
O-GlcNAcylation is a key regulator of multiple cellular metabolic pathways
- Published
- Accepted
- Received
- Academic Editor
- Gwyn Gould
- Subject Areas
- Biochemistry, Cell Biology, Metabolic Sciences
- Keywords
- O-GlcNAc, Nutrient sensing, Glucose uptake, Glycolysis, Mitochondria, Lipid metabolism, Glutamine, Signaling pathway, Homeostasis, TCA
- Copyright
- © 2021 Zhang et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2021. O-GlcNAcylation is a key regulator of multiple cellular metabolic pathways. PeerJ 9:e11443 https://doi.org/10.7717/peerj.11443
Abstract
O-GlcNAcylation modifies proteins in serine or threonine residues in the nucleus, cytoplasm, and mitochondria. It regulates a variety of cellular biological processes and abnormal O-GlcNAcylation is associated with diabetes, cancer, cardiovascular disease, and neurodegenerative diseases. Recent evidence has suggested that O-GlcNAcylation acts as a nutrient sensor and signal integrator to regulate metabolic signaling, and that dysregulation of its metabolism may be an important indicator of pathogenesis in disease. Here, we review the literature focusing on O-GlcNAcylation regulation in major metabolic processes, such as glucose metabolism, mitochondrial oxidation, lipid metabolism, and amino acid metabolism. We discuss its role in physiological processes, such as cellular nutrient sensing and homeostasis maintenance. O-GlcNAcylation acts as a key regulator in multiple metabolic processes and pathways. Our review will provide a better understanding of how O-GlcNAcylation coordinates metabolism and integrates molecular networks.
Introduction
Increasing evidence has suggested that metabolic reprogramming is a key factor in carcinogenesis and tumorigenesis, processes in which many oncogenes and tumor suppressors are metabolic regulators (Jóźwiak et al., 2014). Metabolic reprogramming also occurs in the endometrium during pregnancy (Zuo et al., 2015). In the early 1980s, a new glycosylation modification, O-linked-GlcNAc, was discovered in serine or threonine residues in proteins in the nucleus, cytoplasm, and mitochondria (Yang & Qian, 2017). This process was distinct from traditional O- and N-linked glycosylation which occurred in the endoplasmic reticulum, Golgi, and secretory pathways (Hart, 2019). This discovery generated interest in O-GlcNAc modifications. Research from the past 40 years has indicated that two enzymes regulate this process: O-GlcNAc transferase (OGT) for O-GlcNAc addition, and O-GlcNAcase (OGA) for O-GlcNAc removal (Vocadlo, 2012). O-GlcNAc modifies more than 4,000 cellular proteins and regulates key cellular processes by O-GlcNAcylating proteins, including those involved in signal transduction, mitochondrial activity, cytoskeleton function, and protein degradation (Bacigalupa, Bhadiadra & Reginato, 2018).
O-GlcNAc may also play a role in nutrient sensing and external stress (Hanover, Krause & Love, 2010). UDP-GlcNAc, which precedes O-GlcNAcylation, links glucose, fatty acid, nucleic acid, and nitrogen metabolic pathways. Therefore, the role of O-GlcNAcylation as a nutrient sensor appears crucial for physiological and metabolic processes, such as cell growth, survival, nutritional status sensing, and multiple signaling pathway integration (Slawson, Copeland & Hart, 2010). Similarly, it may have a crucial role in metabolism-related diseases, including diabetes, cancer, cardiovascular disease, and neurodegenerative disease. Thus, O-GlcNAcylation levels may reflect, to some extent, cellular systemic metabolic status. While there is no shortage of information on metabolic regulation by O-GlcNAcylation, information regarding how it regulates coordinating metabolic processes are scarce. We reviewed the effects of O-GlcNAcylation on glucose metabolism, mitochondrial oxidation, lipid metabolism, and amino acid metabolism, and discuss its role in nutrient sensing, homeostatic maintenance, and major signaling pathways in physiological states. Our results yield a comprehensive starting point and general overview for this exciting field.
Survey Methodology
We conducted a literature search using the PubMed search engine with keywords and their combinations: O-GlcNAc; nutrient sensing; glucose uptake; GLUT1; GLUT2; GLUT3; GLUT4; GLUT5; glycolysis; mitochondria; oxidative phosphorylation; TCA; pentose phosphate pathway; glycogen synthesis; glycogenolysis; gluconeogenesis; lipid metabolism; triglyceride; cholesterol; phospholipids; amino acid metabolism; glutamine metabolism; alanine metabolism; AKT; NF-kB; HIF-1α; ERK1/2; β-catenin; c-Myc; Hippo/YAP; SOX2; OCT4; signaling pathway; homeostasis. This range provided objective subject coverage. We filtered articles published within 5- or 10-years based on the number of articles retrieved from various word combinations, as well as some early key articles. Our aim was to describe the regulation of metabolism by O-GlcNAcylation, not the regulation of O-GlcNAcylation by metabolisms, so we excluded articles of low relevance by reading the abstracts. Additionally, some word combinations did not produce results, for example: “O-GlcNAc, GLUT2”; “O-GlcNAc, GLUT5”; “O-GlcNAc, phospholipids”. It is worth noting that this is a comprehensive but not an exhaustive review of the literature.
O-GlcNAc and Nutrient-sensing
Metabolic homeostasis in mammals depends on interactions between nutrient-sensing components and signaling pathway regulation (Sharma, Saluja & Banerjee, 2018). Nutrient-sensing pathways are interrelated and combine nutrition information in different ways. For example, amino acids regulate the target rapamycin (TOR, mTOR) pathway and AMP/ATP levels affect the AMP-activated protein kinase (AMPK) signal pathway (Johnson, Rabinovitch & Kaeberlein, 2013; Ruan et al., 2013). The hexosamine biosynthetic pathway (HBP) is another branch of glycolysis (Love & Hanover, 2005; Chiaradonna, Ricciardiello & Palorini, 2018). In 1991, Marshall et al. observed that approximately 2–5% of glucose consumed by adipocytes entered the HBP (Marshall, Bacote & Traxinger, 1991). However, current studies present different views on flux into the HBP (Gibb et al., 2017; Olson et al., 2020). Evidence has suggested that HBP activation is also involved in ATP, uridine, glutamine, and acetyl-CoA function (Zachara & Hart, 2004; Hanover, Chen & Bond, 2018), and that the end-product of HBP, UDP-GlcNAc, integrates intracellular nutrient flux information, including glucose, nitrogen, nucleotide, and fatty acid metabolism (Vosseller et al., 2002; Hart et al., 2011; Swamy et al., 2016; Ma et al., 2017; Ohashi et al., 2017) (Fig. 1).
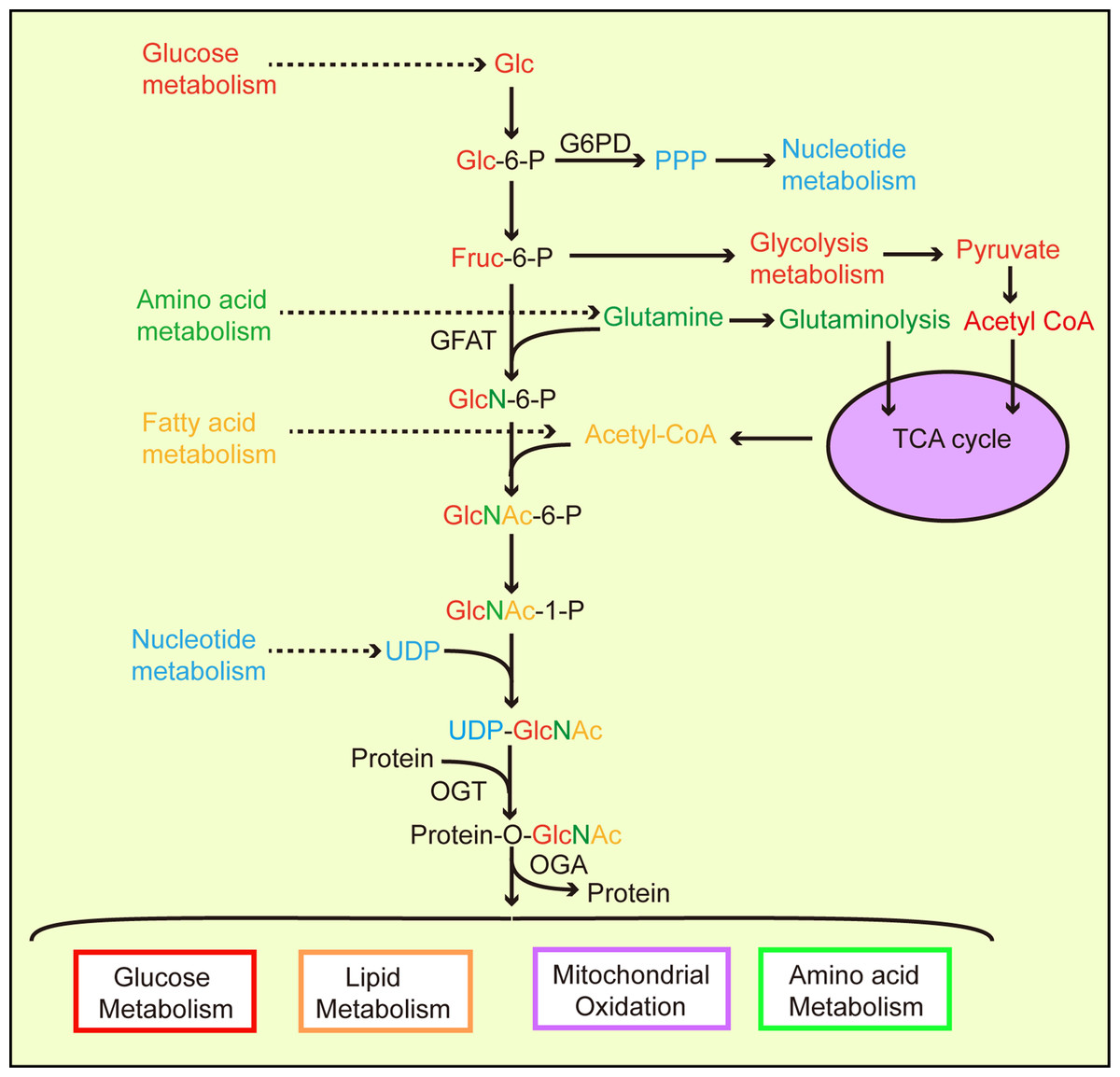
Figure 1: The hexosamine biosynthetic pathway (HBP) links and regulates major biological metabolic processes.
