CCAGGATCCATGGAAAAAACGGTTATAG | |
| pQENR-Ra | (HindIII)TATAAGCTTTTAACAACCACACCGGAG | |
| pQEND-Fa | (BamHI)CCAGGATCCATGAAGAAGATAGAAATTTTTGATCCTG | |
| pQEND-Ra | (HindIII)TATAAGCTTCTATTTTTTCACGTTTAACTTTAAACGTACGACTGG | |
| ProR-F | TTCAGTTGAATATATAAGCG | |
| ProR-R | TATCTAGAGCCATATCTATACCTCCTT | |
| ProN-F | TGAGACTGCTATGAAAG | |
| ProN-R | GTCTAGACTGTTTCTGTGACATC | |
| PED-Rb | ACATTAATACAGGTCCAATAATCCAGTTT | |
Northern analysis
Cell pellets of appropriately treated cultures at mid-exponential growth phase were suspended in NETS buffer (0.1 M NaCl, 10 mM Tris-HCl pH 8.0, 1 mM EDTA, 1% SDS) followed by phenol-chloroform extraction. Total RNA in the aqueous phase was collected by ethanol precipitation and then redissolved in RNase-free water. Probes were produced from pAR27 by PCR amplification using a DIG PCR labelling kit (Roche, Penzberg, Germany) following manufacturer’s protocol. For northern blotting, 10 µg of total RNA was loaded per lane and run in 1.8% (w/v) agarose denaturing formaldehyde gels in MOPS buffer (20 mM MOPS, 5 mM sodium acetate, 10 mM EDTA, pH 7.0) before being transferred onto nylon membranes (Hybond™-N+; Amersham plc, Amersham, UK). Prehybridization, hybridization, washes and film (Kodak, Rochester, New York, USA) image development were conducted as described in the product manuals.
Real-time qPCR and primer extension assay
Total RNA samples isolated above were treated with RQ1 RNase-free DNase (Promega, Madison, Wisconsin, USA) before cDNA synthesis using a Transcriptor First Strand cDNA Synthesis kit from Roche following manufacturer’s instruction. Real-time qPCR to quantify arsD cDNA with primer pair RT-DF/RT-DR was carried out using a SensiFAST™ SYBR No-ROX kit (BioLine, Taunton, Massachusetts, USA) on a LightCycler@480 detection system (Roche, Penzberg, Germany). The PCR conditions were: one cycle of 95 °C for 5 min; 45 cycles of 95 °C for 10 s, 60 °C for 20 s and 72 °C for 10 s. Each run was followed by a melt analysis comprising a 2.2 °C/s ramp rate from 95 °C to 65 °C. The cDNA was quantified using a Nanodrop® ND-1000 spectrophotometer.
Primer PED-R labelled with 5’-end 5-6-carboxyfluorescein (6FAM) was used in reverse transcription before the resultant cDNA products were examined using an ABI® 3130xl Genetic Analyzer (Applied Biosystem, Carlsbad, California, USA) with GeneScan™-500 LIZ® internal size standards (Applied Biosystem, Carlsbad, California, USA) and analysed using GeneMapper® software (Version 4.0).
Construction of hairpin mutant strain
To eliminate the inverted repeats between arsC and arsD, a mutant 638-bp DNA was synthesised (GeneArt, Bavaria, Germany) to replace the original KpnI-RsrII fragment in pAR27. The resultant plasmid pAR27HPΔ was introduced into E. coli AW3110 for RNA assays.
Electrophoretic mobility shift assay
The coding regions of arsR and arsD were amplified from pAR27 and cloned into pQE30 (Qiagen, Hilden, Germany) at BamHI and HindIII sites resulting in His-tag recombinant vectors pQENR and pQEND, respectively. Upon sequence verification, the two constructs were transformed into E. coli strain M15 followed by induction at 37 °C for 4 h using 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cell pellets were lysed by French press (SLM Aminco, Rochester, New York, USA) at 20,000 psi. The target recombinant proteins were isolated and purified from the filtered lysates using HisTrap™ HP, HiLoad16/60 Superdex75 and Vivaspin 10 kDa MWCO columns (GE healthcare, Wauwatosa, Wisconsin, USA) and then stored in small aliquots at −80 °C before use.
A 167-bp fragment covering the promoter region of ars1 was PCR amplified using primer pair ProR-F/ProR-R from plasmid pAR27 and named proR. Another 132-bp fragment within orf7 was also amplified using ProN-F/ProN-R as a non-specific DNA control named proN. After purification by Sephadex G50 spin column chromatography (Sambrook, Fritsch & Maniatis, 1989), the PCR products were incubated with the above purified His-ArsR or His-ArsD proteins in binding buffer (10 mM Tris-HCl pH 7.6, 80 mM KCl, 0.2 mM EDTA, 0.2 mM dithiothreitol, 10% glycerol and 0.75 µM BSA; 30 µL) on ice for 30 min. The samples were then loaded onto a 6% non-denaturing acrylamide gel and run at 120 V for 40 min in TBE buffer (pH 8.3) before staining with ethidium bromide.
