
Characterization of the first vaginal Lactobacillus crispatus genomes isolated in Brazil
- Published
- Accepted
- Received
- Academic Editor
- Hector Mora-Montes
- Subject Areas
- Bioinformatics, Microbiology, Molecular Biology, Gynecology and Obstetrics, Women’s Health
- Keywords
- Lactobacillus, Genomics
- Copyright
- © 2021 Oliveira de Almeida et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2021. Characterization of the first vaginal Lactobacillus crispatus genomes isolated in Brazil. PeerJ 9:e11079 https://doi.org/10.7717/peerj.11079
Abstract
Background
Lactobacillus crispatus is the dominant species in the vaginal microbiota associated with health and considered a homeostasis biomarker. Interestingly, some strains are even used as probiotics. However, the genetic mechanisms of L. crispatus involved in the control of the vaginal microbiome and protection against bacterial vaginosis (BV) are not entirely known. To further investigate these mechanisms, we sequenced and characterized the first four L. crispatus genomes from vaginal samples from Brazilian women and used genome-wide association study (GWAS) and comparative analyses to identify genetic mechanisms involved in healthy or BV conditions and selective pressures acting in the vaginal microbiome.
Methods
The four genomes were sequenced, assembled using ten different strategies and automatically annotated. The functional characterization was performed by bioinformatics tools comparing with known probiotic strains. Moreover, it was selected one representative strain (L. crispatus CRI4) for in vitro detection of phages by electron microscopy. Evolutionary analysis, including phylogeny, GWAS and positive selection were performed using 46 public genomes strains representing health and BV conditions.
Results
Genes involved in probiotic effects such as lactic acid production, hydrogen peroxide, bacteriocins, and adhesin were identified. Three hemolysins and putrescine production were predicted, although these features are also present in other probiotic strains. The four genomes presented no plasmids, but 14 known families insertion sequences and several prophages were detected. However, none of the mobile genetic elements contained antimicrobial resistance genes. The genomes harbor a CRISPR-Cas subtype II-A system that is probably inactivated due to fragmentation of the genes csn2 and cas9. No genomic feature was associated with a health condition, perhaps due to its multifactorial characteristic. Five genes were identified as under positive selection, but the selective pressure remains to be discovered. In conclusion, the Brazilian strains investigated in this study present potential protective properties, although in vitro and in vivo studies are required to confirm their efficacy and safety to be considered for human use.
Introduction
The vaginal microbiota of reproductive-age women is classified into at least five types called community state types (CST). Four of them are dominated by Lactobacillus crispatus (CST-I), L. gasseri (CST-II), L. iners (CST-III), or L. jensenii (CST-V). The CST-IV is characterized as having a significantly lower number of lactobacilli and an increased number and diversity of strict and facultative anaerobes (Ravel et al., 2011). The lactobacilli use the glycogen supplied by the host as a carbon source and create a protective environment against infections or colonization by pathogens and non-indigenous microbes by the production of L- and/or D-lactic acid, bacteriocins, hydrogen peroxide, competition for tissue adhesion, enhancement of the protective mucus layer integrity and modulation of the innate immune system response (Ravel et al., 2011; Smith & Ravel, 2017).
CST-IV and III increase the risk of bacterial vaginosis (BV) due to L. iners being less effective in controlling the vaginal microbiota. BV is a condition characterized by a microbiota similar to CST-IV, vaginal pH > 4.5 and production of amino acid compounds, sometimes associated with clinical symptoms including discharge, fishy odor, and presence of clue cells. These conditions damage the host defenses and favor the development of opportunistic microorganisms that behave like pathogens (Smith & Ravel, 2017; Barrientos-Durán et al., 2020). BV can be asymptomatic or associated with gynecological and obstetric complications, besides increasing the risk of sexually transmitted infections (Barrientos-Durán et al., 2020).
CST-I, dominated by L. crispatus, is most associated with vaginal health. This species is considered a biomarker of a healthy microbiota, and some strains are used as a probiotic to treat BV. Its effect as a probiotic is not entirely clear, but it is believed to involve competitive exclusion strategies (Almeida et al., 2019). It was shown to outcompete Gardinerella vaginallis for tissue adhesion in vitro (Ojala et al., 2014) and, as other lactobacilli, inhibit the growth of pathogens in vivo by the production of lactic acid, but not hydrogen peroxide (Tachedjian, O’Hanlon & Ravel, 2018). The decrease in L. crispatus is associated with BV, but the causes are not well understood. The differences in persistence and protection among strains is probably influenced by genetic differences that, if described, could be applied in the screening of more efficient probiotic strains (Almeida et al., 2019).
Comparative genomic analyses have been performed to identify genomic features of L. crispatus associated with female urogenital tract using samples from North America, Europe and Asia (Ojala et al., 2014; Abdelmaksoud et al., 2016; France, Mendes-Soares & Forney, 2016; Van der Veer et al., 2019; Pan, Hidalgo-Cantabrana & Barrangou, 2020; Petit & Read, 2020; Zhang et al., 2020). Analyses that searched for genomic features associated with lactobacilli-dominated (healthy) or BV microbiota were performed using samples isolated from the USA (Abdelmaksoud et al., 2016) and Netherlands (Van der Veer et al., 2019). Those studies point out mechanisms related to persistence during BV, such as phase variation, rather than protection against this infirmity (Van der Veer et al., 2019). In Latin America, although L. crispatus has been previously studied in Brazil as vaginal isolates (Branco et al., 2010) or as part of the vaginal microbiota (Martinez et al., 2008; Marconi et al., 2020), no complete genome has been sequenced, analyzed, and deposited in GenBank.
In this study, we characterize the first L. crispatus genomes isolated in Brazil from healthy vaginal microbiomes and used genome-wide association study (GWAS) and positive selection analyses to identify genetic mechanisms involved healthy or BV conditions and selective pressures acting in the vaginal microbiome.
Materials and Methods
Genome sequencing, assembly, and annotation
In a previous study, vaginal fluid samples were collected from individuals diagnosed as healthy or with BV, with the approval of the ethics committee in research (COEP) of the Federal University of Minas Gerais (protocols ETIC 062/03 and 212/07) (Branco et al., 2010). L. crispatus strains were identified by cellular morphology (Gram-positive bacilli or coccobacilli), biochemistry test (catalase-negative) and 16S-23S rRNA restriction profiling (Branco et al., 2010). The genomes of strains CRI4, CRI8, CRI10 and CRI17 isolated from four healthy patients were sequenced using HiSeq 2500 (Illumina, San Diego, CA, USA) with paired-end libraries of 2 × 150 bp. The sequencing reads quality was examined using FastQC v0.11.8 (Andrews, 2015). The sequencing reads were mapped to the genomes of 46 L. crispatus vaginal isolates to filter out contaminants (Table 1) using bowtie v2 (Langmead & Salzberg, 2012), and the mapped reads were extracted using Samtools v1.7-2 (Li et al., 2009).