According to cellular physiological needs, the flow of glucose to different metabolic pathways is accordingly adjusted. Glucose participates in glycolysis to provide cellular energy. Glucose also provides nucleotides for cell growth via the pentose phosphate pathway. During UDP-GlcNAc synthesis, the HBP links glucose, amino acids, fatty acids, and nucleotide metabolic pathways through the required synthetic precursors. Then, generated UDP-GlcNAc O-GlcANcylates proteins via OGT catalysis, allowing O-GlcNAcylated proteins to partake in a variety of metabolic processes.{kind=link}
Glutamine fructose-6 phosphate amidotransferase (GFAT) is a rate-limiting enzyme in the HBP which converts fructose 6-phosphate, which is produced by glycolysis and glutamine (from the amino acid synthesis pathway), into glucosamine 6-phosphate (Kornfeld et al., 1964). OGT activity is highly sensitive to UDP-GlcNAc levels (Bacigalupa, Bhadiadra & Reginato, 2018). A difference or change in the nutrient environment alters UDP-GlcNAc levels and the O-GlcNAcylation of intracellular proteins, thereby triggering changes in physiology or cell fate. Previous evidence has suggested that hyperglycemia causes insulin resistance via the HBP (Teo, Wollaston-Hayden & Wells, 2010) resulting in increased UDP-GlcNAc levels in cells (Traxinger & Marshall, 1991). O-GlcNAc levels increased significantly in the liver, heart, erythrocytes, and leukocytes of diabetic mammals, including humans (Ruan et al., 2012, 2013). Studies have shown that AMPK phosphorylates chromatin-associated fumarate hydratase (FH) at Ser75 under glucose deprivation and that FH is involved in promoter activation, thereby promoting growth arrest gene expression (Fricovsky et al., 2012). However, the O-GlcNAcylation of FH at Ser75 inhibits downstream events mediated by FH, especially in cancer cells with high OGT activity (Fricovsky et al., 2012). UDP-GlcNAc and protein O-GlcNAcylation cellular levels fluctuate in relation to glucose, free fatty acid, uridine, and glutamine availability, which suggests that O-GlcNAc senses nutrients (Johnson, Rabinovitch & Kaeberlein, 2013; Hanover, Chen & Bond, 2018) (Fig. 1).
O-GlcNAcylation Regulates Glucose Metabolism
O-GlcNAcylation regulaties glucose uptake
The first step in glucose metabolism involves transporting glucose through the membrane into cells. This process is undertaken by members of the glucose transporter (GLUT) family of proteins. GLUT1 was one of the first family members to be described and is the most widely expressed and studied transporter (Augustin, 2010). Studies have shown that elevated O-GlcNAcylation indirectly increases GLUT1 expression and glucose uptake via hypoxia-induced factor (HIF)-1α in cancer cells (Ferrer et al., 2014). Our previous work showed that elevated GLUT1 and O-GlcNAcylation levels of the endometrium during embryo implantation were involved in the regulation of endometrial receptivity (Han et al., 2019; Zhang et al., 2020). A mutual regulation between the GLUT1 and O-GlcNAcylation may result in: (1) elevated GLUT1 may enhance O-GlcNAcylation by coordinating glucose flow into HBP (2) elevated O-GlcNAcylation may increase GLUT1 expression through a similar molecular mechanism, which may be HIF-1α. Two recent studies provided evidence for this regulatory effect (Wang et al., 2020; Hu et al., 2021). Infection by hepatitis B promoted glucose uptake by upregulating GLUT1 expression in hepatocytes, providing substrates for the HBP synthesis of UDP-GlcNAc, leading to increased O-GlcNAcylation (Hu et al., 2021). Acyl-CoA ligase 4 (ACSL4) enhanced O-GlcNAcylation via GLUT1 to promote hepatocellular carcinoma growth and survival, and in turn, O-GlcNAcylation increased ACSL4 expression and promoted cancer growth, potentially involving downstream GLUT1 regulation by ACSL4 (Wang et al., 2020). These findings suggested that mutual regulation between the GLUT1 and O-GlcNAcylation may be necessary to maintain high metabolic cell demands.
Insulin-sensitive GLUT4 is also modified by O-GlcNAc (Buse et al., 2002). GLUT4 is stored in GLUT4 storage vesicles (GSVs) in the absence of insulin signaling. During insulin stimulation, signaling responses are generated by receptors binding to transport GSVs to cell membranes, fusing vesicle and cell membranes, resulting in GLUT4 transport to the cell membrane (Zorzano et al., 1996), and vesicle proteins are also modified by O-GlcNAc (Buse et al., 2002). Evidence has indicated that elevated O-GlcNAcylation inhibits GLUT4 translocation and 2-deoxy-d-glucose (2-DG) uptake during insulin stimulation (Park, Ryu & Lee, 2005). However, increased O-GlcNAcylation of insulin receptor substrate-1 (IRS-1) and Akt2 reduces their phosphorylation in the insulin signaling pathway, leading to impaired glucose utilization and insulin resistance in adipocytes. Whether this is related to the O-GlcNAcylation of GLUT4 and its vesicles is unclear at present, but it remains a strong hypothesis. Therefore, by regulating GLUTs, O-GlcNAcylation affects cellular glucose intake and plays a crucial role in regulating cell fate. However, whether O-GlcNAcylation directly or indirectly regulates other GLUT family members and affects glucose uptake requires additional study.
O-GlcNAcylation regulates glycolysis
Glucose is phosphorylated by hexokinase (HK) at cell entry to generate glucose-6-phosphate, which remains in the cell, but whose metabolic fate is determined by cellular physiological requirements (Bacigalupa, Bhadiadra & Reginato, 2018). Some studies have shown that increased OGT enhances HK activity, whereas others have shown that HK is merely modified by O-GlcNAc, and positively regulates its expression (Baldini et al., 2016a). When the cell requires more energy, cellular glucose undergoes glycolysis, during which most key enzymes are also modified by O-GlcNAc (Bacigalupa, Bhadiadra & Reginato, 2018). Phosphofructokinase-1 (PFK1) is the first rate-limiting enzyme of glycolysis and catalyzes the conversion of fructose 6-phosphate to fructose 1, 6-bisphosphate. The O-GlcNAcylation of PFK1 at Ser529 in cancer cells was shown to inhibit its kinase activity (Yi et al., 2012), which may lead to elevated upstream glycolytic intermediates, increasing flux through the pentose phosphate pathway (PPP) and the biosynthetic precursors of cell growth. O-GlcNAc modification levels are further improved by increasing the HBP flux. The last committed step of glycolysis is catalyzed by pyruvate kinase M2 (PKM2), which is modified by O-GlcNAc (Chaiyawat et al., 2015). Recent cancer studies have shown that O-GlcNAcylation destabilizes the active tetramer PKM2 and reduces pyruvate kinase activity, but O-GlcNAcylation increases glucose uptake and consumption and promotes lactate production, and lipid and DNA synthesis (Wang et al., 2017). This potentially shifts the glycolytic flux to anabolic pathways to promote Warburg effects (Wang et al., 2017). Similarly, Singh et al. (2020) reported that PKM2 O-GlcNAcylation inhibited its activity and promoted aerobic glycolysis in cancer cells. Furthermore, these authors found that OGA was highly expressed in several cancers and that PKM2 was a target of OGA-associated acetyltransferase activity. OGA was also found to enhance PKM2 acetylation and facilitate O-GlcNAcylation of PKM2 by OGT (Singh et al., 2020). This study posits the idea that OGA exhibits acetyltransferase activity, which is supported by earlier studies (Toleman et al., 2004), and is now more widely accepted that while there are structural similarities to histone acetyltransferases, OGA is not catalytically active as an acetyltransferase. Indeed, recent structural studies found that human OGA lacks the residues necessary for binding to acetyl-CoA and described this region as a “catalytically incompetent pseudo-AT domain” as reported by Rao et al. (2013). These studies help us understand the OGA’s pathogenic role, but not OGT’s. Similarly, the cooperation of the two antagonistic enzymes, OGT and OGA, may shed new mechanistic insights on glycolytic metabolism regulation. Previous studies indicated that OGT mediated metabolic reprogramming in cancer cells (Yi et al., 2012; Ferrer et al., 2014), yet OGA may also play a key role in this process. Furthermore, coordinated OGT and OGA actions may provide explanations for the cellular maintenance of the O-GlcNAc cycle in response to metabolic demands.
In their study, Zhang et al. (2017) observed that cells from patients with pre-B acute lymphocytic leukemia (pre-B-ALL) exhibited increased O-GlcNAcylation levels, elevated glycolysis, and O-GlcNAcylation regulation via the PI3K/AKT/c-Myc pathway, which affected cellular glycolysis levels. Furthermore, OGT knockdown in breast cancer cells reduced glycolysis and PPP pathway metabolites (Ferrer et al., 2014). These observations suggested that O-GlcNAcylation induced changes in glycolysis processes and was likely involved in reprogramming the cellular metabolic network (Figs. 2, 3). Proteomics identified that fructose bisphosphate aldolase, triosephosphate isomerase, and glyceraldehyde-3-phosphate dehydrogenase were modified by O-GlcNAc for other glycolysis pathway enzymes (Cieniewski-Bernard et al., 2004; Bacigalupa, Bhadiadra & Reginato, 2018). This data indicated that O-GlcNAc modification was directly or indirectly related to glycolytic processes. However, whether O-GlcNAcylation of some enzymes affects their structure, activity, and function remains unclear.