Results and Discussion
Transcript analysis of CDB3 ars1
To examine the transcription pattern of CDB3 ars1, total RNA extracts from arsenite-stressed and control bacterial cells were first analyzed by northern blot analysis using three probes, covering different regions of the cluster (Fig. 1A). While there was no hybridization signal detected in the control RNA sample, arsenite treatment induced the expression of ars1 and a 5.8-kb transcript corresponding to the full cluster length was detected by all three probes (Fig. 1B). This suggests that arsRYCDATorf7orf8 can be transcribed as a single polycistronic mRNA and that there is only one promoter at the 5’-end of the cluster. In addition, probe 1 (covering region RY), but not probes 2 and 3 (covering A and Torf7, respectively), detected two other RNA fragments of approximately 1.9 kb and 1.5 kb. A further northern blot assay (Fig. 1C) demonstrated that probes R and C (covering internal fragments of arsR and arsC, respectively) can both hybridize to the 1.9-kb RNA band, corresponding to the length of arsRYC but the 1.5-kb RNA band was only detected when probe C was applied. It was also observed that the amount of 1.5-kb RNA increased with treatment time, which is evident in the 4 mM arsenite samples: at 3 min the 1.5-kb RNA was not detected but was remarkably abundant after 30 min (Fig. 1D). This suggests that the first three genes, arsRYC, can also be transcribed as a polycistronic unit during ars1 expression and that the inverted repeat between arsC and arsD may function as a read-through transcriptional attenuator. Additionally, the 1.5-kb RNA band was found to lack the arsR gene compared with the 1.9-kb RNA fragment. Thus, it was assumed to be a possible differential mRNA degradation product spanning the YC gene region.
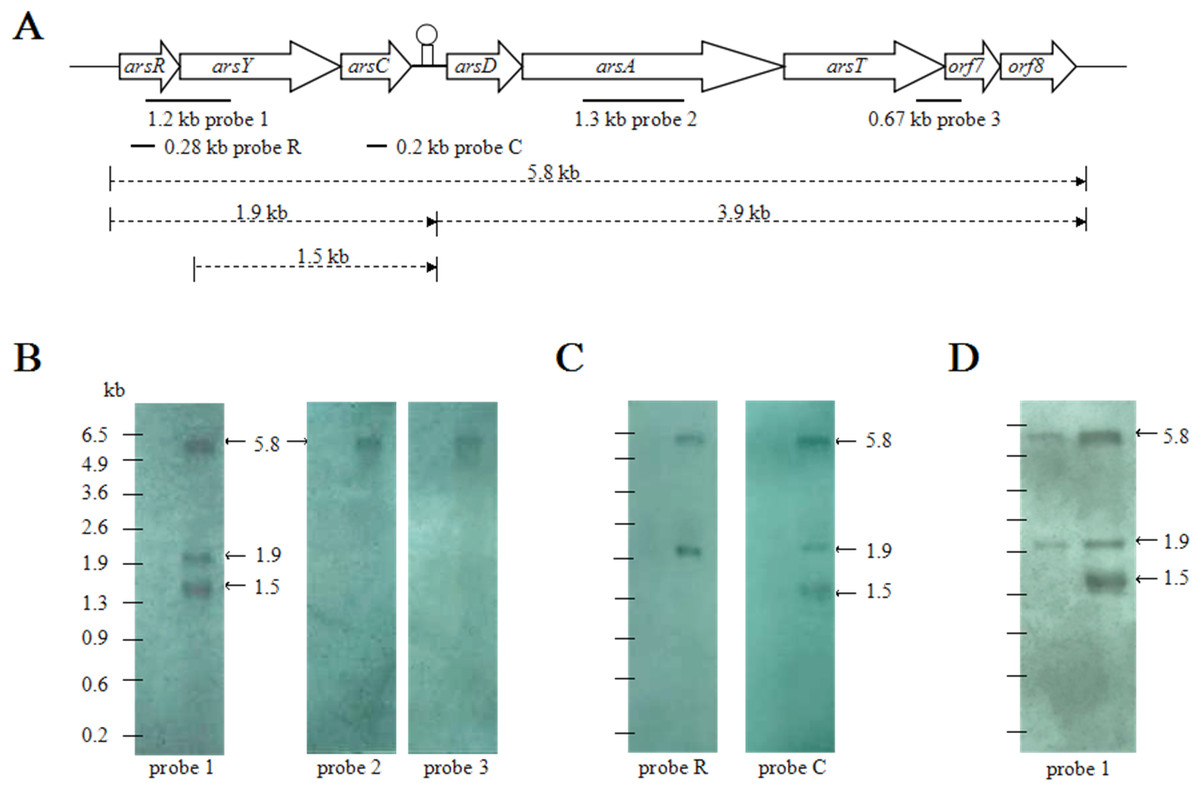
Figure 1: Northern blotting analyses of CDB3 ars1 expression.