| Strain | Condition | Microbiome | Metadata | Country | GenBank | Reference |
|---|---|---|---|---|---|---|
| RL02 | BV | DVM | BV-positive | Netherlands | NKLR01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL07 | BV | DVM | BV-positive | Netherlands | NKLN01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL13 | BV | DVM | BV-positive | Netherlands | NKLI01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL14 | BV | DVM | BV-positive | Netherlands | NKLH01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL15 | BV | DVM | BV-positive | Netherlands | NKLG01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL17 | BV | DVM | BV-positive | Netherlands | NKLE01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL19 | BV | DVM | BV-positive | Netherlands | NKLD01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL20 | BV | DVM | BV-positive | Netherlands | NKLC01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL21 | BV | DVM | BV-positive | Netherlands | NKLB01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL23 | BV | DVM | BV-positive | Netherlands | NKLA01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL24 | BV | DVM | BV-positive | Netherlands | NKKZ01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL25 | BV | DVM | BV-positive | Netherlands | NKKY01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL28 | BV | DVM | BV-positive | Netherlands | NKKV01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL30 | BV | DVM | BV-positive | Netherlands | NKKT01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL31 | BV | DVM | BV-positive | Netherlands | NKKS01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL33 | BV | DVM | BV-positive | Netherlands | NKKQ01 | Dols et al. (2016), Van der Veer et al. (2019) |
| VMC1 | BV | DVM | History of BV. <50% lactobacilli and >50% of BV-associated taxa | USA | LJCZ01 | Abdelmaksoud et al. (2016) |
| VMC2 | BV | DVM | History of BV. <50% lactobacilli and >50% of BV-associated taxa | USA | LJDA01 | Abdelmaksoud et al. (2016) |
| VMC3 | BV | DVM | History of BV. <50% lactobacilli and >50% of BV-associated taxa | USA | LJGP01 | Abdelmaksoud et al. (2016) |
| VMC4 | BV | DVM | History of BV. ~86% L. crispatus and ~12% BV-associated taxa | USA | LJGQ01 | Abdelmaksoud et al. (2016) |
| CRI4 | Healthy | – | Healthy | Brazil | JABERN01 | This study |
| CRI8 | Healthy | – | Healthy | Brazil | JABERO01 | This study |
| CRI10 | Healthy | – | Healthy | Brazil | JABERP01 | This study |
| CRI17 | Healthy | – | Healthy | Brazil | JABERQ01 | This study |
| 2029 | Healthy | – | Healthy, probiotic strain | Russia | AVFH2 | Abramov et al. (2014) |
| 125-2-CHN | Healthy | – | Healthy | China | ACPV01 | Ojala et al. (2014), www.beiresources.org |
| AB70 | Healthy | – | Healthy | South Korea | CP026503, CP026504 | Chang et al. (2019) |
| CIP 104459 | Healthy | – | Healthy | France | VOMA01 | Clabaut et al. (2020) |
| CTV-05 | Healthy | – | Healthy, probiotic strain | – | ADML01 | Hemmerling et al. (2010), Ojala et al. (2014) |
| JV-V01 | Healthy | – | Normal human vaginal flora | – | ACKR01 | Witkin et al. (2013), Ojala et al. (2014), www.beiresources.org |
| MV-1A-US | Healthy | – | Healthy | USA | ACOG02 | Witkin et al. (2013), Ojala et al. (2014), www.beiresources.org |
| MV-3A-US | Healthy | – | Healthy | USA | ACQC01 | Witkin et al. (2013), Ojala et al. (2014), www.beiresources.org |
| RL03 | Healthy | LVM | BV-negative | Netherlands | NKLQ01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL05 | Healthy | LVM | BV-negative | Netherlands | NKLP01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL06 | Healthy | LVM | BV-negative | Netherlands | NKLO01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL08 | Healthy | LVM | BV-negative | Netherlands | NKLM01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL09 | Healthy | LVM | BV-negative | Netherlands | NKLL01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL10 | Healthy | LVM | BV-negative | Netherlands | NKLK01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL11 | Healthy | LVM | BV-negative | Netherlands | NKLJ01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL16 | Healthy | LVM | BV-negative | Netherlands | NKLF01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL26 | Healthy | LVM | BV-negative | Netherlands | NKKX01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL27 | Healthy | LVM | BV-negative | Netherlands | NKKW01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL29 | Healthy | LVM | BV-negative | Netherlands | NKKU01 | Dols et al. (2016), Van der Veer et al. (2019) |
| RL32 | Healthy | LVM | BV-negative | Netherlands | NKKR01 | Dols et al. (2016), Van der Veer et al. (2019) |
| SJ-3C-US | Healthy | – | Healthy | Iran | ADDT01 | Eslami et al. (2016) |
| V4 | Healthy | – | Healthy | France | SRLG01 | Clabaut et al. (2019) |
| VMC5 | Healthy | LVM | No history of BV. >90 % L. crispatus and <10 % BV-associated bacterial taxa | USA | LJOK01 | Abdelmaksoud et al. (2016) |
| VMC6 | Healthy | LVM | No history of BV. Dominated by L. crispatus and L. jensenii | USA | LJOL01 | Abdelmaksoud et al. (2016) |
| VMC7 | Healthy | LVM | No history of BV. >90 % L. crispatus and <10 % BV-associated bacterial taxa | USA | LJOM01 | Abdelmaksoud et al. (2016) |
| VMC8 | Healthy | LVM | No history of BV. >90 % L. crispatus and <10 % BV-associated bacterial taxa | USA | LJON01 | Abdelmaksoud et al. (2016) |
Note:
LVM, Lactobacilli-dominated vaginal microbiota; DVM, Dysbiotic vaginal microbiota.
Ten different assemblies were generated for each genome using SPAdes v3.14.0 (Bankevich et al., 2012) (assembly 1), Unicycler v0.4.5 (Wick et al., 2017) with SPAdes v3.14.0 (2), ABySS v2.0 (Simpson et al., 2009) (3), MaSuRCA v3.4.0 (Zimin et al., 2013) (4) and an in house pipeline (https://github.com/engbiopct/assembly-hiseq) that generates six other assemblies (5–10). In the in-house pipeline, six strategies combine different software. The best k-mer values were identified using KmerStream v1.1 (Melsted & Halldórsson, 2014). Adapters were removed using AdapterRemoval v2.3.1 (Schubert, Lindgreen & Orlando, 2016). The genome assemblers were Edena v3.131028 (Hernandez et al., 2008) and SPAdes v3.13.0 (Bankevich et al., 2012). The six assembly strategies were: Edena (5), KmerStream and SPAdes (6), KmerStream and SPAdes, using Edena assembly as trusted contigs (7), AdapterRemoval, KmerStream and SPAdes (8), reads processed by AdapterRemoval and the raw reads as input, KmerStream, and SPAdes, using Edena assembly as trusted contigs (9), and AdapterRemoval, KmerStream, and SPAdes, using Edena assembly as trusted contigs (10).