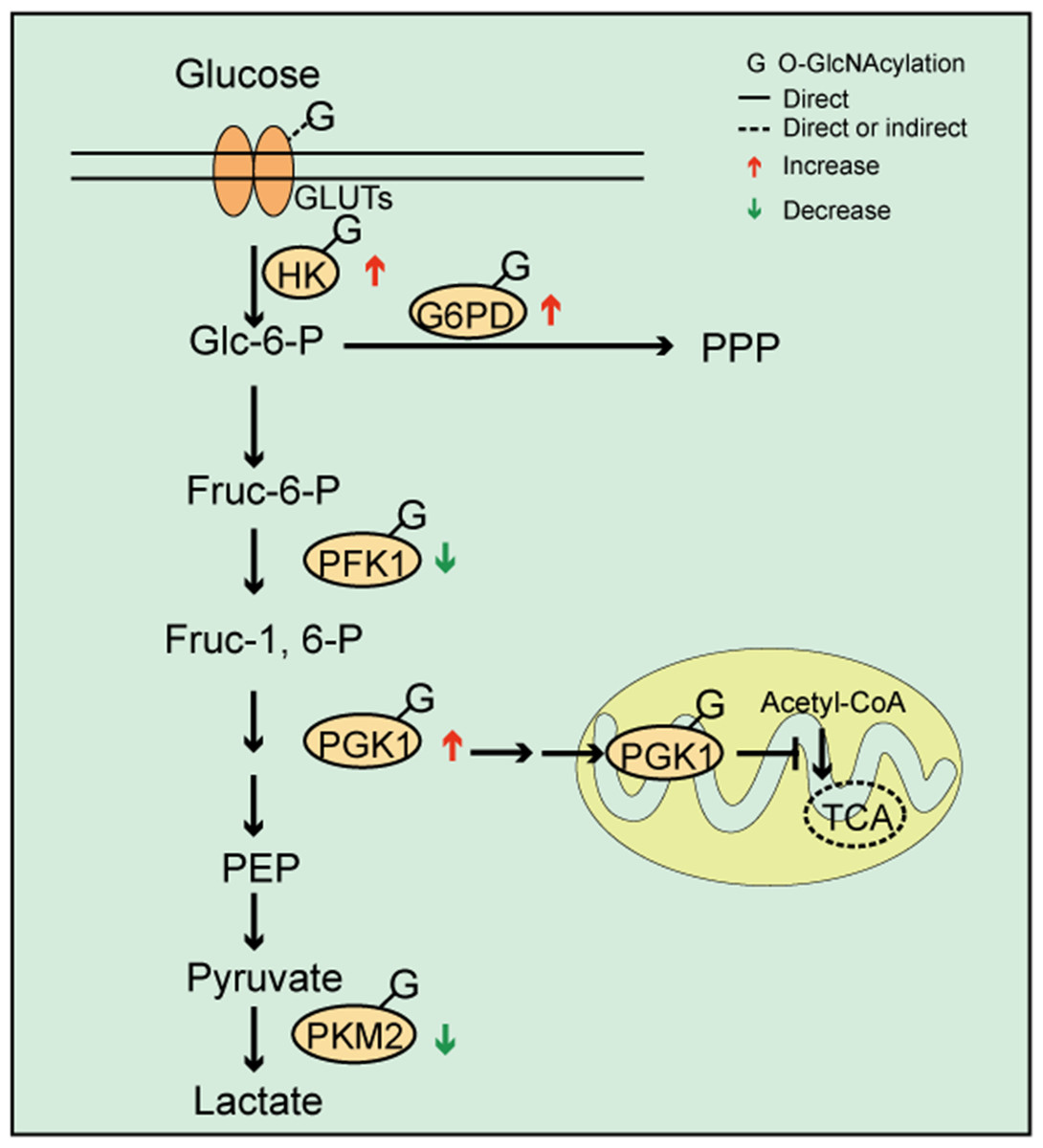
Figure 2: O-GlcNAc modifications and cellular metabolism.
O-GlcNAcylation of key enzymes involved in coordinating glycolysis, pentose phosphate pathways (PPP), and the tricarboxylic acid cycle. The O-GlcNAcylation of PFK1 and PKM2 inhibits enzyme activity, which may lead to increased upstream glycolytic intermediates. The O-GlcNAcylation of G6PD activates its activity and increases PPP flow. O-GlcNAcylation promotes PGK1 activity and induces PGK1 translocation to the mitochondria, which inhibits mitochondrial oxidative phosphorylation.{kind=link}
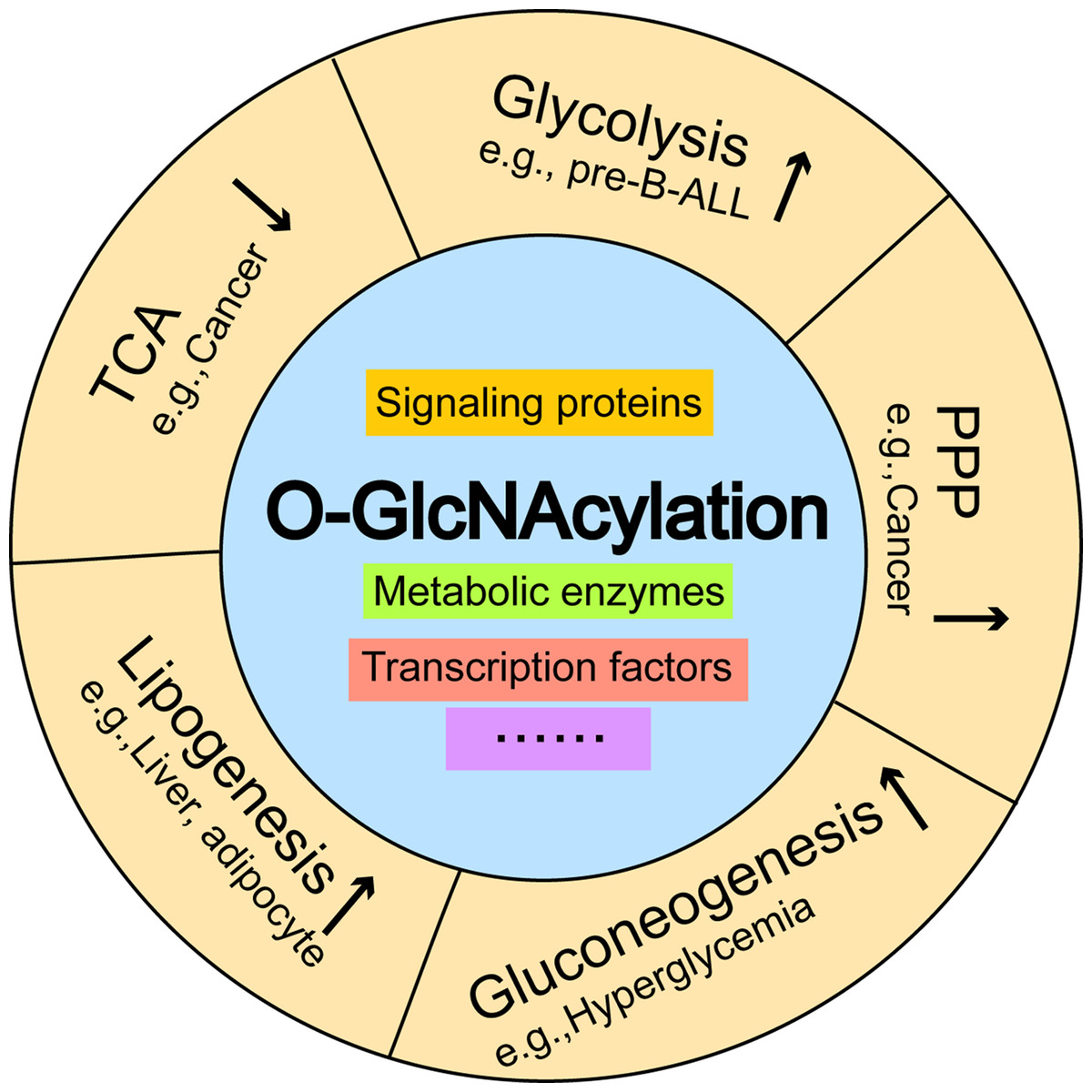
Figure 3: The effects of O-GlcNAcylation on representative metabolic pathways.