(A) A diagram of CDB3 ars1 labelled with locations of probes used and possible transcript sizes (kb). (B) Images of northern blot ting using probes 1, 2 and 3. On each blot the left lane is untreated control and right is arsenite-treated at 2 mM/10 min. (C) Images of northern blot ting using probes R and C. On each blot the left lane is untreated control and right is arsenite-treated at 0.5 mM/5 min. (D) Images of northern blot ting using probe 1 of samples treated with 4 mM arsenite for 3 min (left) and 30 min (right).{kind=link}
Function of the internal hairpin structure
To examine the role of inverted repeats between arsC and arsD, which can form a hairpin structure, the intergenic DNA sequence was altered by mutagenesis to eliminate the repeats (Fig. 2A). A transcription study was carried out in E. coli AW3110, which was shown to produce the two RNA transcripts when transformed by CDB3 ars1 (data not shown). RNA samples isolated from AW3110 strains harbouring pAR27 (ars1) or pAR27HPΔ (hairpin knockout mutant) with or without arsenite treatment were reverse transcribed and analysed using qPCR assay. The results showed that in the mutant strain, expression levels of the long eight-gene transcript increased more than one fold over that in the control strain (Fig. 2B), suggesting that the hairpin structure does function as a transcriptional terminator which can be read through under some conditions.

Figure 2: mRNA secondary structure and long-transcript levels.
(A) Predicted mRNA secondary structure of the C–D intergenic region in CDB3 ars1 and altered nucleotides to abolish the hairpin structure. The four regions of the two stem-loops are indicated by symbols I, II, III and IV. (B) Expression levels of the long transcript in untreated (control) and arsenite-treated E. coli AW3110 strains harbouring pAR27 or pAR27HPΔ. The arsenite treatment was at 0.5 mM for 5 min and qPCR was carried out using RT-DF/RT-DR primer pairs. Each data point corresponds to average copies of ArsD cDNA (copies/µL) and the error bars indicate standard deviation of three independent measurements.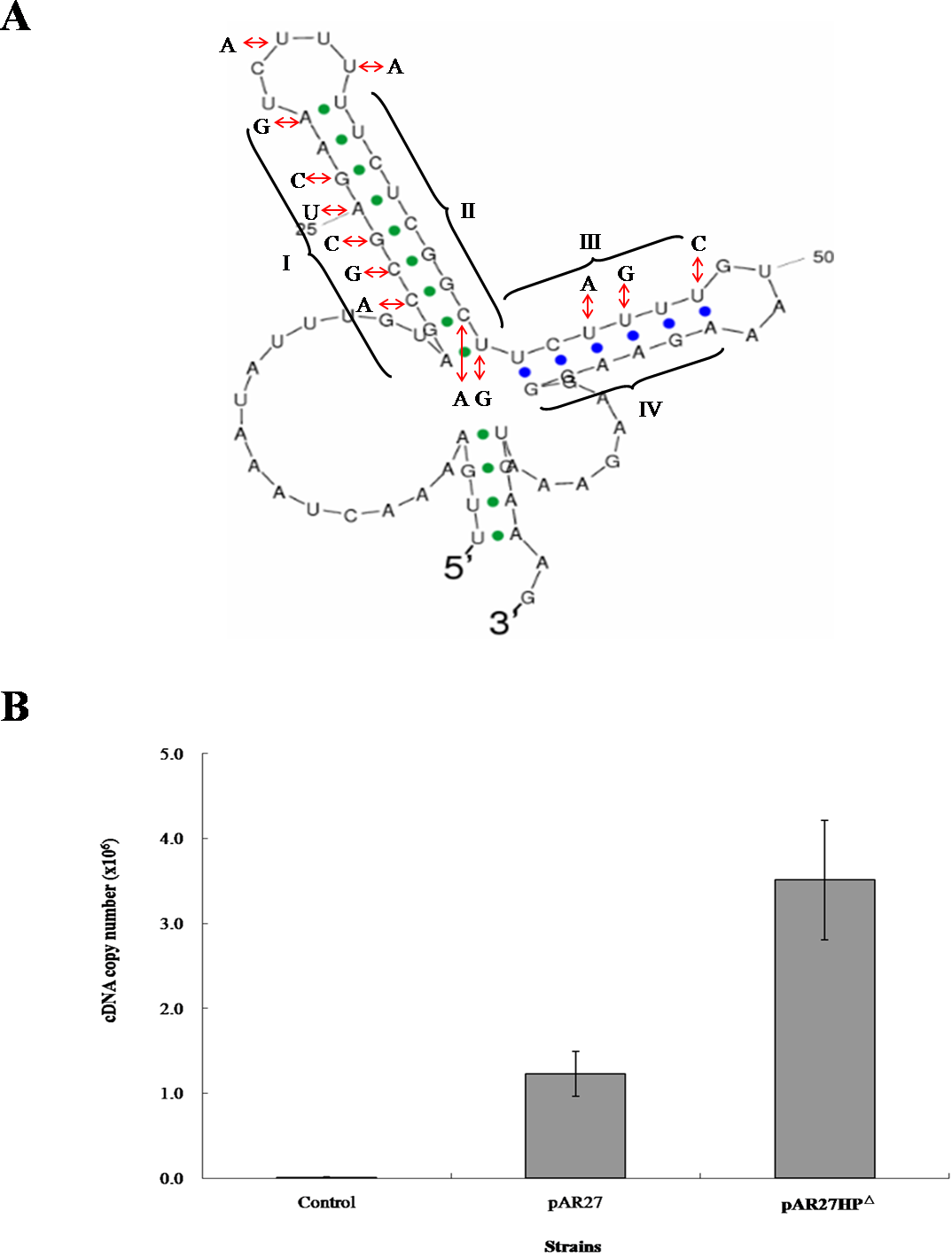{kind=link}
Transcription attenuation has been recognised as a strategy in bacteria to modulate expression of operons through transcription pausing or termination, often in response to extracellular environmental changes. The region between arsC and arsD was predicted to form two stem-loops after transcription due to the presence of inverted repeat sequences. The first one (23-bp) displays features of a Rho-independent terminator, where GC is rich in the repeat followed by concentrated U nucleotides (Fig. 2A) which is common in Bacillus but also in E. coli (Naville & Gautheret, 2010). Terminator