The best assembly for each genome was determined using QUAST v5.0.2 (Gurevich et al., 2013). Then, the genome’s paired-read sequencing data was used for scaffolding with SSPACE v3.0 (Boetzer et al., 2011) and contig extension and gap filling with GapFiller v1.1.1 (Boetzer & Pirovano, 2012). Finally, more gaps were closed using contigs from the other nine assemblies and the chromosome of L. crispatus strain AB70 (CP026503.1) (Chang et al., 2019) as a reference, using GFinisher (Guizelini et al., 2016). The presence of plasmids was investigated using PlasmidFinder 2.1 (Carattoli et al., 2014). The four genomes were identified from the species L. crispatus using the Type (Strain) Genome Server (Meier-Kolthoff & Göker, 2019). The assemblies completeness was evaluated using BUSCO v4.0.6 (Seppey, Manni & Zdobnov, 2019), based on the presence of 402 single-copy orthologous genes shared within Lactobacillales. The genomes were annotated using Prokka v1.11 (Seemann, 2014). The GenBank accession numbers of the genomes from strains CRI4, CRI8, CRI10 and CRI17, isolated from healthy patients, are JABERN01, JABERO01, JABERP01 and JABERQ01, respectively (Table 2). Type Strain Genome Server (Meier-Kolthoff & Göker, 2019) was used to confirm the taxonomic classification of the 50 samples as L. crispatus strains.
| CRI4 | CRI8 | CRI10 | CRI17 | |
|---|---|---|---|---|
| SRA accession | SRR13201099 | SRR13201098 | SRR13201097 | SRR13201096 |
| Replicon accession | JABERN01 | JABERO01 | JABERP01 | JABERQ01 |
| Completeness (%) | 99.5 | 99.2 | 99.0 | 99.0 |
| Size (bp) | 2,376,268 | 2,330,310 | 2,418,420 | 2,384,332 |
| Contig | 100 | 65 | 65 | 79 |
| N50 (bp) | 44.691 | 69.313 | 75.243 | 58.032 |
| L50 | 19 | 10 | 11 | 14 |
| CDS | 2.438 | 2.329 | 2.478 | 2.393 |
| Plasmid | None | None | None | None |
Criteria of public genomes selection
We included in the analysis genomes available in public databases using the following criteria: (i) vaginal isolate, (ii) the metadata explicitly informs the health condition of the individual, and/or the microbiome classification. A total of 46 samples were classified as belonging to “healthy” (26) or “BV” (20) condition groups and were used in previous studies (Hemmerling et al., 2010; Ojala et al., 2014; Abramov et al., 2014; Abdelmaksoud et al., 2016; Eslami et al., 2016; Dols et al., 2016; Chang et al., 2019; Clabaut et al., 2019, 2020; Van der Veer et al., 2019). Table 1 shows the genomes list, including the microbiome classification, when available, and the terms in metadata used to classify the sample.
Probiotic features
Bacteriocins and linear azol(in)e-containing peptides (LAPs) were predicted using BAGEL4 (Van Heel et al., 2018). The enzymes involved in the production of L- and D-lactate and hydrogen peroxide were identified using KEGG Mapper/BlastKOALA, under the pyruvate metabolism pathway. Adhesins were predicted using eggNOG-mapper v2 (Huerta-Cepas et al., 2017). Protein IDs were identified using BLASTp (Camacho et al., 2009) using the GenBank non-redundant (nr) database, selecting hits with 100% identity and coverage. Pathways involved in the biosynthesis of antimicrobial drugs with clinical importance (Chokesajjawatee et al., 2020) were predicted using KEGG Mapper.
Safety assessment of the strains for probiotic applications
Detection of plasmids, insertion sequences, prophages, and CRISPR-Cas elements was performed using PlasmidFinder (Carattoli et al., 2014), ISEScan (Xie & Tang, 2017), PHASTER (Arndt et al., 2016), and CRISPRCasFinder (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index), respectively. The domains of the identified Cas proteins were predicted using InterProScan (Jones et al., 2014) and NCBI’s Conserved Domain Database (Lu et al., 2020). Multiple alignments of sequences of interest were performed using the Clustal Omega web service (Larkin et al., 2007). Local alignment of sequences of interest across genomes was performed using BLASTn (Camacho et al., 2009) implemented in PATRIC (Davis et al., 2020).
Virulence factor genes were detected using the databases VFDB (Liu et al., 2019) and Ecoli_VF (https://github.com/phac-nml/ecoli_vf), while antimicrobial resistance genes were detected using the databases ARG-ANNOT (Gupta et al., 2014), CARD (Alcock et al., 2020), MEGARes (Doster et al., 2019), NCBI AMRFinderPlus (Feldgarden et al., 2019) and ResFinder (Bortolaia et al., 2020). The screening using these databases was performed using ABRicate (https://github.com/tseemann/abricate) with default parameters.
The presence of the toxins hemolysins, enzymes involved in the synthesis of biogenic amines and other undesirable genes, as listed by Chokesajjawatee et al. (2020), were manually screened using KEGG Mapper (Kanehisa & Sato, 2020) implemented in BlastKOALA v2.2 (Kanehisa, Sato & Morishima, 2016).
In vitro detection of phages
One representative strain (L. crispatus CRI4) was subjected to the induction of lysogenic bacteriophages according to previous studies (Kiliç et al., 1996; Raya & H’bert, 2009). A total of 200 µL of the culture was initially inoculated in 5 mL of Man–Rogosa–Sharpe medium. The optical density (OD) was measured until reaching DO600 = 0.2. Then, 0.4 µg/mL of Mitomycin C (Sigma, St. Louis, MO, USA) was added in the culture and incubated at 37 °C overnight. Afterward, the supernatant was collected and filtered in sterile 0.22 µm membrane. A 10 µL sample of the filtered lysate was applied to a 200 mesh grid at the UFMG electron microscopy center (CM-UFMG) and visualization was performed in Tecnai G2-12 Transmission Electron Microscope, SpiritBiotwin FEI, 120 kV.
Comparative analyses using public genomes
We performed comparative analysis including the for strains from Brazil and 46 public genomes (Table 1) to reconstruct their phylogeny and to identify adaptations that could be related to the colonization of the vaginal niche by testing for association between gene presence/absence and features of interest, and detection of positive selection in protein-coding genes.
For phylogenomic analysis, L. helveticus DSM20075 genome (GenBank accession ACLM01) was used as an outgroup, the conserved genes across all 51 genomes were estimated by Roary v3.6.0, and their nucleotide sequences were aligned using MAFFT (Katoh et al., 2005) implemented in Roary. The alignment was used as input for IQ-Tree v1.6.12 (Nguyen et al., 2015) for phylogenetic inference using the Maximum Likelihood. The confidence values were estimated using 1,000 rounds of bootstrapping. The tree was edited using iTOL (Letunic & Bork, 2019).