Aberrant increases in O-GlcNAcylation promote glycolysis in pre-B-ALL, and activates the pentose phosphate pathway, which inhibits the tricarboxylic acid cycle in breast cancer. O-GlcNAcylation induces hyperglycemia by activating gluconeogenesis and contributes to the excessive deposition of hepatic triglycerides by stimulating de novo lipogenesis. Pre-B-ALL: pre-B acute lymphocytic leukemia.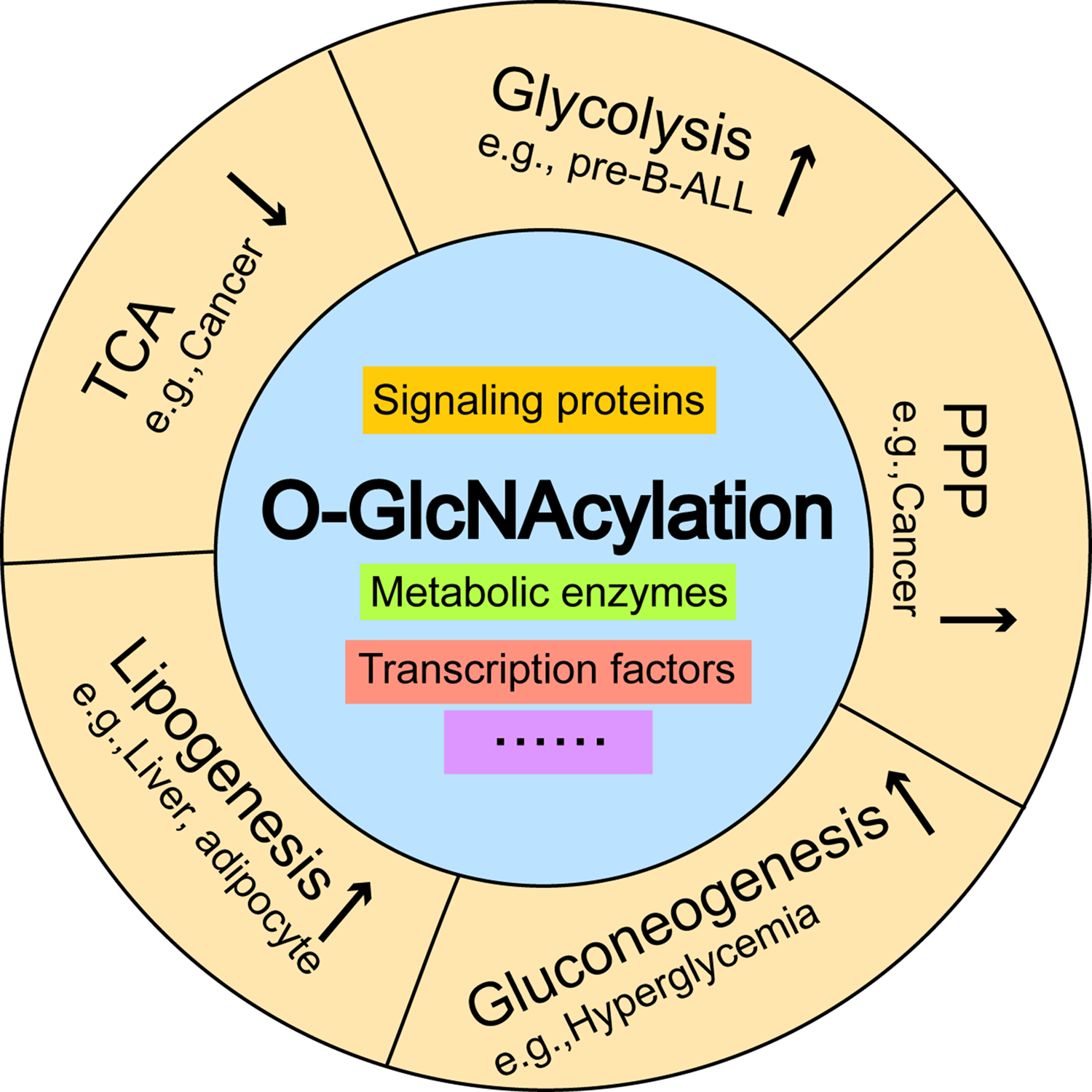{kind=link}
O-GlcNAcylation regulates PPP
As a branch of glucose metabolism, the PPP redirects glucose-6-phosphate to produce pentose and nicotinamide adenine dinucleotide phosphate (NADPH). Thus, one of the consequences in the pathway is to provide ribose for nucleic acid biosynthesis and to support lipid synthesis. Few studies have focused on PPP regulation by O-GlcNAcylation. Yehezkel et al. reported that glucose-6-phosphate dehydrogenase (G6PD) expression was increased and PPP enhanced in OGA-silenced colon cancer cells (Yehezkel et al., 2012). Similarly, OGT knockdown in breast cancer cells reduced PPP metabolites (Ferrer et al., 2014). These data suggested that PPP levels were consistent with total O-GlcNAcylation. However, this evidence was generated in cancer cells, therefore, the question remains whether different cell types exhibit different regulatory mechanisms. It was also shown that G6PD was modified by O-GlcNAc under hypoxic conditions and that O-GlcNAcylation activated G6PD activity and increased PPP flow, thereby providing a precursor for nucleotide and lipid biosynthesis (Rao et al., 2015). These data suggested that O-GlcNAcylation directly regulated PPP through G6PD, which is a key PPP enzyme (Rao et al., 2015) (Fig. 2).
O-GlcNAcylation regulates glycogen synthesis and glycogenolysis
Glycogen is the stored form of glucose. Glycogen is stored in the liver and skeletal muscle. Hepatic glycogen is the main source of blood glucose, and muscle glycogen produces ATP to supply energy by rapid anaerobic glycolysis. Glycogen synthase (GS), which controls the final step in glycogen synthesis, was reportedly modified by O-GlcNAc (Parker et al., 2003). Recent studies indicated that total O-GlcNAcylation was increased in hepatoma cells under glucose deprivation which elevated O-GlcNAcylation of GS and decreased its enzyme activity (Taylor et al., 2008), potentially leading to blood glucose retention (Parker et al., 2003). Taylor et al. (2008) suggested that O-GlcNAcylation elevation was caused by increased OGT and decreased OGA levels. A study by Kang et al. (2009) observed that glycogen degradation via glucose deprivation was the source of UDP-GlcNAc for increased O-GlcNAcylation in cancer cells (Kang et al., 2009). The inhibition of glycogen phosphorylase (GP, the rate-limiting enzyme of glycogenolysis) prevented the increase of O-GlcNAcylation, and GFAT increased under glucose deprivation, suggesting that glycogen was the source of the increased O-GlcNAcylation.
GSK-3β inhibited GS activity, and GSK-3β phosphorylation was self-inactivating, thus promoting glycogen synthesis. GSK-3β was modified by O-GlcNAc (Wang, Pandey & Hart, 2007), however, the role of O-GlcNAcylation towards GSK-3β remains unknown. Due to competition between phosphorylation and O-GlcNAcylation, we believe that O-GlcNAcylation inhibits GSK-3β phosphorylation, thereby inhibiting glycogen synthesis, indirectly contributing to glycogenolysis. However, it is unclear whether GP is modified by O-GlcNAc in terms of the direct regulation of glycogenolysis.
O-GlcNAcylation regulates gluconeogenesis
Abnormal gluconeogenesis leads to hyperglycemia, which is a feature of the diabetic liver. During the insulin response, FOXO1 phosphorylation by AKT promotes its exit from the nucleus, thereby inhibiting gluconeogenic gene transcription by FOXO1. Studies have reported that FOXO1 was modified by O-GlcNAc (Ruan et al., 2013). Other gluconeogenic transcription factors and cofactors are similarly modified by O-GlcNAc, including cAMP response element-binding protein (CREB), regulated transcription coactivator 2 (CRTC2), peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α), and host cell factor C1 (HCF-1) (Fig. 4). OGA overexpression in the liver of high-fat diet fed and db/db mice decreased O-GlcNAcylation levels of CRTC2 and hindered the effects of glucose on gluconeogenesis (Dentin et al., 2008). Studies have also found that PGC-1α promoted FOXO modification and OGT activation (Housley et al., 2009). Ruan et al. (2013) reported that OGT formed a nutrition-sensing complex with HCF-1 and that glycosylation modified PGC-1α via OGT (Ruan et al., 2012). Overall, O-GlcNAcylation promoted gluconeogenesis, thus, O-GlcNAcylation suppression towards key gluconeogenic transcription factors and co-factors could be a therapeutic strategy for type 2 diabetes.
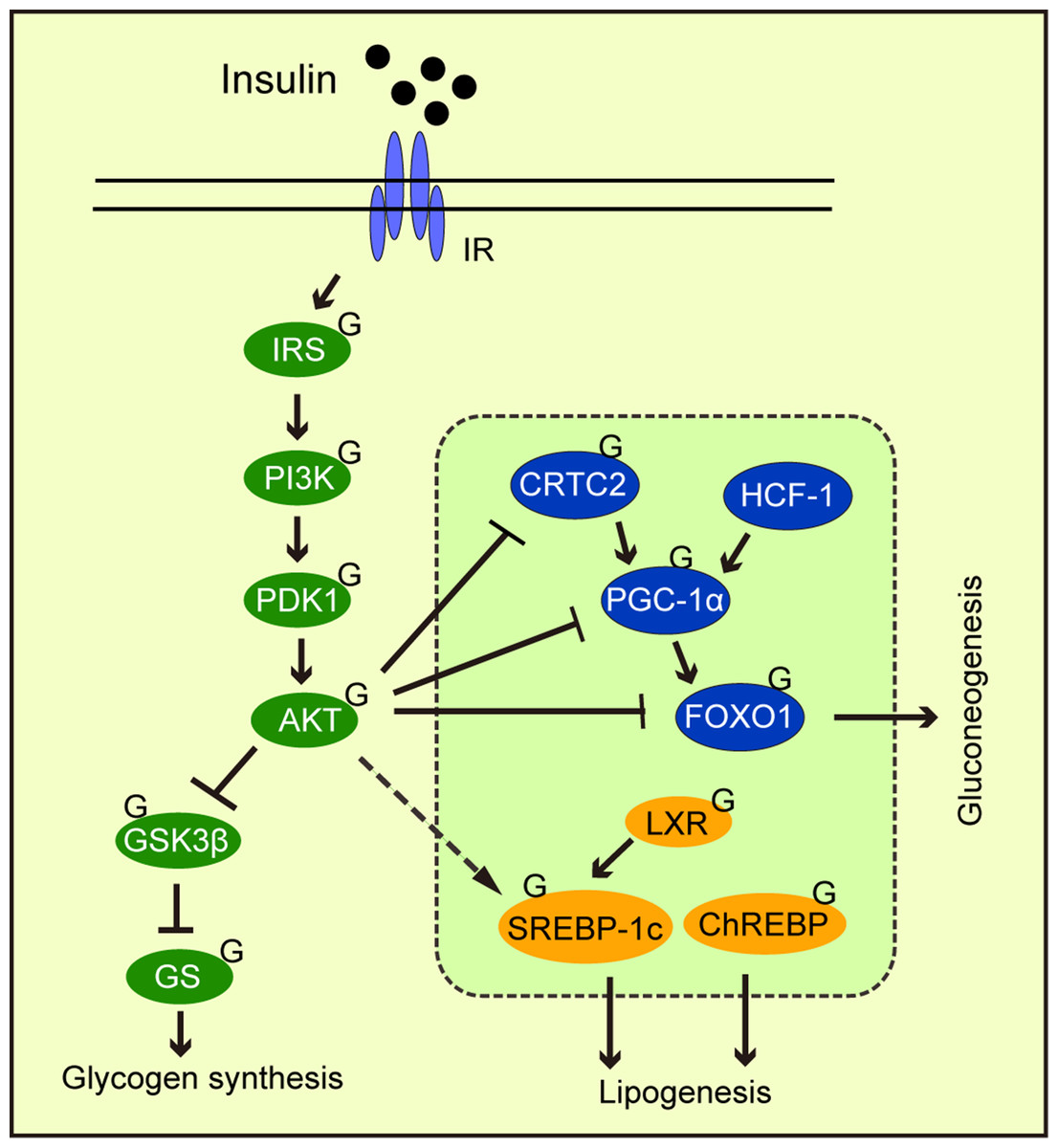
Figure 4: The effects of O-GlcNAcylation on insulin signaling, glycogen synthesis, lipogenesis, and gluconeogenic metabolism.