read-through via anti-termination has been demonstrated as a mechanism of transcriptional attenuation (Naville & Gautheret, 2010). In the case of CDB3 ars1, the predicted internal terminator structure may be tackled by an anti-terminator protein, which would stabilise the anti-terminator structure allowing production of the long eight-gene transcript. Alternatively, the anti-termination read-through may be via a riboswitch mechanism (Naville & Gautheret, 2010). Hence, the formation of a hairpin structure to terminate transcription after arsC in CDB3 ars1 RNA might be interfered with by some small molecule(s). This type of anti-termination usually involves an anti-terminator (Naville & Gautheret, 2010); however, our sequence analysis did not identify an anti-terminator structure in the RNA region. One other possibility is a weak termination without actual regulation by an anti-terminator or protein. Such a terminator has been found in the E. coli lac operon between lacZ and lacY. However, switch control function of the terminator was only speculated under some specific physiological circumstances with no experimental evidence obtained (Murakawa et al., 1991). Since ArsY and ArsC produced from the short transcript arsRYC can provide quite significant resistance, along with the downstream gene products conferring additional tolerance (Zheng et al., 2013), it is reasonable to assume that employing a switch to regulate the two alternative transcription processes would benefit the host bacteria in terms of energy consumption. Further investigation is warranted. A database search also revealed that a number of Bacillus strains, such as Bacillus cereus ATCC10987 and Bacillus thuringiensis BM1904.1B171, harbor a similar seven-gene operon (arsRYCDAOrf7,8, only lack of arsT). They all contain a hairpin sequence in the intergenic region between arsC and D so assumingly have the similar transcription pattern as CDB3 ars1. Such internal terminator-like structures my also exist in other types of ars operons.
Site specific cleavage of arsY RNA
To confirm the identity of the 1.5-kb RNA, a primer extension assay was conducted to map the 5’-end. Fluorescent labeled primer PED-R (29-bp) was annealed from 295 bp downstream of the arsY coding region. The electropherogram of extension products showed a predominant peak corresponding to a 261-bp cDNA fragment (Fig. 3A), demonstrating that the 5’-end of 1.5-kb RNA was the 35th bp of arsY. This nucleotide (uridine) is immediately downstream of an inverted repeat region in Y RNA sequence (Fig. 3B). This further supports the assumption that the smaller RNA resulted from degradation.
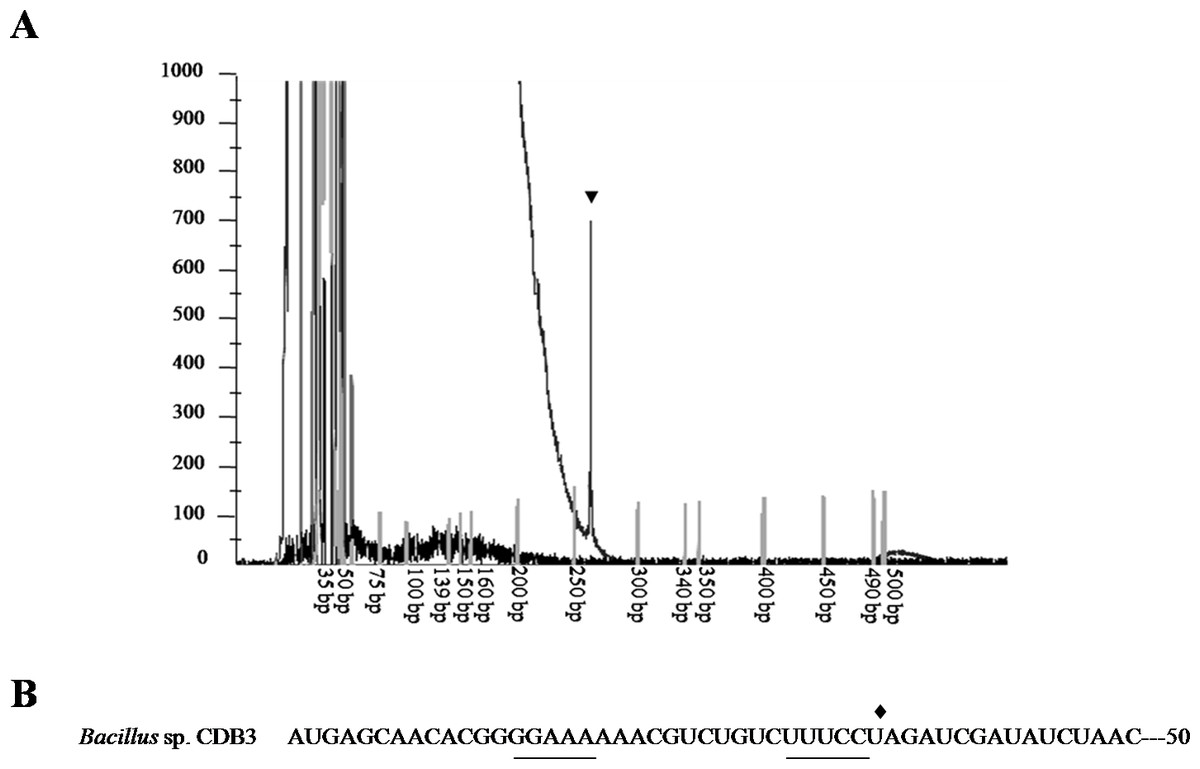
Figure 3: Mapping of RNA degradation product.