We tested the associations of gene presence/absence with a health condition and geographical locations suggested by the phylogenetic tree. A GWAS based on gene presence/absence was performed using Scoary v1.6.16 (Brynildsrud et al., 2016). Scoary estimates association by pairwise comparisons on a phylogeny (Maddison, 2000) to correct population structure and permutation. The input was a gene presence-absence matrix from the 50 L. crispatus genomes estimated using Roary and a phylogenetic tree generated by IQ-Tree v1.6.12, utilizing the core gene alignment calculated by Roary, and a matrix containing the presence-absence of the features across the samples.
A genome-scale positive selection analysis was performed using POTION v1.1.2 (Hongo et al., 2015). To generate the input, FastOrtho (https://github.com/PATRIC3/FastOrtho) was used to identify ortholog groups across the 50 L. crispatus genomes. The file containing the orthologous group’s information and multifasta files containing nucleotide sequences of protein-coding genes were used as input for POTION v1.1.2. The genome-scale positive selection analysis used site tests with the models M1 and M2, and M7 and M8 (Yang & Nielsen, 2002). The POTION configuration file is available as Data S1. The function of the identified proteins was annotated using eggNOG-mapper (Huerta-Cepas et al., 2017), the subcellular localization using SufG+ v1.2.1 (Barinov et al., 2009), and the GenBank protein ID using BLASTp (Agarwala et al., 2016).
Results
Genome sequencing and taxonomy
The four sequenced genomes were assembled as drafts, and no plasmid was found. Table 2 shows the statistics of genome assembly and annotation. The genomes completeness ranged from 99% to 99.5%, while the reference strain AB70, available as a complete genome sequenced using PacBio RS II platform (Chang et al., 2019), had its completeness estimated as 99%. The four sequenced genomes and the 46 public genomes were classified as L. crispatus by TYGS, with dDDH > 70% and G+C content divergence of less than 1% to the strain JCM 1185T (Data S2).
Probiotic features
Features associated with probiotic effects in the four strains from Brazil are shown in Table 3. We identified genes involved in the biosynthesis of D-lactate (1), L-lactate (3), hydrogen peroxide (1), bacteriocins (9), LAPs (1) and adhesins (10, five classes) across the four genomes. No pathway for biosynthesis of antimicrobial drugs of clinical importance was found.
| Strain | |||||||
|---|---|---|---|---|---|---|---|
| Feature | Product (Gene) | KEGG ID | Protein ID | CRI4 | CRI8 | CRI10 | CRI17 |
| Lactic acid synthesis | |||||||
| D-lactate | 1 | 1 | 1 | 1 | |||
| D-lactate dehydrogenase [EC:1.1.1.28] (ldhA) | K03778 | WP_005720611 | 1 | 1 | 1 | 1 | |
| L-lactate | 3 | 3 | 3 | 3 | |||
| L-lactate dehydrogenase [EC:1.1.1.27] (ldh) | K00016 | WP_005721100 | 1 | 1 | 1 | 1 | |
| K00016 | WP_005720302 | 1 | WP_170080485 | 1 | WP_005721074 | ||
| L-2-hydroxyisocaproate dehydrogenase (hicDH) | K00016 | WP_005727148 | WP_005719855 | 1 | 1 | 1 | |
| Bacteriocin | |||||||
| Class: 210.2; SakT_alpha | 2 | 2 | 3 | 2 | |||
| ggmotif; ComC; Bacteriocin_IIc; | – | WP_005721006 | 1 | 1 | 1 | 1 | |
| ComC; L_biotic_typeA; Bacteriocin_IIc; 20.2; bacteriocin_LS2chaina | WP_005721005 | 1 | 1 | 1 | 1 | ||
| ComC; L_biotic_typeA; Bacteriocin_IIc; | – | WP_005720990 | – | – | 1 | – | |
| Class: 70.3; Helveticin-J | 1 | 2 | 2 | 2 | |||
| 70.3; Helveticin-J | – | WP_005729773 | – | 1 | 1 | 1 | |
| – | WP_005720754 | 1 | 1 | 1 | 1 | ||
| Class: 64.3; Enterolysin_A | 1 | 2 | 2 | 2 | |||
| 64.3; Enterolysin_A | – | WP_005728076 | – | 1 | 1 | 1 | |
| – | WP_005719715 | 1 | 1 | 1 | 1 | ||
| Class: 163.2; Penocin_A | – | – | 2 | 1 | |||