O-GlcNAc protein modification in the insulin signaling pathway weakens signaling. O-GlcNAcylation of the transcription factors and cofactors, CRTC2, PGC-1α, and FOXO1, promote gluconeogenesis. O-GlcNAcylation promotes the transcriptional regulation of SREBP-1c by LXPs and stabilizes ChREBP, thereby regulating lipogenesis.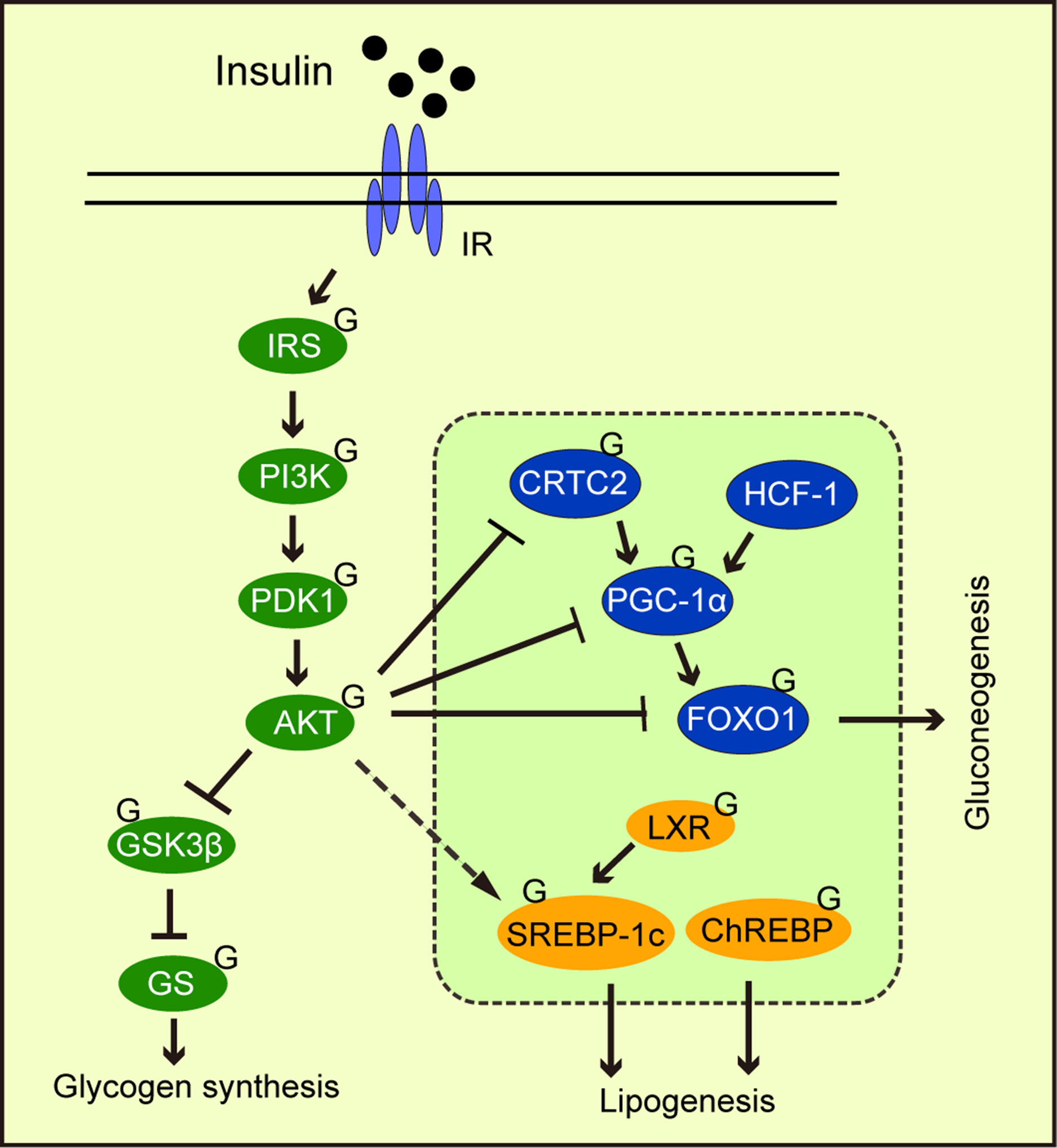{kind=link}
O-GlcNAcylation Regulates Mitochondrial oxidation
O-GlcNAcylation regulates oxidative phosphorylation
Mitochondria are essential organelles for nutrient status signaling and energy metabolism regulation. The regulatory role of O-GlcNAcylation in mitochondria has gained considerable traction in recent years. In 2003, Hanover et al. (2003) identified an OGT splice variant, mOGT, which targeted the mitochondrial inner membrane, suggesting that O-GlcNAcylation was present in mitochondria. This increased research interest in O-GlcNAcylation functions in mitochondria suggest that O-GlcNAc protein modifications are highly prevalent in this organelle (Arvanitis et al., 2005; Hu et al., 2009; Gu, Ande & Mishra, 2011). A 2005 study reported that the antibody, mAbH, recognized epitope H containing O-GlcNAc residues, which were highly-expressed in mitochondria-rich cells, suggesting the presence of O-GlcNAcylation in the components of respiratory chain and energy transduction in mitochondria (Arvanitis et al., 2005). Subsequently, Clark et al. (2008) identified two mitochondrial ATP synthase α and β subunits, and two complex I components (Nduf-1 and Nduf-2) modified by O-GlcNAc via a chemoenzyme strategy for the selective labeling of O-GlcNAc-glycosylated proteins. Hu et al. (2009) reported that NDUFA9 of complex I, subunits core 1 and 2 of complex III, and mitochondrial DNA-coding subunit I of complex IV (Cox I) in the respiratory chain, were all O-GlcNAcylated in cardiomyocytes. High glucose (30 mM) levels increased O-glycosylation of mitochondrial proteins, impaired the activity of complexes I, III, and IV, and decreased mitochondrial calcium and cellular ATP levels. However, these effects were restored by OGA overexpression (Hu et al., 2009). These observations suggested that increased O-GlcNAcylation of mitochondrial proteins in cardiomyocytes led to impaired mitochondrial functions, by impairing oxidative phosphorylation. Interestingly, Ma et al. (2015) investigated rats that were acutely treated with the OGA inhibitor thiamet-G, using comparative proteomic and O-GlcNAcomic analyses, and identified over 88 O-glycosylated proteins of cardiac mitochondrial function, nearly half of which were localized to the oxidative phosphorylation system. They observed that acute O-GlcNAcylation elevation increased cardiac mitochondrial respiratory capacity, increased oxygen consumption, and regulated oxidative phosphorylation at multiple sites of the respiratory chain, thereby promoting ATP production rates and enhanced Ca2+ uptake. Mitochondrial dysfunction due to elevated O-GlcNAcylation caused by high glucose (Hu et al., 2009) may be the etiology of cardiomyopathy, which is one of the complications of diabetes. Since acute TMG treatment did not alter blood glucose levels, the acute increase in O-GlcNAcylation may have exerted a protective effect on cardiac mitochondria.
Another study reported that O-GlcNAcylation of ATP synthase subunit-α (ATP5A) was reduced in the brains of patients with Alzheimer’s disease (AD), AD mouse models, and amyloid β (Aβ)-treated cells (Cha et al., 2015). Aβ reduced the O-GlcNAcylation of ATP5A by inhibiting the direct interaction of mOGT with ATP5A, thereby decreasing ATPase activity and ATP production (Cha et al., 2015). Recently, Sacoman et al. (2017) proteomically identified 84 candidate mitochondrial glycoproteins, including two mitochondrial DNA-encoded proteins, cytochrome c oxidase subunit 2, and NADH-ubiquinone oxidoreductase chain 4. In addition, by reducing endogenous mOGT levels, membrane potential was lost, which led to mitochondrial content loss. Unexpectedly, OGT knockdown throughout the cell exerted no effects on mitochondrial contents and interconnectivity, suggesting that mOGT contributed to mitochondrial morphology (Sacoman et al., 2017). Furthermore, endogenous mOGT reduction was associated with increased mitochondrial respiration per mitochondrion, suggesting that a compensatory mechanism overcomes reduced mitochondrial contents. Dynamin-related protein 1 (DRP1), which drives mitochondrial fusion and fission, was identified at O-GlcNAc modification sites, and interestingly, elevated O-GlcNAcylation triggered DRP1 translocation from the cytoplasm to the mitochondria (Gawlowski et al., 2012). Moreover, Sacoman et al. (2017) also reported that mitochondrial fragmentation induced by mOGT down-regulation was dependent on DRP1. Banerjee, Ma & Hart (2015) observed OGA (mitochondrial OGA, mOGA) activity in mitochondria, leading to the possibility that mOGT/mOGA was involved in the O-GlcNAcylation regulation of DRP1, and whether mOGT affects DRP1 localization by O-GlcNAcylation. In addition, the protein nucleoplasmic transfer to generate mitochondrial O-GlcNAcylation has been recognized (Trapannone et al., 2016), but there are subunits encoded by mitochondrial DNA that never leave the mitochondria, suggesting that the O-GlcNAcylation of some mitochondrial proteins requires mOGT (Sacoman et al., 2017). Furthermore, UDP-GlcNAc were transported into the mitochondrial matrix via a mitochondrial inner membrane transporter (Banerjee, Ma & Hart, 2015). This suggests that an intra-mitochondrial O-GlcNAc cycle exists and that it has a vital role in mitochondrial metabolism and function. This cycle may also be influenced by intracytoplasmic O-GlcNAc (Fig. 5). The above data demonstrated that the O-GlcNAcylation was involved in mitochondrial metabolism regulation (Lozano et al., 2014), and supported the association of O-GlcNAcylation in mitochondrial oxidative phosphorylation.

Figure 5: O-GlcNAc cycling in mitochondria.