(A) Electropherogram showing the result of primer extension assay. The extension product is indicated by arrowhead and size standards are labelled below. (B) The first 50 nucleotides sequence of CDB3 arsY RNA coding region indicating an inverted repeat (underlined) and 5’-end of 1.5-kb RNA (pointed by diamond).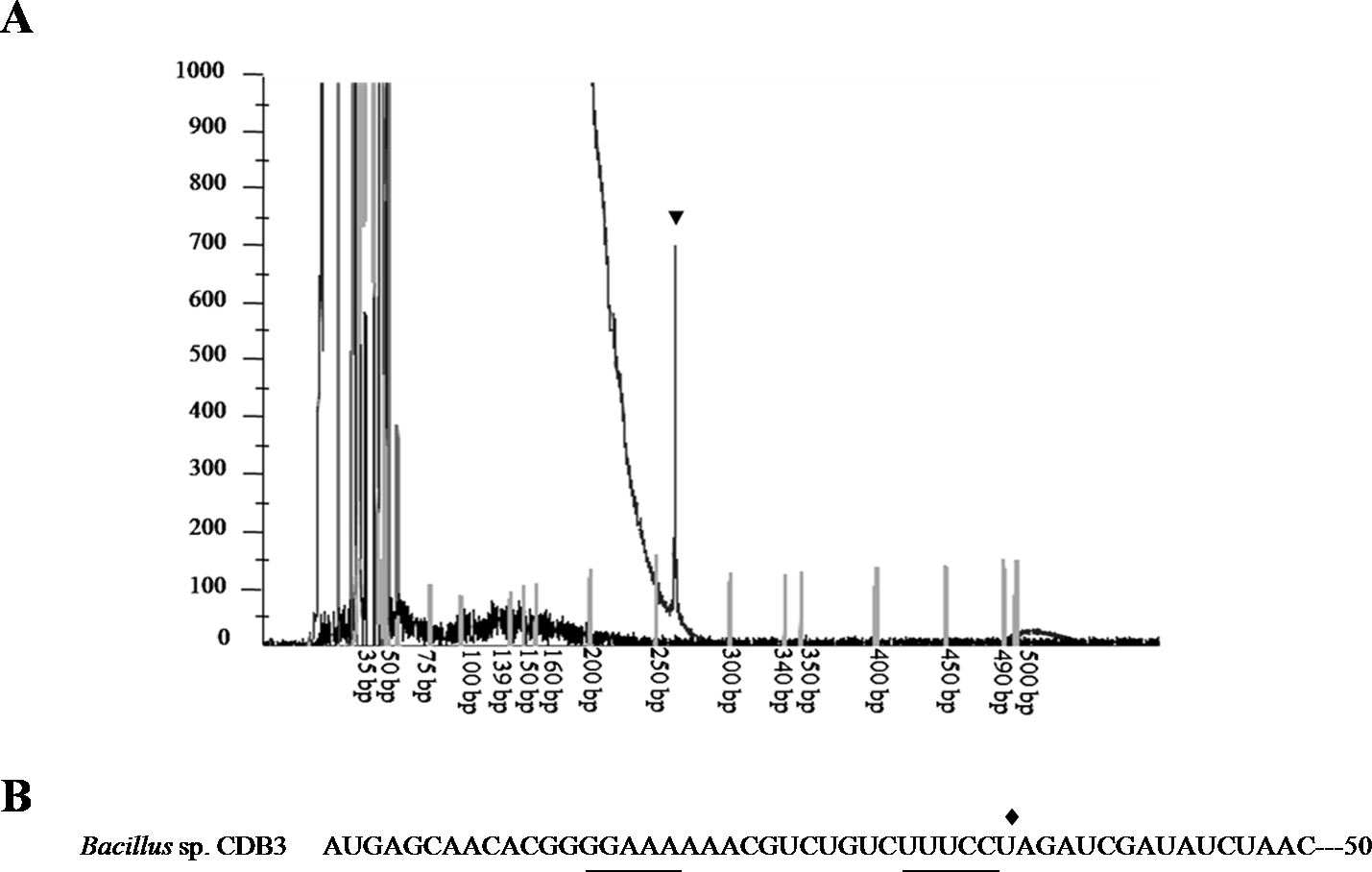{kind=link}
Differential rate of RNA degradation is a post-transcriptional regulation strategy and has been studied in different living organisms including bacteria (Houseley & Tollervey, 2009). A previous report on the E. coli plasmid R773 arsRDABC operon found rapid degradation of ArsB messenger and proposed that two RNA hairpins, one in the intergenic region before arsB and the other in the 5’ end of arsB, are responsible (Owolabi & Rosen, 1990). Cai & DuBow (1996) also communicated a similar case for the E. coli chromosomal arsRBC operon. Our finding that the CDB3 arsY RNA also cleaved in the 5’ region adjacent to a hairpin supports the notion that RNA secondary structure is important for degradation (Owolabi & Rosen, 1990). Compared with E. coli RNA, Bacillus sp. CDB3 RNA has only one hairpin located between the 5th and 12th codons of arsY which is equivalent to the second hairpin present in the E. coli counterpart (from 3rd codon of arsB RNA) (Owolabi & Rosen, 1990), with a free energy of −7.0 kcal/mol. This secondary RNA structure may limit the translation of ArsY as suggested for the E. coli hairpin on ArsB translation (Owolabi & Rosen, 1990). However, in the absence of a second upstream hairpin (as found in E. coli mRNA), it also assumes a main role in the specific degradation. The 5’ end of a degradation product has been identified as U35 which is straight after the hairpin, not before, suggesting a cleavage point between C34 and U35 (Fig. 3). Although only the prominent band of arsYC RNA fragment (1.5 bp) was identified, it is reasonable to assume that this specific cleavage also took place in the full operon transcript, which was not observed due to co-migration of the cleaved (∼5.4 kb) and intact RNA (∼5.8 kb) on our RNA gels (Fig. 1). The upstream part of transcript after cleavage (∼400 bp: arsR plus 34 bp of Y) was not detectable by northern analysis, indicating that it had been rapidly degraded. Unlike the first hairpin in E. coli R773 (Owolabi & Rosen, 1990), the 5-bp repeat stem at the 3’ end of the degradation product is probably not strong enough to act as a decay terminator. Nevertheless, our results demonstrate another case of specific degradation for ars polycistronic RNA in the transporter gene regions, suggestive of differential rate of RNA degradation, which helps prevent toxic levels of hydrophobic transporter proteins (Owolabi & Rosen, 1990). This regulatory pathway controlling intracellular protein concentration at the expression level may be common to a variety of bacteria.