| bacteriocinII; Bacteriocin_II; ComC; Bacteriocin_IIc; 163.2; Penocin_A | – | WP_005723822 | – | – | 1 | 1 | |
| Bacteriocin_IIc; | – | WP_005727428 | – | – | 1 | – | |
| Class: 6.3; Bacteriocin_helveticin_J | 1 | 1 | 1 | 1 | |||
| 6.3; Bacteriocin_helveticin_J | – | WP_005728268 | WP_005718134 | 1 | 1 | 1 | |
| Class: LAPs | - | - | 1 | - | |||
| Putative nitr oreductase MJ1384 | – | WP_005721909 | – | – | 1 | – | |
| Hydrogen peroxide synthesis | |||||||
| Pyruvate oxidase [EC:1.2.3.3] (poxL) | K00158 | WP_005723618 | 1 | 1 | 1 | 1 | |
| Adhesin | |||||||
| Putative adhesin | WP_005728236 | 1 | 1 | 1 | 1 | ||
| WP_005729490 | – | 1 | 1 | 1 | |||
| Antimicrobial production | – | – | – | – | – | – | – |
Safety assessment of the strains for probiotic applications
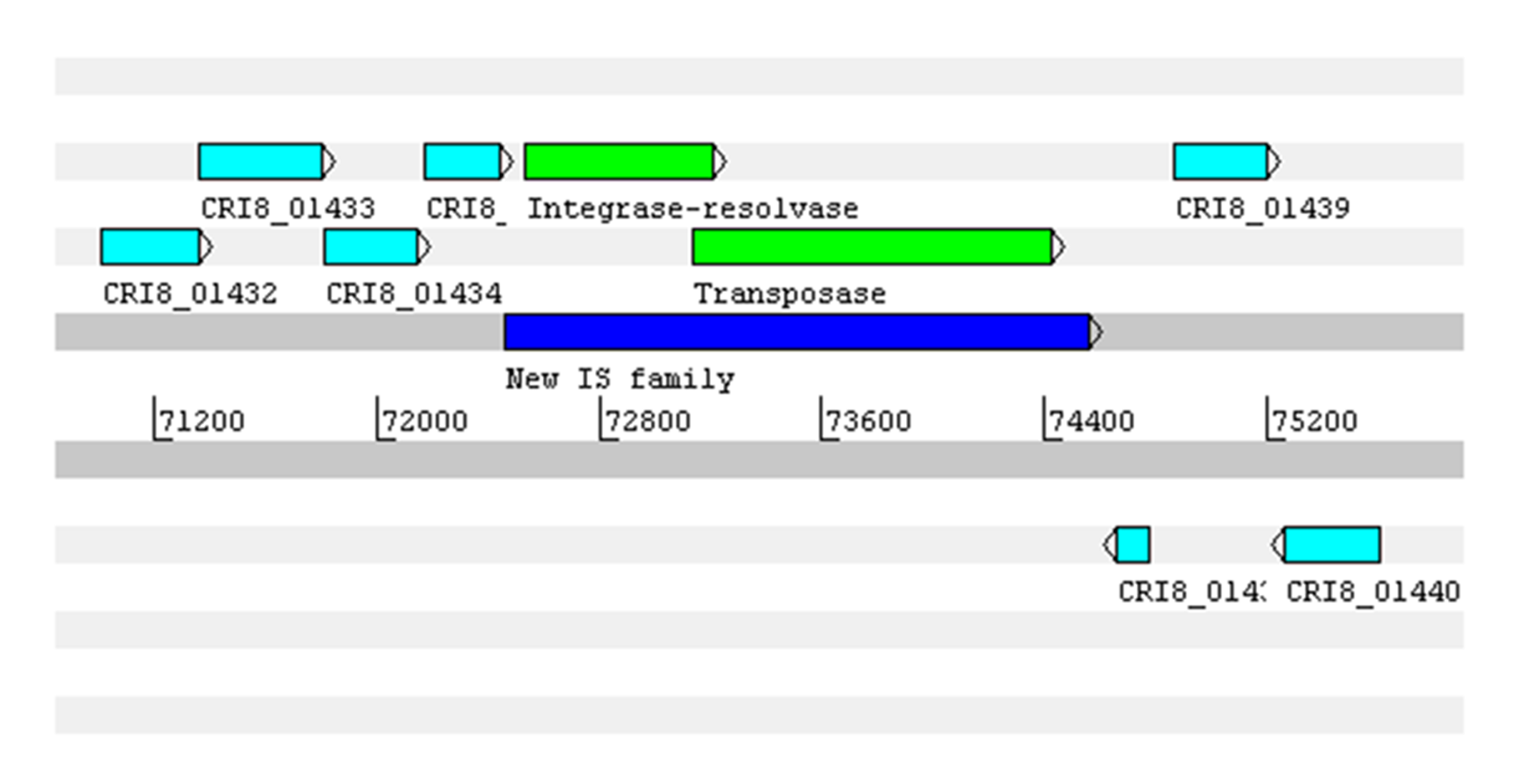
Features associated with safety in the four strains from Brazil are shown in Table 4. About mobile elements, no plasmid was predicted, as previously stated. A total of 131 to 184 IS from 14 known families were identified in the four genomes, and one new family was predicted across CRI8 (2 copies), CRI10 (2) and CRI17 (1) (Data S3). The new IS has a size of 2,088 bp and harbors two genes coding a site-specific integrase-resolvase (IS607-like family, GenBank protein ID WP_005728427.1) and a transposase (IS605 family, AZR15009.1) (Data S4). The multiple alignments of the five copies show the IS is fragmented in the contig 44 of strain CRI10, and the difference among the complete sequences is a SNP (T to G) in position 1,350 (Data S5). A BLASTn against the four strains from Brazil and other 123 public L crispatus genomes database identified hits with ≥99% identity and ≥98% coverage with 42 genomes from strains isolated from the female human urogenital tract, 39 of them public genomes (Data S3).
| Strain | |||||||
|---|---|---|---|---|---|---|---|
| Feature | Product (Gene) | KEGG ID | BLASTp hit | CRI4 | CRI8 | CRI10 | CRI17 |
| Mobile elements | |||||||
| Plasmid | – | – | – | – | |||
| Insertion sequences | 131 | 171 | 178 | 184 | |||
| Prophages | 2 questionable, 4 incomplete | 5 incomplete | 1 questionable, 5 incomplete | 1 questionable, 4 incomplete | |||
| CRISPR-Cas system | CAS-TypeIIA | CAS-TypeIIA | CAS-TypeIIA, CAS-TypeIIC | CAS-TypeIIA | |||
| Bacterial toxins | |||||||
| Hemolysin A (tlyA) | K06442 | WP_005723149 | – | 1 | 1 | 1 | |
| Putative hemolysin (tlyC) | K03699 | WP_005727867 | 1 | 1 | 1 | 1 | |
| Hemolysin-III related (hlyIII) | K11068 | WP_005720215 | 1 | 1 | 1 | 1 | |
| Bile salt deconjugation | |||||||
| Choloylglycine hydrolase/bile salt hydrolase (cbh) | K01442 | WP_005718943 | 1 | 1 | 1 | 1 | |
| Biogenic amine formation | |||||||
| Ornithine decarboxylase (odcI) | K01581 | WP_005727730 | 1 | 1 | 1 | 1 | |
| Antimicrobial resistance | – | – | – | – | – | – | – |
Five or six prophages were predicted across the four genomes, classified as questionable (score 70–90) or incomplete (score < 70), with most of them close to contig ends. In strains CRI8 and CRI17, some of the predicted prophages harbored the new IS (Fig. 1; Data S6). We identified CRISPR-Cas systems subtype II-A in the four strains and subtype II-C only in strain CRI10 (Table 4; Data S7). A closer examination of the subtype II-A system reveals that all four genomes contain CRISPR loci and Cas proteins-encoding genes cns2, cas2, cas1, and cas9. However, a transposase is inserted in csn2 in each strain, resulting in two gene fragments. The gene cas9 is also fragmented in all strains, with a transposase inserted between fragments in strains CRI4 and CRI8. CRI17 has an extra copy of cas9. The subtype II-C system is represented in CRI10 by a single cas9 gene (Fig. 2). All the cas9 CDSs lack one or more domains.
