A standardized, extensible framework for optimizing classification improves marker-gene taxonomic assignments
- Published
- Accepted
- Subject Areas
- Bioinformatics, Computational Biology, Ecology, Microbiology, Taxonomy
- Keywords
- bioinformatics, microbiome, executable paper, qiime, rRNA, ITS, microbial ecology, fungal diversity, bacterial diversity
- Copyright
- © 2015 Bokulich et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ PrePrints) and either DOI or URL of the article must be cited.
- Cite this article
- 2015. A standardized, extensible framework for optimizing classification improves marker-gene taxonomic assignments. PeerJ PrePrints 3:e934v1 https://doi.org/10.7287/peerj.preprints.934v1
Abstract
Background: Taxonomic classification of marker-gene (i.e., amplicon) sequences represents an important step for molecular identification of microorganisms.
Results: We present three advances in our ability to assign and interpret taxonomic classifications of short marker gene sequences: two new methods for taxonomy assignment, which reduce runtime up to two-fold and achieve high precision genus-level assignments; an evaluation of classification methods that highlights differences in performance with different marker genes and at different levels of taxonomic resolution; and an extensible framework for evaluating and optimizing new classification methods, which we hope will serve as a model for standardized and reproducible bioinformatics methods evaluations.
Conclusions: Our new methods are accessible in QIIME 1.9.0, and our evaluation framework will support ongoing optimization of classification methods to complement rapidly evolving short-amplicon sequencing and bioinformatics technologies. Static versions of all of the analysis notebooks generated with this framework, which contain all code and analysis results, can be viewed at http://bit.ly/srta-010.
Author Comment
This paper is being submitted to Microbiome for peer review.
Supplemental Information
Supplementary Figure 1. Mock community datasets analyzed in this study
Supplementary Figure 2. Mock community A composition
Supplementary Figure 3. Mock community B composition
Supplementary Figure 4. Mock community C composition
Supplementary Figure 5. Mock community D composition
Supplementary Figure 6
Taxonomy classifier configuration and mock community composition alter assignment accuracy at family-level.
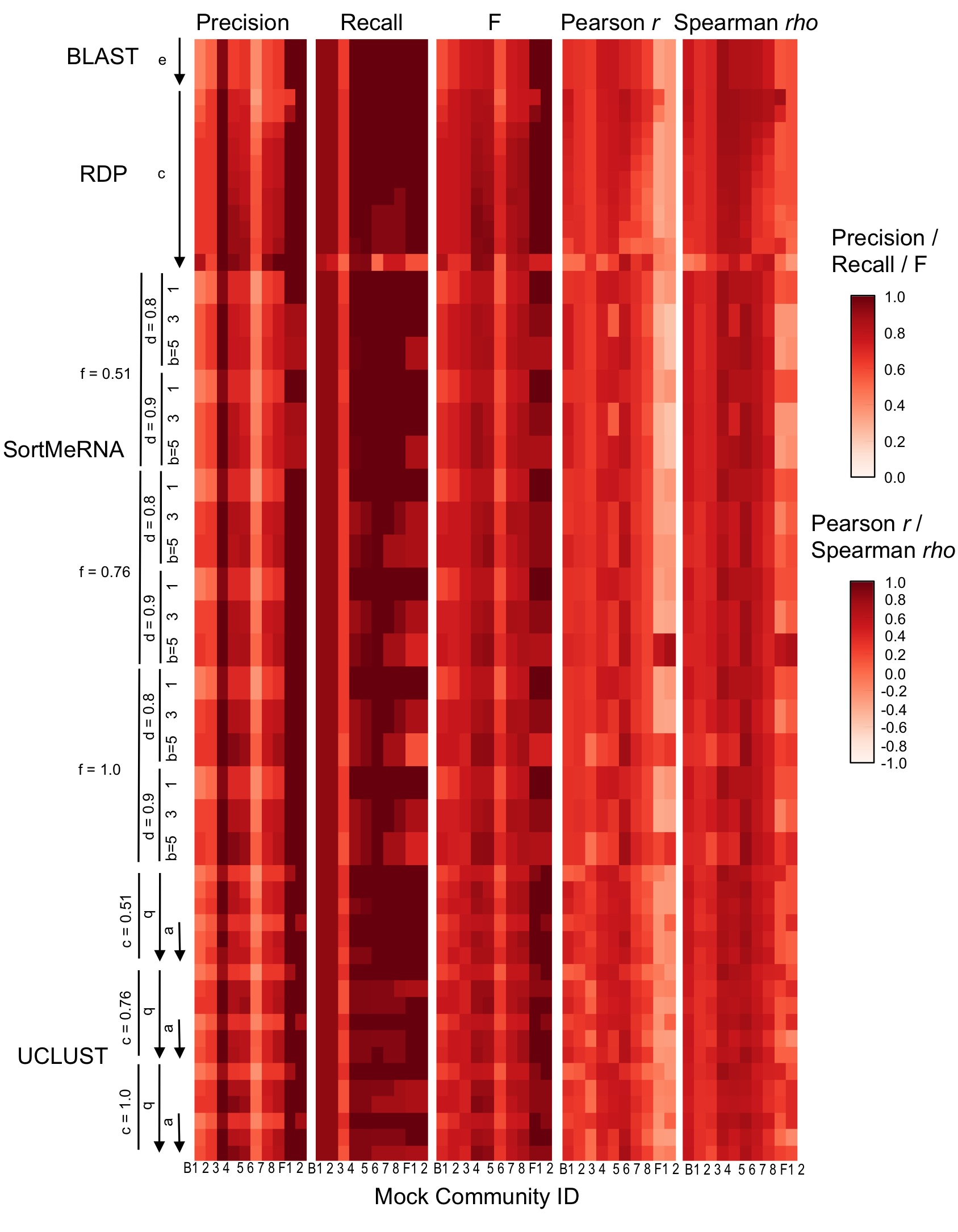{kind=link}
Supplementary Figure 7
Taxonomy classifier configuration and mock community composition alter assignment accuracy at species-level.