Confirmation of ArsR as a repressor of ars1
The CDB3 ArsR protein is homologous to the ars operon regulator of Bacillus subtilis Skin element with 89% similarity (Bhat et al., 2011), suggesting a potential repressor function in Bacillus sp. CDB3. Sequence analysis of the ars1 promoter region identified a 22-bp inverted repeat (Fig. 4A), probably acting as repressor binding site. With the recombinant CDB3 ArsR protein produced and purified from E. coli, an electrophoretic mobility shift assay (EMSA) demonstrated that the ArsR protein retarded the mobility of a 167-bp DNA fragment (proR) containing the promoter region of ars1 by forming a DNA-protein complex (Fig. 4B left). At a fixed concentration (0.1 mM) of DNA, the binding was enhanced along with increasing concentrations (0.37–1.5 mM) of ArsR. However, there was no interaction observed when the protein was incubated with a non-specific DNA fragment (proN) even at 25.5 mM (255-fold concentration as that used for proR) (Fig. 4B right), which confirmed the binding specificity between ArsR and the ars1 promoter. Since the transcription of ars1 can be induced by arsenic, interruption of the binding by arsenic compounds was assessed. As shown in Fig. 4C (left), with the concentration of arsenite increased the protein-DNA complex was gradually disrupted and a complete separation occurred at 50 mM. However, arsenate did not affect the ArsR-DNA complex even at a concentration of 80 mM (Fig. 4C right). Therefore, arsenite, but not arsenate, is capable of binding CDB3 ArsR, leading to dissociation from the ars1 promoter.
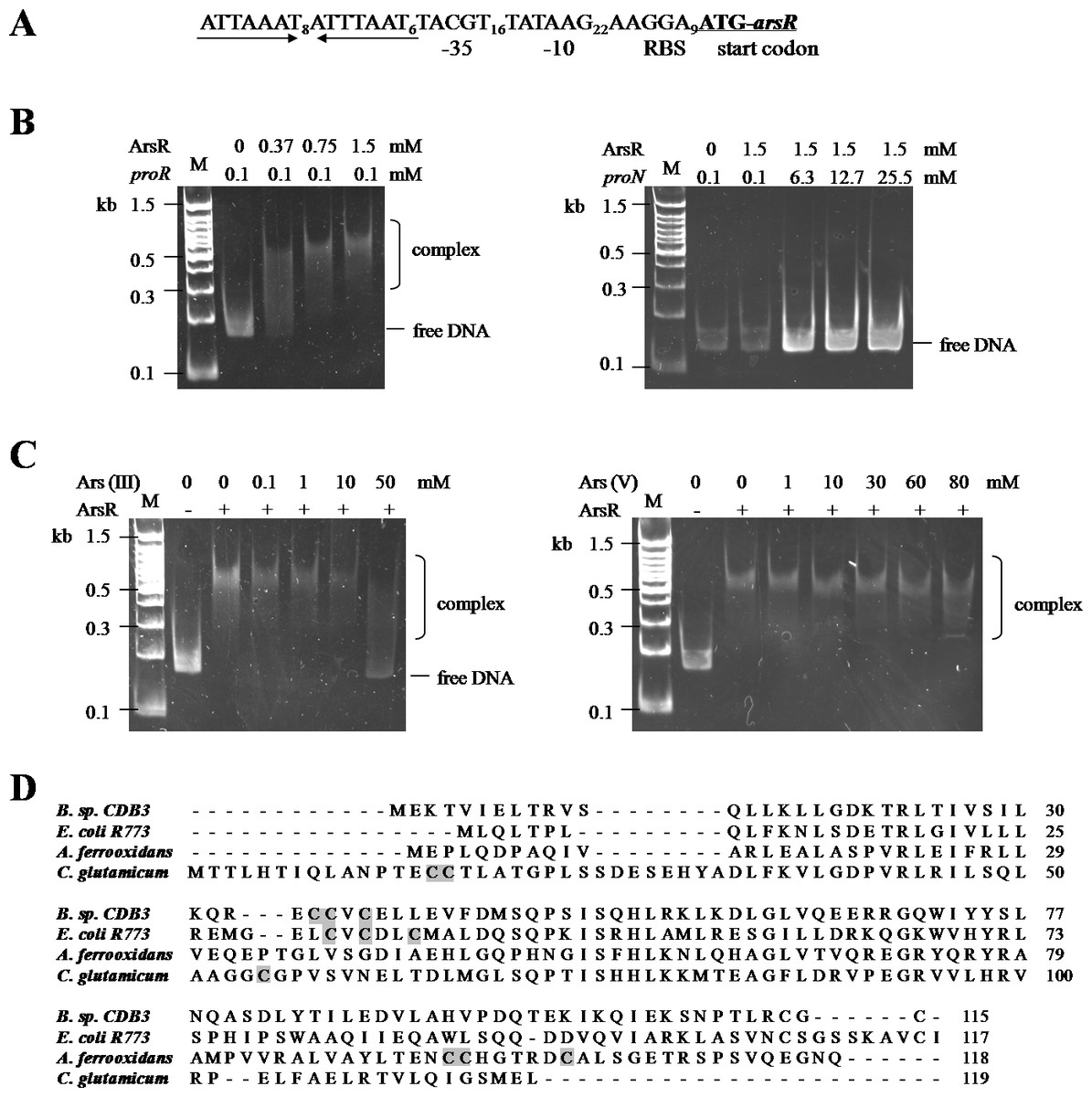
Figure 4: Binding motifs and mobility shift assays of ArsR.
(A) Putative promoter region in ars1. The inverted repeat is marked by inverted arrows lines . The putative ribosomal binding site (RBS), −35 and −10 boxes and start codon of arsR are indicated. (B) Examination of ArsR binding. Left: ArsR binding with proR. The amounts of DNA fragment and protein used were indicated above the panel. Right: ArsR binding with proN. ArsR was incubated with proN at equal and much higher concentrations than that used for