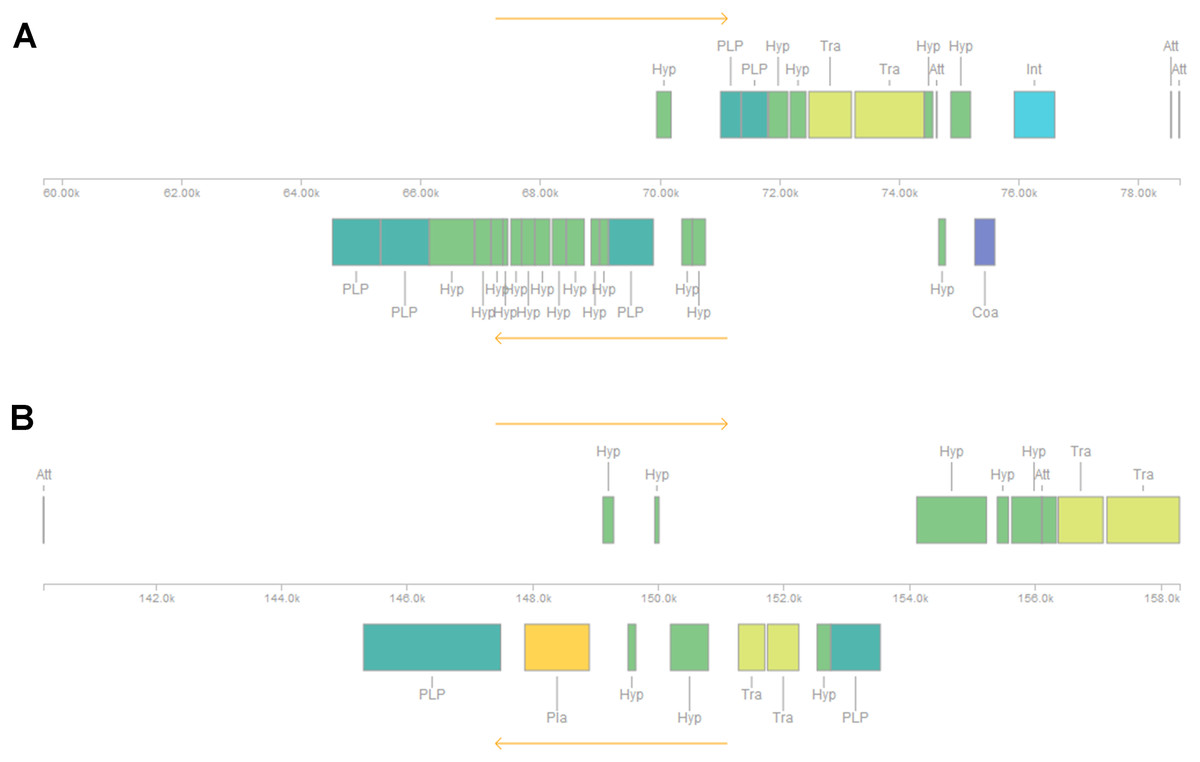
Figure 1: Insertion sequence from IS607-like family located in two prophages (regions) in Lactobacillus crispatus strain CRI8.
(A) Region 2 and (B) Region 3. The insertion sequences from the IS607-like family are two subsequent transposases, located in region 2 and at the end of region 3. Att, Attachment Site; Coa, Coat protein (purple); Hyp, Hypothetical protein (green); Int, Integrase (blue); Pla, Plate protein (orange); PLP, Phage-like Protein (cyan); Tra, Transposase (olive).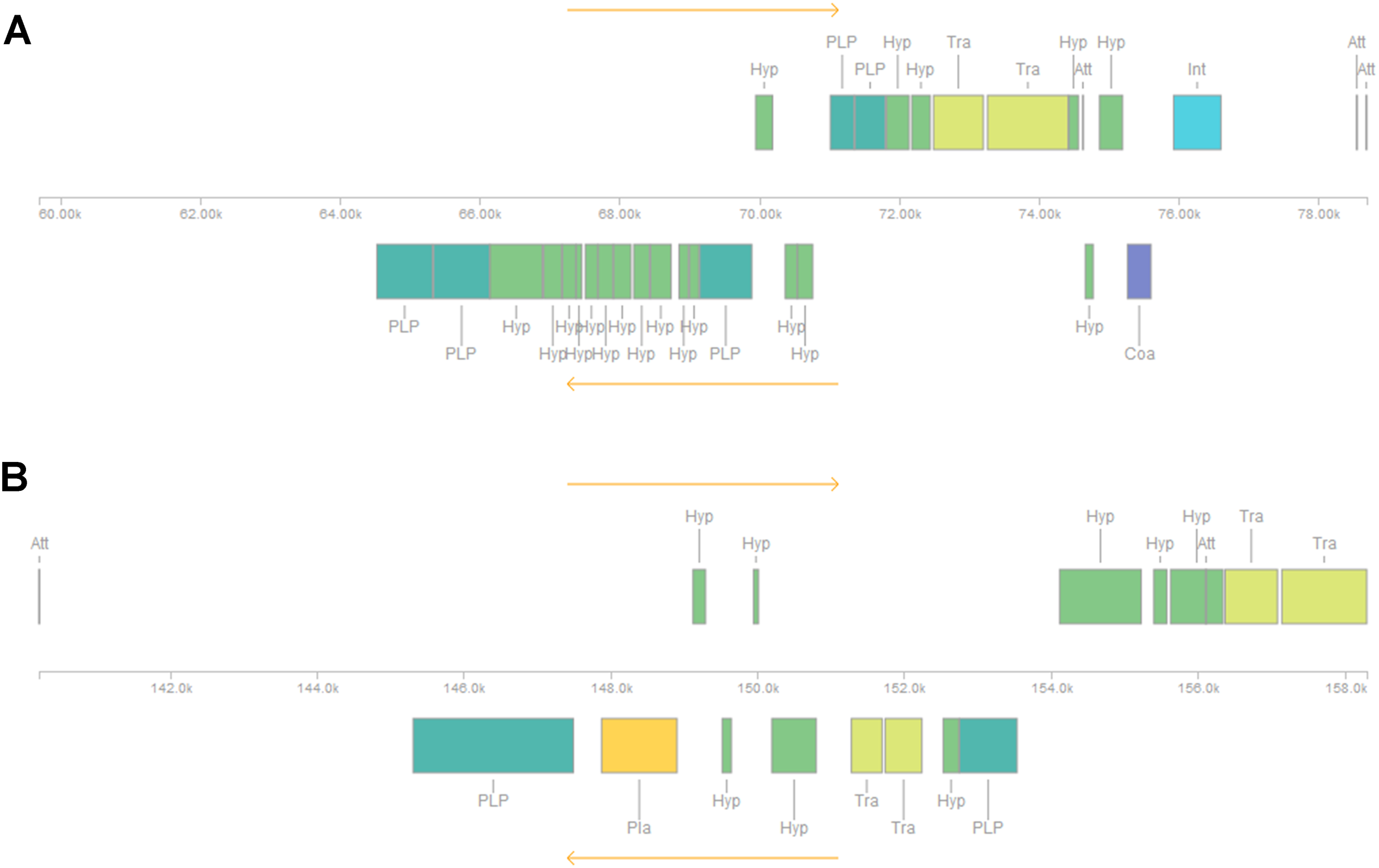{kind=link}
Figure 2: CRISPR-Cas systems subtype II-A predicted in four Lactobacillus crispatus strains from Brazil.
(A) Strain CRI4, (B) CRI8, (C) CRI10 and (D) CR17. The genes csn2 and cas9 are fragmented in the four genomes. Colors of the protein coding sequences green—csn2, cas2 and cas1; red—cas9 fragments; pink—transposase.{kind=link}
No virulence or antimicrobial resistance gene was predicted using ABRicate. With the search for virulence and undesirable genes using KEGG Mapper/BlastKOALA, we identified two hemolysins (tlyC and hlyIII), one bile salt hydrolase (cbh), and one enzyme involved in the biosynthesis of putrescine (Ornithine decarboxylase, odcI). Manual screening of Prokka annotation showed a third hemolysin, Hemolysin A. The search for toxin biosynthesis identified an “Ornithine decarboxylase” (odcI) (WP_005727730.1) involved in the synthesis of the biogenic amine putrescine, in all for strains.