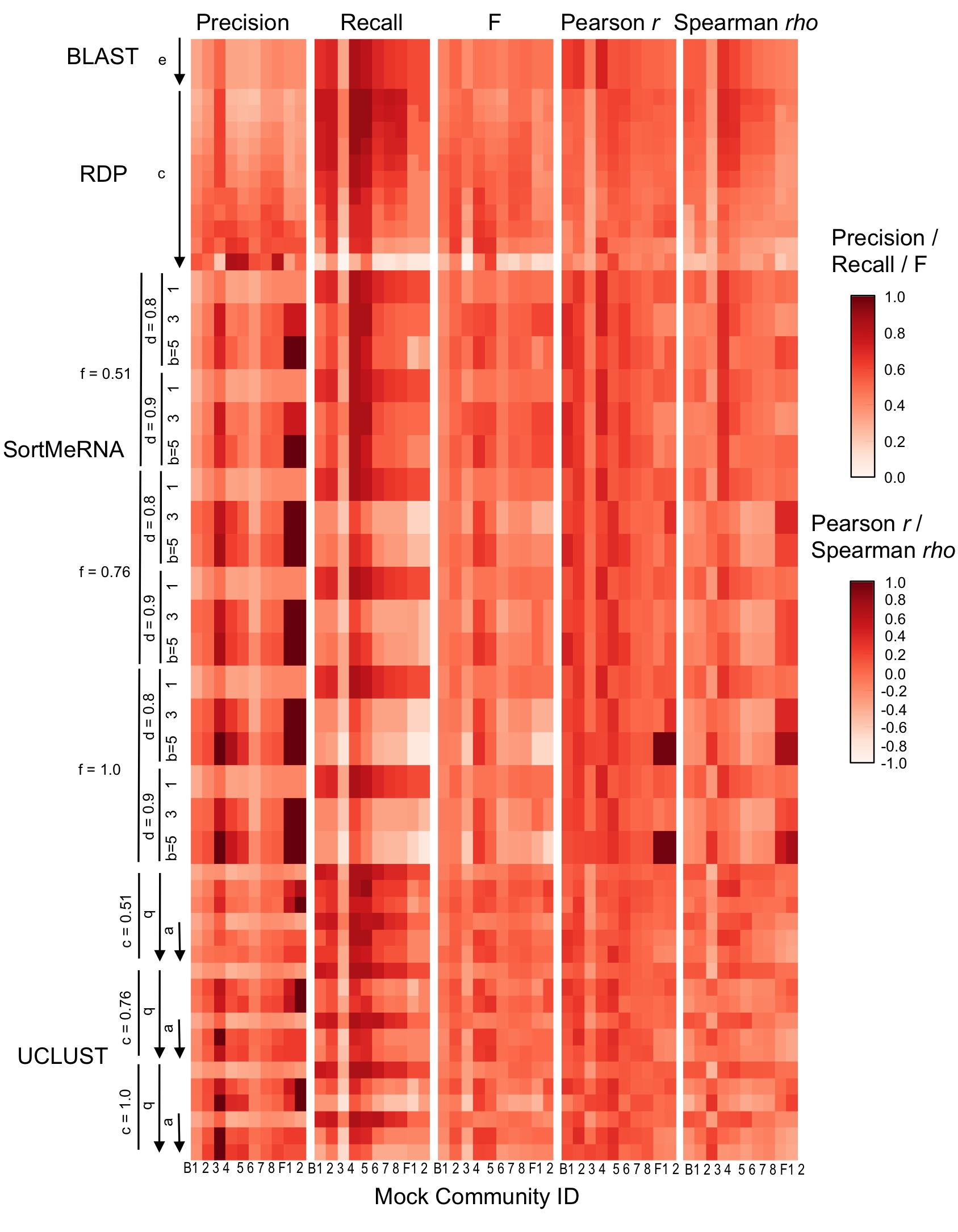{kind=link}
Supplementary Figure 8
Taxonomy classifier selection critically shapes assignment accuracy of simulated communities. Violin plots illustrate the distribution of precision, recall, and F-measure values across all simulated communities and all parameter configurations for a given method for family-level (left), genus-level (middle), or species-level taxonomy assignments (right). Heavy dashed lines indicate median values, fine dashed lines indicate quartiles.