proR. (C) Effect of arsenite (Left) and arsenate (Right) on ArsR-DNA complex. Each reaction contains 0 .1 mM proR and 0.75 mM of ArsR (+) or 0 mM of ArsR (−).The arsenic concentrations in each reaction are shown on top of panel. Lane M is a 100 bp DNA ladder with representative sizes indicated. (D) Multiple ArsR sequences alignment . Representative homologues (accession numbers in parentheses) are from E. coli R773 (P15905), Bacillus sp. CDB3 (AF178758), Acidithiobacillus ferrooxidans (AAF69241) and Corynebacterium glutamicum (YP_225794.1). The identified or predicted metalloid binding cysteines are highlighted in shadow.{kind=link}
ArsR is a member of ArsR/SmtB family identified from an extensive range of microbes, responding to transition metals, heavy metals and metalloids (Pennella & Giedroc, 2005). Investigations of this transcriptional repressor suggest that they share a winged helix protein as ancestor but their inducer binding sites are diverse and appear to evolve independently (Osman & Cavet, 2010). The Bacillus CDB3 ArsR possesses three cysteine residues: Cys35, Cys36 and Cys38 in the ELCVCDL binding motif, which vary in positions from other known ArsRs (Fig. 4D). The typical ELCVCDL motif of metal(oid)-binding site consists of Cys32, Cys34 and Cys37 adjacent to a DNA binding domain, represented by E. coli R773 ArsR (Shi, Wu & Rosen, 1994). By contrast, the metalloid binding site of ArsR in Acidithiobacillus ferrooxidans is composed of Cys95, Cys96 and Cys102 contiguous to the C-terminal dimer interface (Qin et al., 2007) and that in Corynebacterium glutamicum consists of Cys15, Cys16 and Cys55 (Ordóñez et al., 2008). The three sulfur thiolates have been proposed to form a pyramidal cage to specifically bind trivalent arsenite which is three-coordinate (Shi et al., 1996).
ArsD may not function as a repressor of ars1
An EMSA was also carried out using recombinant CDB3 ArsD protein to test its ability to bind DNA. Such a binding affinity was not detected so the results did not show a mobility shift of proR DNA after reaction with the ArsD protein (Fig. 5A) indicating that CDB3 ArsD is probably not a repressor. A sequence alignment between CDB3 ArsD and two biochemically characterised repressor ArsDs from E. coli R773 (Wu & Rosen, 1993) and Acidiphilium multivorum AIU 301 pKW301 (Suzuki et al., 1998) showed a distinct dissimilarity (Fig. 5B). Compared with the other two proteins, CDB3 ArsD does not possess the two C-terminal vicinal cysteine pairs, Cys112–Cys113 and Cys119–Cys120.