In vitro detection of phages
Several rod-shaped particles of varying length and thickness were observed by electron microscopy, some of them similar to Myoviridae bacteriophages. This pattern was repeated throughout the slide (Fig. 3).

Figure 3: Transmission electron microscopy of the strain CRI4 filtrate.
(A) Presence of stick-shaped structures. (B) Structure similar to a capsid with contractile tail (Myoviridae).{kind=link}
Comparative analyses using public genomes
The strains did not cluster according to a health condition. However, strains from Brazil were the closest to strains from the Netherlands, even in different clusters (Fig. 4). The GWAS based on gene presence/absence implemented in Scoary did not identify an association with a health condition (p > 0.05). Due to the clustering of strains from Brazil and the Netherlands, we tested the association with these two geographical locations and the result was also negative. A total of 8 protein-coding genes were identified as under positive selection (q < 0.05). After manual curation for false positives caused by alignment artifacts, five genes were obtained: a surface exposed “S-layer protein precursor,” three phage related proteins, and a hypothetical protein (Table 5; Data S8).
| Product (Gene) | COG | Location | Sequences/genomes | PS sites | Positions | Protein ID |
|---|---|---|---|---|---|---|
| Integrase core domain protein | L | CYTOPLASMIC | 53/50 | 1 | 160 | EKB63650 |
| S-layer protein precursor | S | SECRETED | 42/50 | 1 | 130 | EKB61518 |
| Hypothetical protein | – | CYTOPLASMIC | 15/50 | 11 | 31, 39, 49, 48, 52, 58, 94, 95, 103, 107, 126 | WP_126708926 |
| ORF6N domain protein | K | CYTOPLASMIC | 10/50 | 2 | 240, 256 | WP_133463822 |
| Tyrosine recombinase XerS (xerS) | L | CYTOPLASMIC | 4/50 | 1 | 231 | WP_133475995 |
Note:
PS, positively selected sites.
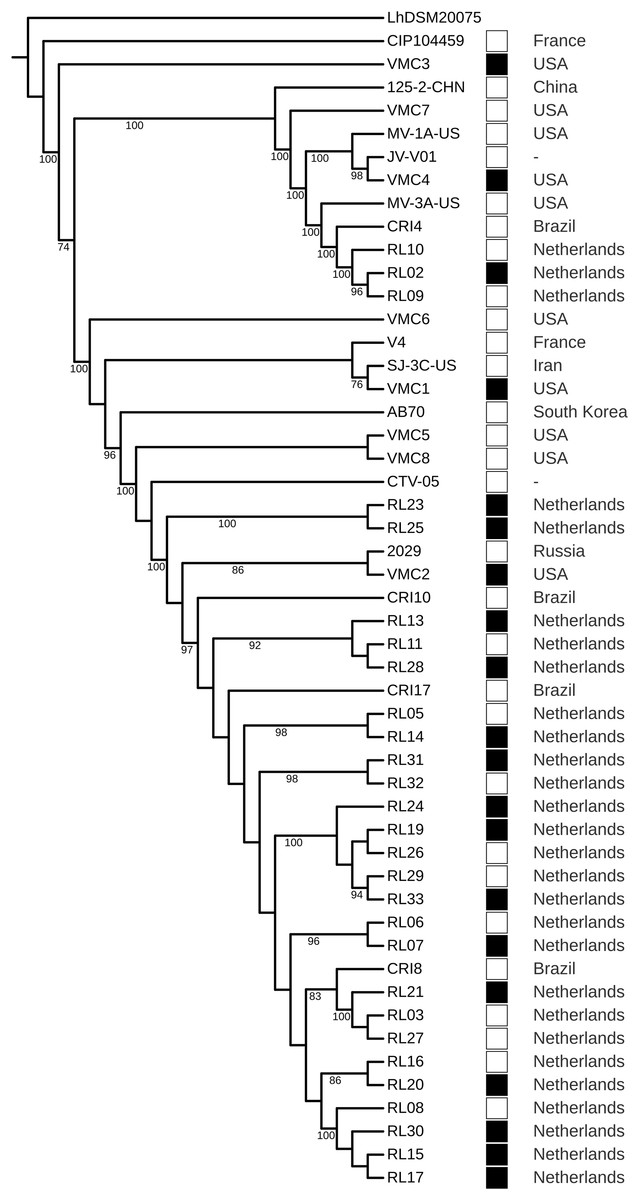
Figure 4: Phylogenomic tree of Lactobacillus crispatus strains from vaginal isolates.
The tree was built using the nucleotide sequences of 118 core genes predicted by Roary and aligned by MAFFT, 1,000 rounds of bootstrapping, and Maximum Likelihood phylogenetic inference implemented in IQ-TREE. Filled squares—bacterial vaginosis samples, Empty squares—samples from healthy individuals.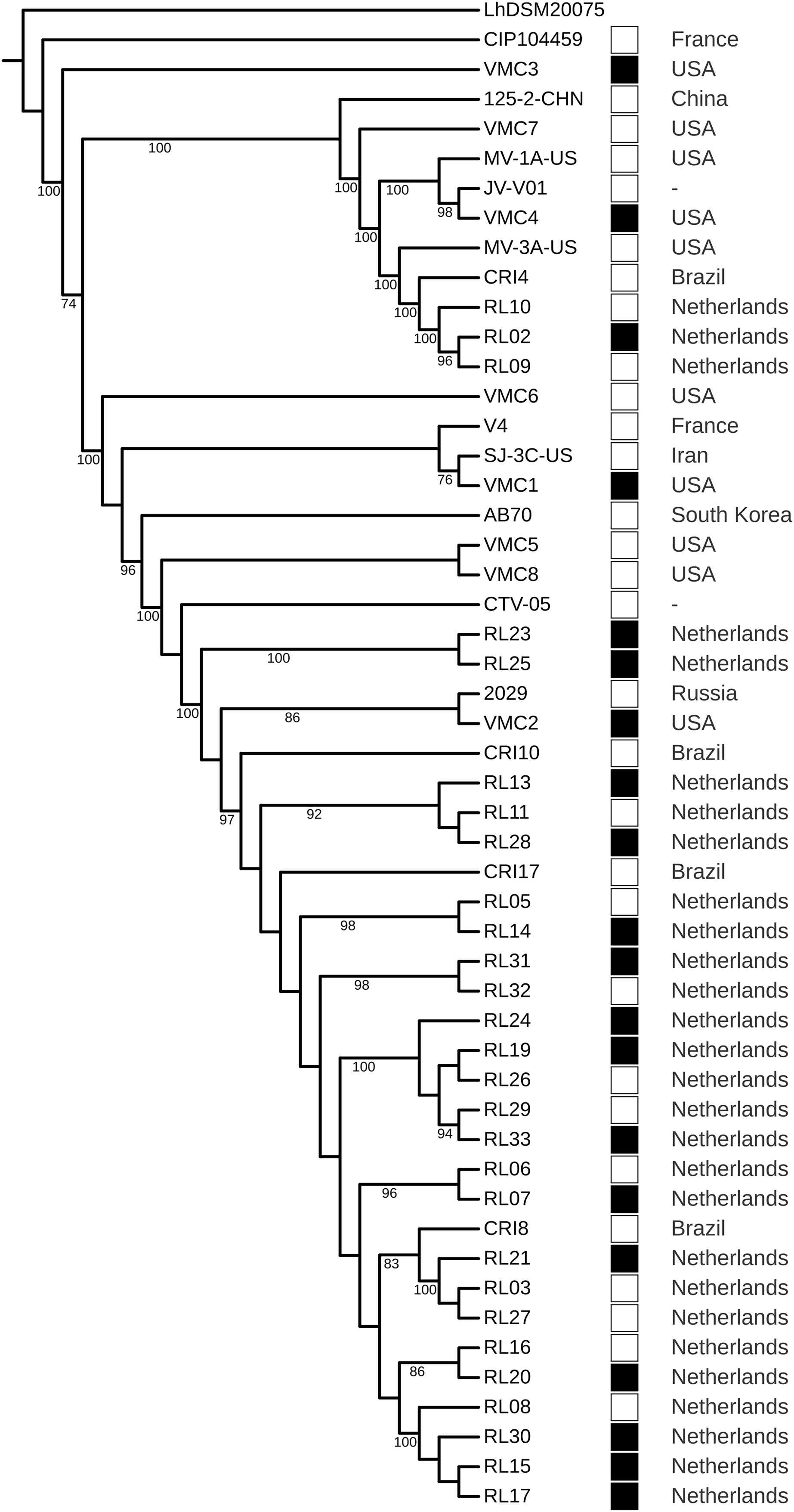{kind=link}
Discussion
In the first four L. crispatus sequenced genomes from Brazil, we identified genes involved in the protective properties of vaginal lactobacilli (Smith & Ravel, 2017) such as the production of lactic acid, bacteriocins, hydrogen peroxide and adhesion (Table 3).