UDP-GlcNAc in the cytoplasm is transported to mitochondria where mitochondrial proteins are dynamically O-GlcNAcylated via mOGT and mOGA catalysis. In addition, O-GlcNAc modified proteins in the cytoplasm are also translocated to mitochondria.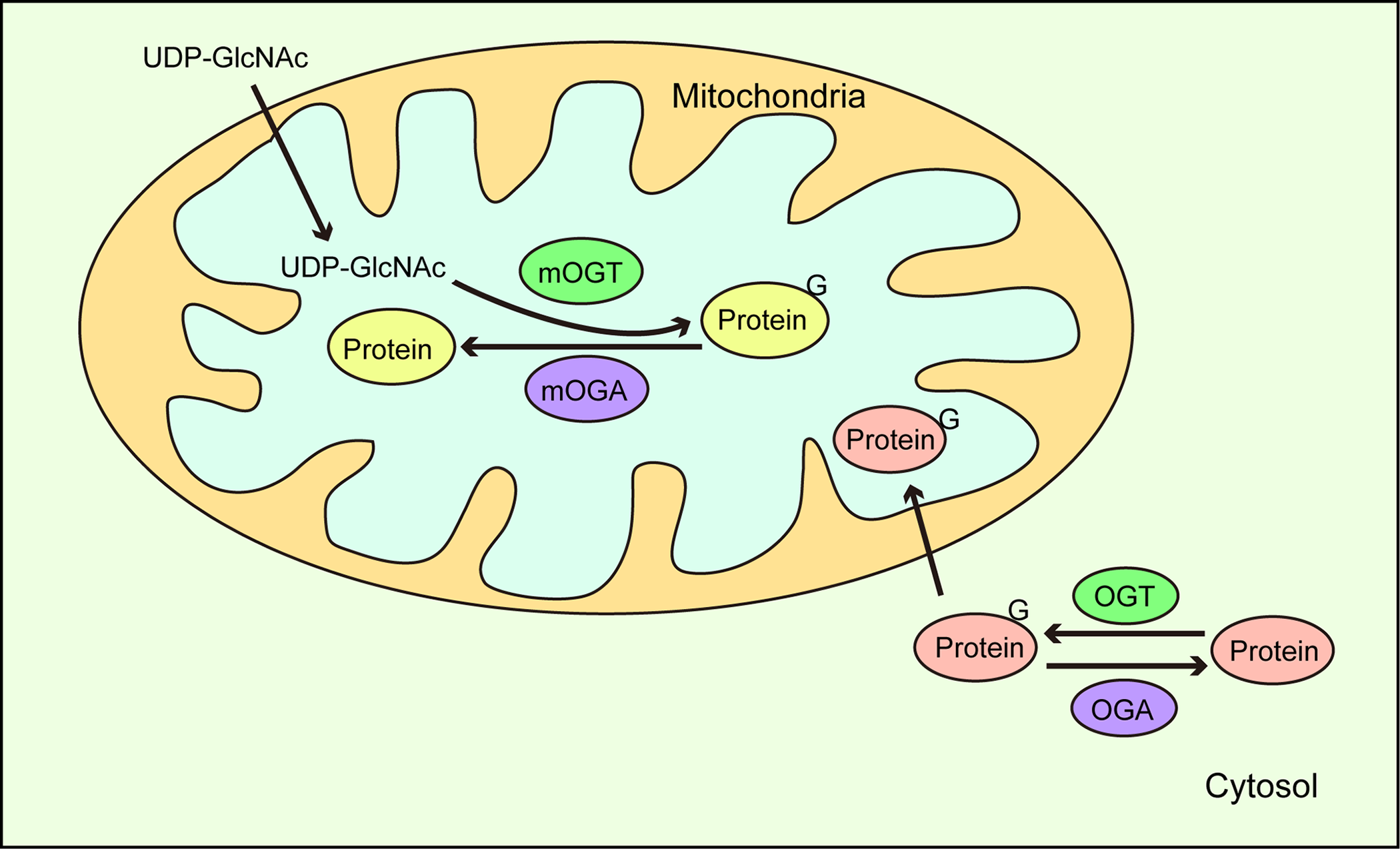{kind=link}
O-GlcNAcylation regulates the tricarboxylic acid cycle (TCA)
The TCA cycle is a common pathway for sugar, fat, and amino acid catabolism. These major nutrients can be transformed into each other through this cycle under certain conditions. However, there is not much research on O-GlcNAcylation in TCA cycle regulation. In 2008, Clark et al. reported that both the α and β subunits of the pyruvate dehydrogenase complex were O-GlcNAc modified. This was followed by Cao et al. (2013) who observed that malate dehydrogenase was a substrate for O-GlcNAcylation. In 2014, a proteomic analysis of mitochondrial protein changes, observed that protein abundance in mitochondrial respiratory chain and TCA cycle processes were decreased, regardless of OGT or OGA overexpression (Tan et al., 2014), suggesting O-GlcNAcylation effects on the TCA cycle. Interestingly, Tan et al. (2014) reported that adenovirus-mediated OGT/OGA overexpression in SY5Y neuroblastoma cells showed similar changes in the mitochondrial pathway. Subsequently, Ferrer et al. (2014) used liquid chromatography-mass spectrometry to measure TCA cycle intermediates in triple negative breast cancer cells (MDA-MB-231) and observed that O-GlcNAcylation inhibition by short-hairpin OGT increased TCA cycle metabolites. In 2016, Ma et al. (2016) using comparative proteomics techniques, observed that the vast majority of enzymes in the TCA cycle were O-GlcNAcylated and that some had multiple modification sites. It will be interesting to note if the O-GlcNAcylation of these enzymes translates to regulatory roles in the TCA cycle. A recent study reported that phosphoglycerate kinase 1 (PGK1), the first enzyme to produce ATP during glycolysis, was modified by O-GlcNAc at Thr255 (Nie et al., 2020). This modification promoted PGK1 activity, increased lactate production, induced PGK1 translocation to the mitochondria, and reduced mitochondrial oxidative phosphorylation by inhibiting the pyruvate dehydrogenase complex. However, blocking PGK1 Thr255 O-GlcNAcylation appeared to inhibit glycolysis, enhancing the TCA cycle (Fig. 2) and suppressing cancer cell proliferation (Nie et al., 2020). This study provided important insights into coordinated metabolic signals between glycolysis and the TCA cycle in O-GlcNAcylation (Figs. 2, 3). Associations between the O-GlcNAc cycle, mitochondrial biogenesis, and target proteins are being increasingly reported (Nugent & Bale, 2015; Ohashi et al., 2017; Tan et al., 2017). This evidence has suggested that O-GlcNAcylation plays important roles in the mitochondria. Indeed, O-GlcNAcylation regulation could be mechanistically exploited to respond to the reprogramming of mitochondrial metabolism.
O-GlcNAcylation regulates lipid metabolism
Lipids are widely distributed in cells and are important structural components of membranes and biofilms. They also serve as second messengers of intracellular signal transmission, and important energy sources during nutrient deficiency or cell growth (van Meer, Voelker & Feigenson, 2008; Holthuis & Menon, 2014; Efeyan, Comb & Sabatini, 2015). Lipid metabolism dysregulation is associated with the progression of a variety of metabolic diseases (Cohen, Horton & Hobbs, 2011; Schwartz et al., 2013). In 2005, Hanover et al. (2005) reported that a Caenorhabditis elegans OGT homozygous deletion mutant exhibited striking metabolic changes, exemplified by increased glycogen storage and decreased triglyceride levels. However, it was unclear whether triglyceride reduction was due to decreased synthesis or increased triglyceride mobilization. Fülöp et al. (2008) studied the effects of age on tissue O-GlcNAc levels and observed increased O-GlcNAcylation with age in all tissues examined, while serum triglyceride levels increased. This could indicate a link between O-GlcNAcylation and triglycerides. In 2011 Guinez et al. observed that O-GlcNAcylation of carbohydrate response element binding protein (ChREBP) increased self-stabilization, and transcriptional activity on the lipogenic genes, acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), and stearoyl-CoA desaturase 1 (SCD1). OGT overexpression increased ChREBP levels in the mouse liver, enhancing lipogenic gene expression, and the excessive deposition of hepatic triglycerides. OGA overexpression reduced ChREBP levels and decreased adipogenic protein levels of ACC and FAS, and ChREBP deglycosylation reduced lipid droplet accumulation in hepatocytes (Guinez et al., 2011). Similarly, O-GlcNAcylation of ChREBP induced lipid accumulation in mesangial cells (Park et al., 2014a). This evidence indicated that O-GlcNAcylation may induce increased triglyceride synthesis via increased ChREBP levels. In 2018, Sodi et al. reported that OGT downregulation in breast cancer cells decreased unsaturated fatty acid biosynthesis, pantothenate/coenzyme A biosynthesis, and fatty acid biosynthesis (Sodi et al., 2018). OGT inhibition degraded sterol regulatory element-binding protein 1 (SREBP-1) via phosphorylation of the AMPK pathway, and reduced expression of its transcriptional targets, ATP-citrate lyase (ACLY) and FAS, thereby decreasing lipid synthesis and causing cancer cell death (Sodi et al., 2018). Thus, this study identified a key role for the O-GlcNAcylation regulation of lipid metabolism in tumor cells.
Further studies revealed that liver X receptors (LXRs) maintained the expression of SREBP-1c, ChREBP-α, and the newly identified short isoform, ChREBP-β (Bindesbøll et al., 2015). LXRs were O-GlcNAcylated (Anthonisen et al., 2010). Bindesbøll et al. (2015) reported that LXRs and OGT interacted and were localized to the nucleus; they also discovered that LXRs deletion reduced nuclear O-GlcNAc signal transduction, including the O-GlcNAcylation of ChREBP-α (Bindesbøll et al., 2015) (Fig. 4). As the key to de novo fatty acid synthesis, FAS is also modified by O-GlcNAc; increased O-GlcNAcylation prevented FAS degradation and elevated its expression (Baldini et al., 2016b). Interestingly, Groves et al. (2017) observed that OGA interacted with FAS and decreased self-activity, whereas FAS overexpression increased stress-induced O-GlcNAcylation in U2OS cells (Groves et al., 2017). This suggests that there is a feedback mechanism for the FAS regulation of O-GlcNAcylation via OGA inhibition.
In addition to regulating glucose nutrient substrates, lipid synthesis is also controlled by insulin. SREBP-1c is one such transcription factor. Recent studies indicated that O-GlcNAcylation was a negative regulatory mechanism of insulin signaling transduction (Yang et al., 2008b; Whelan et al., 2010; Zhang, Yin & Yang, 2014). Increased OGT levels in mouse livers led to insulin resistance and dyslipidemia (Yang et al., 2008b). In addition, hepatic lipogenic pathways in animals and humans with diabetes were hyperactivated (Ruan et al., 2013) and associated with the abnormality of O-GlcNAcylation of the key regulatory factor (ACC, FAS, and SCD1) of lipogenesis. Furthermore, in adipose cells OGT caused the accumulation of N-arachidonyl ethanolamine, an endogenous appetite-inducing cannabinoid, and also activated de novo lipid desaturation, causing hyperorexia and promoting obesity (Li et al., 2018). In the mechanism, this regulation is related to the O-GlcNAcylation of PPARγ.