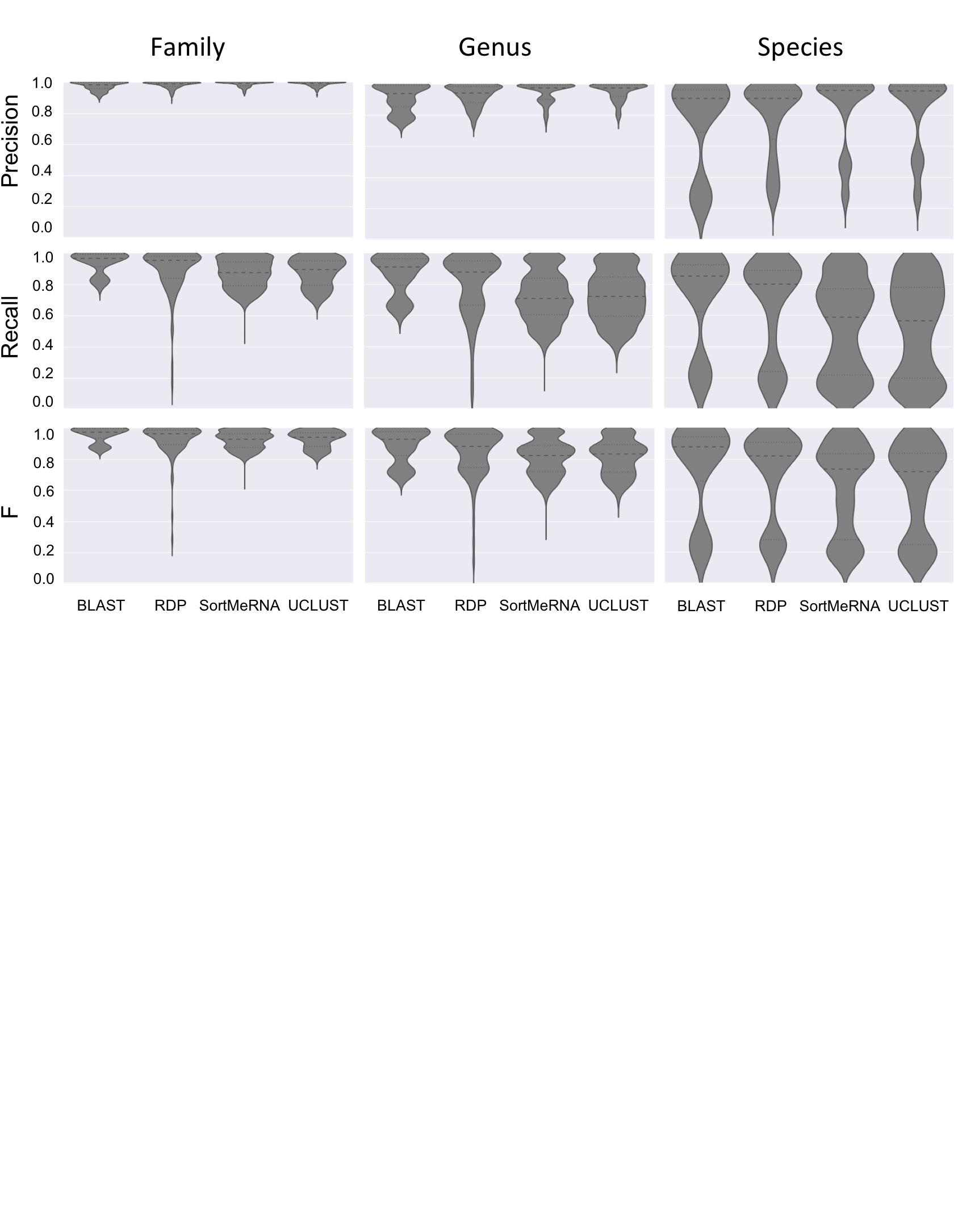{kind=link}