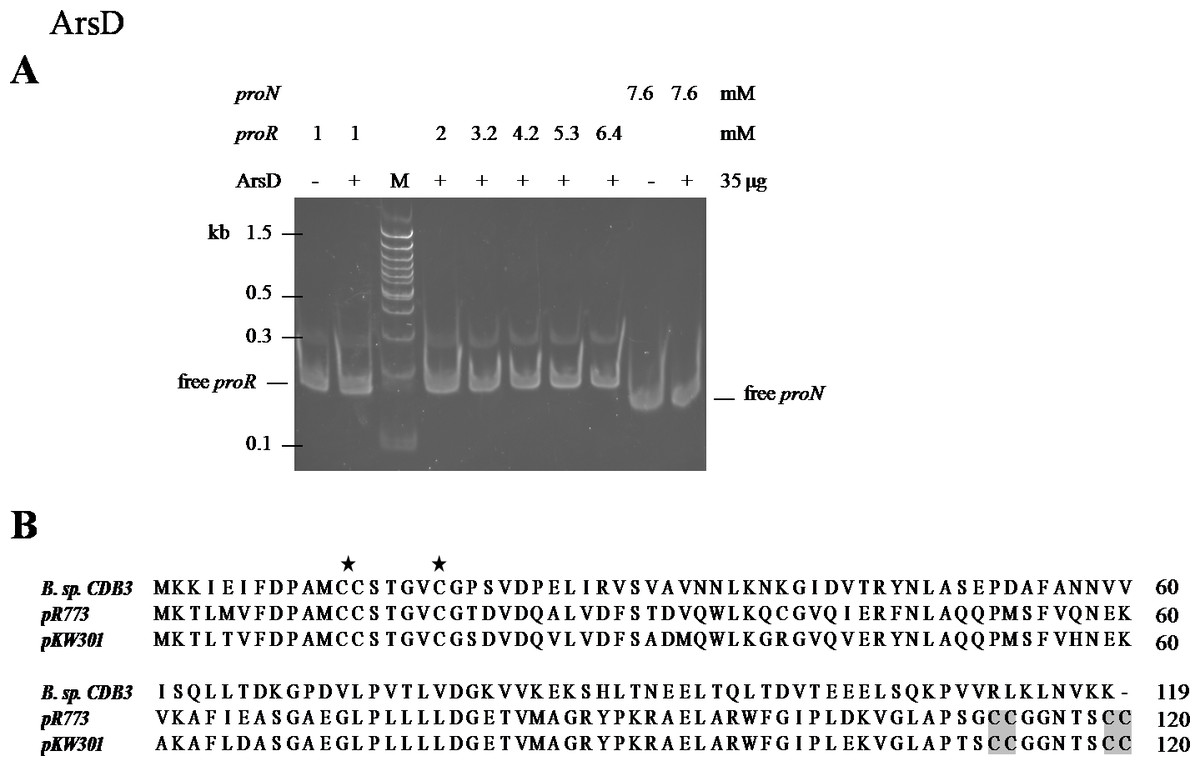
Figure 5: Mobility shift assay of ArsD.
(A) EMSA of ArsD binding to proR and proN. Indicated amount of each DNA fragment binds with (+) or without (−) ArsD. M is a 100 bp DNA ladder with representative sizes indicated. (B) Sequence alignment of ArsDs from Bacillus sp. CDB3 (AAD51848.1), E. coli pR773 (AAA93060) and Acidiphilium multivorum pKW301 (BAA24821). Accession numbers are in parentheses. The stars indicate conserved Cys in all three sequences and shadow indicates those not present in Bacillus sp. CDB3 ArsD.{kind=link}
The regulatory function of ArsD in arsenic defence has been demonstrated in at least two species, E. coli and A. multivorum AIU 301 (Suzuki et al., 1998; Li, Chen & Rosen, 2001). However, the ArsD encoded by Bacillus sp. CDB3 ars1 did not show any interactions with the operon promoter in our mobility assay. Li, Chen & Rosen (2001) predicted that the two vicinal cysteine pairs (Cys112,113 and Cys119,120) in the C-terminal region are involved in regulatory function in E. coli R773. Our result supports the proposition. Conservation of the three N-terminal cysteine residues (Cys12, Cys13 and Cys18), which were shown to be essential for the metallochaperone function of ArsD in E. coli R773 (Lin, Yang & Rosen, 2007), points to its function as a chaperone. The involvement of arsD in arsenic resistance of Bacillus sp. CDB3 ars1 has been demonstrated (Bhat et al., 2011) and the role of its product as a chaperone waits to be confirmed.
Conclusions
The eight-gene operon of Bacillus sp. CDB3 ars1 has been revealed to express its genes in a more complicated way than other known ars operons. It represents another example of specific mRNA degradation in the transporter gene region and possibly the first case of transcription attenuation in ars operons. The possible coordination of the two transcripts and other factors involved in both the transcriptional and post-transcriptional processes warrant further investigation.
Supplemental Information
Raw data for qPCR
Figure S1. Raw data for qPCR results in Figure 2B