O-GlcNAcylation Regulates Amino Acid Metabolism
O-GlcNAcylation regulates glutamine metabolism
The amino acid glutamine is widely-distributed in mammalian physiology (Bergström et al., 1974; Wishart et al., 2013) and acts as a nitrogen donor for cellular growth. Glutamine is also used as an energy substrate for cell metabolism in the TCA cycle of the mitochondria. It participates in amino acid and lipid synthesis (DeBerardinis et al., 2007; Dang, Le & Gao, 2009), and is a key precursor for UDP-GlcNAc synthesis (Altman, Stine & Dang, 2016). The glutamine-driven HBP leads to the O-GlcNAc modification of intracellular target proteins (Swamy et al., 2016; Abramowitz et al., 2019; Akella, Ciraku & Reginato, 2019). Petrus et al. (2020) reported that high glutamine levels decreased glycolysis and intra-nuclear O-GlcNAcylation in human adipocytes. This observation exemplified the diversity and complexity of O-GlcNAcylation regulation by glutamine. However, there is little direct evidence on O-GlcNAcylation regulation towards glutamine metabolism. Studies have indicated some enzymes are modified by O-GlcNAc during glutamine metabolism, including glutamine synthetase and glutamine dehydrogenase (Clark et al., 2008). However, it is unclear if these modifications directly affect enzyme activity. Another recent study indicated that c-Myc regulated glutamine absorption and metabolism (Wise et al., 2008), and that c-Myc itself was modified and regulated by O-GlcNAc, suggesting an indirect regulatory role for O-GlcNAcylation in glutamine metabolism. Glutamine metabolism was also regulated by HIF-1α and SIRT1, both of which were modified by O-GlcNAc (Stegen et al., 2016; Ren et al., 2017; Bacigalupa, Bhadiadra & Reginato, 2018), potentially indicating another indirect regulatory link to glutamine metabolism.
Itkonen et al. (2016) recently reported that alanine levels decreased in prostate cancer cells treated with OGT inhibitors, suggesting that O-GlcNAcylation was involved in alanine metabolism. Few studies have reported O-GlcNAcylation regulation in amino acid metabolism, and future studies should be conducted to further our understanding of the O-GlcNAcylation regulatory mechanism on protein, glucose, and lipid metabolism.
O-GlcNAcylation regulates signaling pathways
Intracellular signal transduction coordinates physiological activities in response to different stimuli. Signal integration not only requires multiple signal inputs from different molecules, but also the triggering of multiple signal outputs via spatiotemporal regulation (Ong, Han & Yang, 2018). Given the ongoing interest in O-GlcNAcylation, it is theorized O-GlcNAcylation regulates various cellular processes, and functions as a key signal junction, or integrator (Hart, Housley & Slawson, 2007; Bond & Hanover, 2015). A typical example involves insulin signaling. After we eat, insulin binds to its receptor, phosphorylating downstream PI3K, and activating AKT. Then, AKT phosphorylates and inactivates GSK3 (Fang et al., 2000), promoting glycogen synthesis. AKT inhibits gluconeogenesis by phosphorylating FOXO, which promotes gluconeogenic gene transcription (Manning & Cantley, 2007) from the nucleus. In adipocytes, elevated O-GlcNAcylation promoted insulin signaling attenuation through key signaling O-GlcNAcylation proteins, including IRS-1 (Whelan et al., 2010), PI3K (Yang et al., 2008b), and AKT (Wang et al., 2012) (Fig. 4). O-GlcNAcylation at Thr305/312 reduced AKT activity by suppressing Thr308 phosphorylation (Wang et al., 2012). Interestingly, Onodera, Nam & Bissell (2014) reported that the downregulation of O-GlcNAcylation levels in three-dimensional cultured breast cancer cells inhibited AKT signaling. This observation suggested that AKT signaling regulation by O-GlcNAcylation may depend on cell fate. PI3K/AKT signaling was shown to regulate glucose uptake and utilization (Vander Heiden, Cantley & Thompson, 2009) and stimulated lipogenic gene expression (Chang et al., 2005). Another major downstream molecule of AKT is mTOR, which is a serine/threonine kinase. mTOR has been shown to be influenced by OGT stability and thus regulates O-GlcNAc modification (Park et al., 2014b). This may represent a feedback regulatory mechanism that responds to high O-GlcNAcylation levels.
HIF responds to oxygen changes in physiological and disease environments. HIF-1α is transcribed to activate GLUT1, 3, and glycolytic enzyme expression (Semenza, 2010). Under a normoxic state, hydroxylated HIF-1α is recognized by the E3 ligase von Hippel-Lindau (VHL) and then ubiquitinated and degraded in the proteasome. Under hypoxic conditions, the hydroxylation of HIF-1α with oxygen and α-ketoglutarate as substrates is inhibited, and its expression stabilized. A recent study by Ferrer et al. (2014) showed that reduced O-GlcNAcylation promoted HIF-1α hydroxylation and its interaction with VHL, causing HIF-1α degradation and a reduction in GLUT1 expression, thereby affecting metabolic reprogramming. The transcription factor, c-Myc was shown to participate in cell glycolysis and anabolism regulation (Soga, 2013). The O-GlcNAcylation of c-Myc stabilizes itself and thus affects cell metabolism (Chou, Hart & Dang, 1995; Chou & Hart, 2001). Similarly, the transcription factor, NF-κB promoted tumor cell switching from oxidative phosphorylation to glycolytic metabolism (Wang & Jin, 2010), with O-GlcNAcylation regulating its activation (Ramakrishnan et al., 2013). The O-GlcNAcylation of NF-κB p65 by high glucose was shown to inhibit the interaction of NF-κB with IκB, leading to the nuclear translocation of NF-κB and the activation of its target genes (Yang et al., 2008a). The O-GlcNAcylation promoted NF-κB activation in intestinal epithelial cells (Sun et al., 2020), and similarly, the activation of NF-κB by O-GlcNAcylation has also been confirmed in the placenta of hyperglycemia rats (Dela Justina et al., 2017). Elevated O-GlcNAcylation in the tumor microenvironment also promoted cancer progression via the downregulation of p38 MAPK activity and upregulation of the ERK1/2 signaling pathway (Moriwaki & Asahi, 2017). O-GlcNAcylation also played a role in myocardial hypertrophy by activating ERK1/2 (Ding et al., 2013). β-catenin was also O-GlcNAcylated, with elevated O-GlcNAcylation increasing β-catenin stability, nuclear accumulation, and transcriptional activity (Zhou et al., 2016; Harosh-Davidovich & Khalaila, 2018; Gao et al., 2019). β-catenin overexpression upregulated O-GlcNAcylation levels (Gao et al., 2019) to reveal a reciprocal regulatory mechanism between both molecules. Several examples of this phenomenon have been cited in the literature, e.g., O-GlcNAcylation influenced tumorigenesis by regulating the Hippo/YAP pathway (Peng et al., 2017), and cellular development and differentiation by regulating SOX2 and OCT4 activity (Jang et al., 2012). This evidence strongly supports the role of O-GlcNAcylation in regulating cellular metabolic processes, through a pervasive role in signaling pathways (Fig. 6).
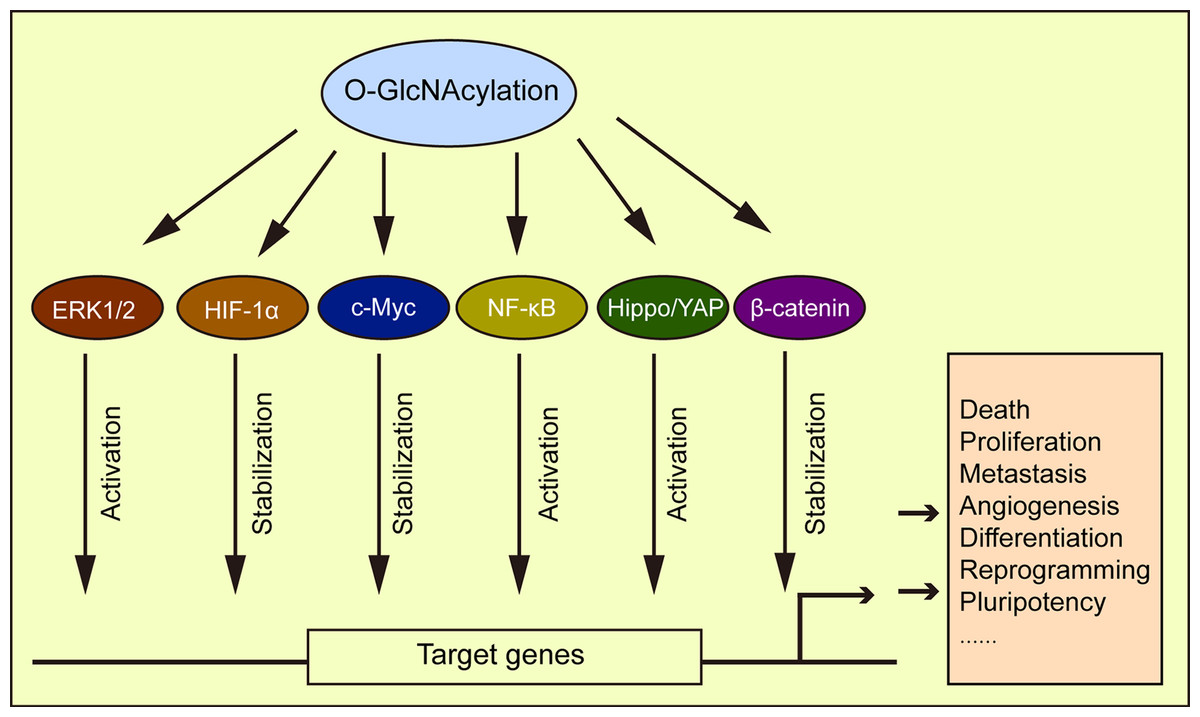
Figure 6: O-GlcNAcylation is associated with multiple signaling pathways.