For safety reasons, a probiotic strain should not present features associated with virulence and antimicrobial resistance, as well as mobile elements that could transfer those features to other microorganisms from the host microbiome (Pariza et al., 2015; Chokesajjawatee et al., 2020). No antimicrobial resistance gene was found, and only three hemolysins were predicted as virulence factors, which have also been previously identified in other Lactobacillus probiotic strains (Zafar & Saier, 2020) (Table 4). The hemolysin-III is widespread across Lactobacillus species, including strains considered as safe and commercially available (Chokesajjawatee et al., 2020). This result implies these genes should not be a safety concern. However, the hemolytic activity of those proteins from Lactobacillus must be verified in further studies. We predicted the synthesis of the toxic biogenic amine putrescine by the ornithine decarboxylase pathway in all four strains (Table 4). This is one of the pathways in which the decarboxylation of amino acids and organic acids generates a proton motive force that can regulate intracellular pH (acidic stress response) and generate ATP (Romano et al., 2014; Del Rio et al., 2018). The production of biogenic amines is an important safety issue when screening probiotic strains as they may cause intoxication when consumed in high concentrations (García-Villar, Hernández-Cassou & Saurina, 2009; Linares et al., 2011).
Concerning mobile elements, as expected for probiotic strains, no plasmids were found. Despite of numerous ISs from 14 known families (Table 4; Data S3) and one from a new family-specific of vaginal isolates (Datas S3, S4, and S5), none of them were associated with antibiotic resistance genes.
Four to six incomplete prophages across the genomes were revealed (Table 4; Data S6). Some incomplete prophages could be the result of an assembly artifact, as the genomes were assembled as drafts and most prophages were located close to a contig end. The possible role of the newly described IS, and the associated prophages in the genome evolution (Siguier, Gourbeyre & Chandler, 2014) or adaptation to the urogenital niche should be investigated. Moreover, the presence of complete phages was confirmed by electron microscopy in CRI4 lysate after Mitomycin C induction (Fig. 3). Therefore, our results suggest the possibility of lysogeny of this strain.
The presence of a functional CRISPR-Cas system could prevent the infection by bacteriophages (Crawley et al., 2018) that could influence the vaginal microbiota (Macklaim et al., 2013). We detected the subtype II-A in the four genomes and II-C in CRI10 (Table 4; Data S7). However, all csn2 and cas9 genes across the genomes are fragmented (Fig. 2), lacking one or more domains. Also, the subtype II-C in CRI10 is probably a prediction artifact, as a single subtype with two sets of cas genes can be detected as multiple subtypes (Crawley et al., 2018) and only the subtypes I-B, I-E, and II-A were reported for this species (Petit & Read, 2020). The result suggests that the CRISPR-Cas systems are not functional due to gene fragmentation which could compromise the prevention of phage infections.
In the phylogenomic tree, health-related condition does not show clustering (Fig. 4), a result also found in other studies (Abdelmaksoud et al., 2016; Pan, Hidalgo-Cantabrana & Barrangou, 2020). However, Brazil-Netherlands clusters were formed. The GWAS performed using Scoary did not find gene presence/absence associated with the health condition or the Brazil-Netherlands clusters. This result is different from a previous study, which has shown an association of transposases and a glycosyltransferase being more abundant in BV strains (Van der Veer et al., 2019). The differences obtained between association studies could be due to the multifactorial characteristic of BV (Barrientos-Durán et al., 2020; Marconi et al., 2020).
The positive selection analysis identified five genes (Table 5; Data S8). The S-layer precursor has the surface layer A protein (SLAP) domain described in slpB from L. acidophilus ATCC 4356 (Boot, Kolen & Pouwels, 1995). S-layer proteins form symmetric, porous, lattice-like layers that cover the cell surface with poorly known functions that can involve mediation of bacterial adherence to host cells, extracellular matrix proteins, or protective or enzymatic functions (Hynönen & Palva, 2013). Surface exposed proteins are located in the interface with the environment and can be under positive selection due to interaction with several factors such as antimicrobial compounds, viruses, hosts, and other bacteria (Petersen et al., 2007). The three phage related proteins have domains for DNA biding, integration, and recombination. The selective pressures acting in those proteins have yet to be identified.
Conclusions
The first L. crispatus genomes from vaginal isolates from Brazil presented several genes associated with probiotic characteristics. Although mobile genetic elements were detected, they do not present antimicrobial resistance genes that could be transmitted to other bacteria. For safety issues, the functionality of the hemolysin related genes must be further experimentally confirmed. No genomic feature was associated with healthy and BV conditions, and the positive selection was predicted in an S-layer protein and phage related genes but have yet to be investigated.
Supplemental Information
Taxonomic classification of 50 Lactobacillus crispatus genomes.
Insertion sequences in the Lactobacillus crispatus genomes isolated in Brazil.
New insertion sequence from IS607-like family predicted in Lactobacillus crispatus CRI8, CRI10, and CR17.
Shows the complete sequence in strain CRI8.
{kind=link}