O-GlcNAcylation directly or indirectly regulates different signaling pathways affecting a variety of cellular processes.{kind=link}
Maintaining O-GlcNAc homeostasis
O-GlcNAcylation is important for the spatiotemporal regulation of cell metabolism, and cellular O-GlcNAcylation homeostasis is vital for the maintenance of cell functions and the continuation of normal physiological processes. Thus, imbalanced homeostasis is directly implicated in the pathogenesis of many human diseases. However, the conditions under which imbalanced O-GlcNAcylation arise, and the mechanism for the maintenance of O-GlcNAcylation homeostasis are still unknown.
A buffer system has been proposed to maintain this intracellular O-GlcNAcylation balance through the mutual expression and activity regulation between OGT and OGA (Yang & Qian, 2017). Increased OGT activity leads to a concomitant increase in OGA activity and vice versa, thus buffering cells from drastic O-GlcNAcylation transformation (Burén et al., 2016). Furthermore, it was suggested that OGA functioned as a coactivator to upregulate OGT gene transcription by binding to the transcription factor, C/EBP-β (Qian et al., 2018) and the absence or inhibition of OGT reduced OGA expression (Kazemi et al., 2010). OGA’s pharmacological inhibition increased OGA gene transcription (Zhang et al., 2014). Park et al. (2017) reported that under high O-GlcNAcylation conditions, OGT introns were retained and OGT expression was suppressed, whereas, under low O-GlcNAcylation conditions, OGT introns were removed and OGT expression increased. A conservative OGT intron clipping muffler (ISS) was necessary for the retention of OGT introns, whereas the absence of an ISS disrupted OGT intron retention, and responses to metabolic conditions, thereby altering O-GlcNAcylation levels in cells (Park et al., 2017). This suggested a self-compensating mechanism to maintain the vital balance, however, autoregulation may be limited to tolerating fluctuations in moderate or acute stimuli (Yang & Qian, 2017) (Fig. 7). Typically the exposure to prolonged, severe, and chronic stress causes the O-GlcNAc homeostatic balance to be disrupted, generating physiological and metabolic cellular changes, and potentially impacting survival or resulting in specific cellular damage.
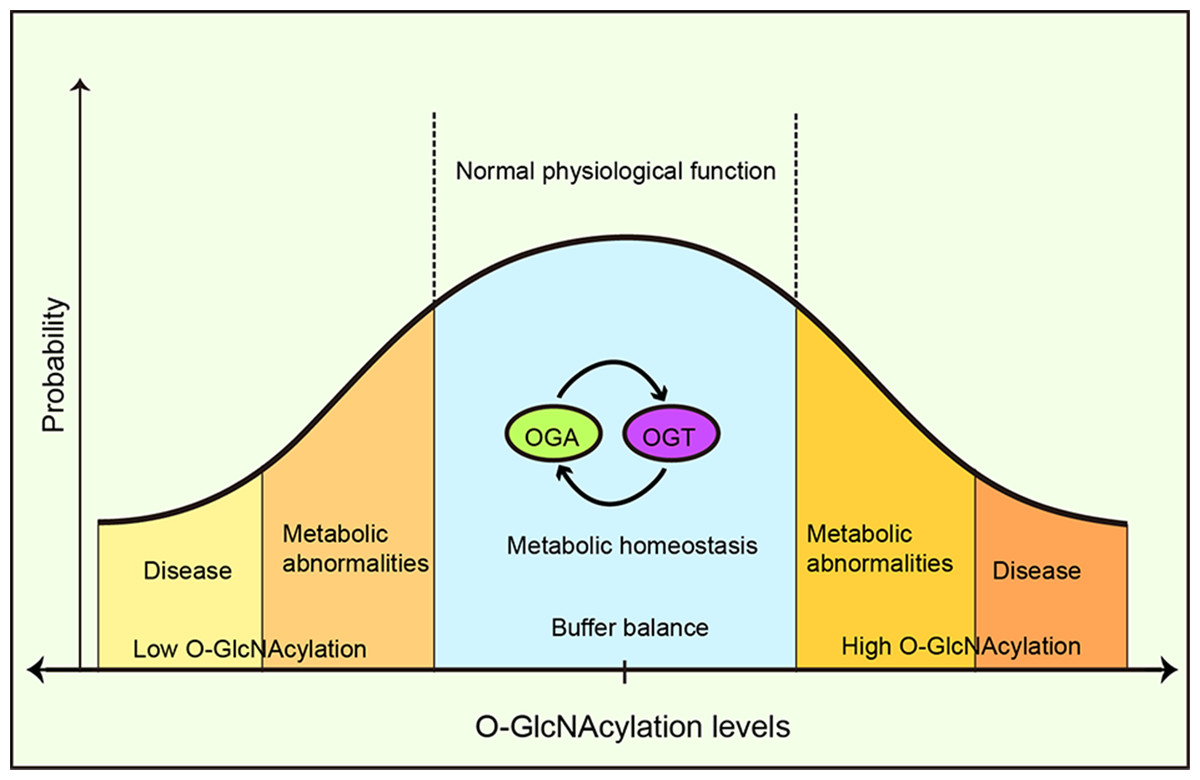
Figure 7: O-GlcNAcylation metabolic homeostasis is critical for normal cellularphysiology.
Balanced O-GlcNAcylation is maintained via the reciprocal regulation of OGT and OGA, ensuring metabolic homeostasis and normal cellular physiological functions. High or low OGlcNAcylation levels lead to metabolic abnormalities and/or disease.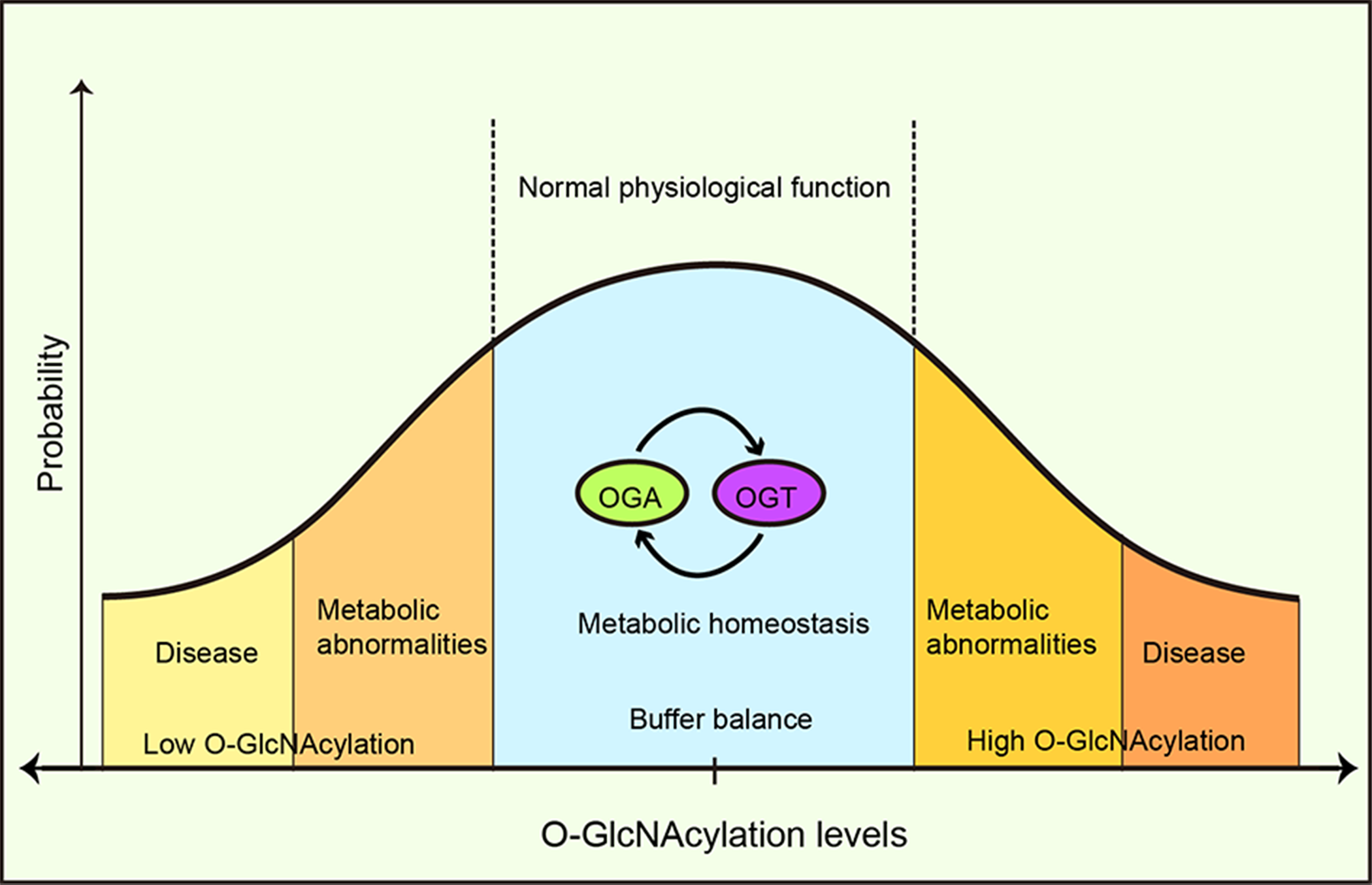{kind=link}
Conclusions
Remarkable advances in research have been made over the past 40 years in our understanding of the various biological functions underpinning O-GlcNAcylation. It is our current understanding that O-GlcNAcylation regulates some aspects of cellular metabolism. Our understanding of the cellular metabolic regulation in normal and disease states is increased by identifying O-GlcNAcylation mechanisms in functional proteins. There is considerable evidence to suggest that O GlcNAcylation is implicated in metabolic reprogramming, however, several molecular regulatory mechanisms require elucidation. Common O-GlcNAc signals in pathological states may become viable therapeutic targets. In this review, we summarized the roles of O-GlcNAcylation in glucose, lipid, biological oxidation, and amino acid metabolism, and discussed its role in physiological processes, such as cellular nutrient sensing, and homeostasis maintenance. O-GlcNAcylation typically acts as a regulator in multiple metabolic processes and pathways, signifying an influencing molecular interconnectivity between these processes. This notion provides us with an integrated understanding and foundation of potential communication exchanges between these metabolic pathways, all mediated by O-GlcNAcylation. O-GlcNAcylation response to stress may not be limited to key proteins, but more to the regulation of signaling and metabolism. Additional studies are needed to determine how O-GlcNAcylation coordinates and integrates related cellular and molecular networks.