Evaluating multi-locus phylogenies for species boundaries determination in the genus Diaporthe
- Published
- Accepted
- Subject Areas
- Microbiology, Molecular Biology, Mycology
- Keywords
- Diaporthe, phylogeny, maximum likelihood, maximum parsimony, Phomopsis, multi-locus
- Copyright
- © 2017 Santos et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Preprints) and either DOI or URL of the article must be cited.
- Cite this article
- 2017. Evaluating multi-locus phylogenies for species boundaries determination in the genus Diaporthe. PeerJ Preprints 5:e2822v1 https://doi.org/10.7287/peerj.preprints.2822v1
Abstract
Background. Species identification is essential for controlling disease, understanding epidemiology, and to guide the implementation of phytosanitary measures against fungi from the genus Diaporthe. Accurate Diaporthe species separation requires using multi-loci phylogenies. However, defining the optimal set of loci that can be used for species identification is still an open problem. Methods. Here, we addressed that problem by identifying five loci that have been sequenced in 142 Diaporthe isolates representing 96 species: TEF1, TUB, CAL, HIS and ITS. We then used every possible combination of those loci to build, analyse, and compare phylogenetic trees. Results. As expected, species separation is better when all five loci are simultaneously used to build the phylogeny of the isolates. However, removing the ITS locus has little effect on reconstructed phylogenies, identifying the TEF1-TUB-CAL-HIS four loci tree as almost equivalent to the five loci tree. We further identify the best 3-loci, 2-loci, and 1-locus trees that should be used for species separation in the genus. Discussion. Our results question the current use of the ITS locus for DNA barcoding in the genus Diaporthe and suggest that TEF1 might be a better choice if one locus barcoding needs to be done.
Author Comment
This is a submission to PeerJ for review.
Supplemental Information
Symmetric Difference Distance between Maximum Parsinomy trees
Symmetric Difference Distance between Maximum Likelihood trees
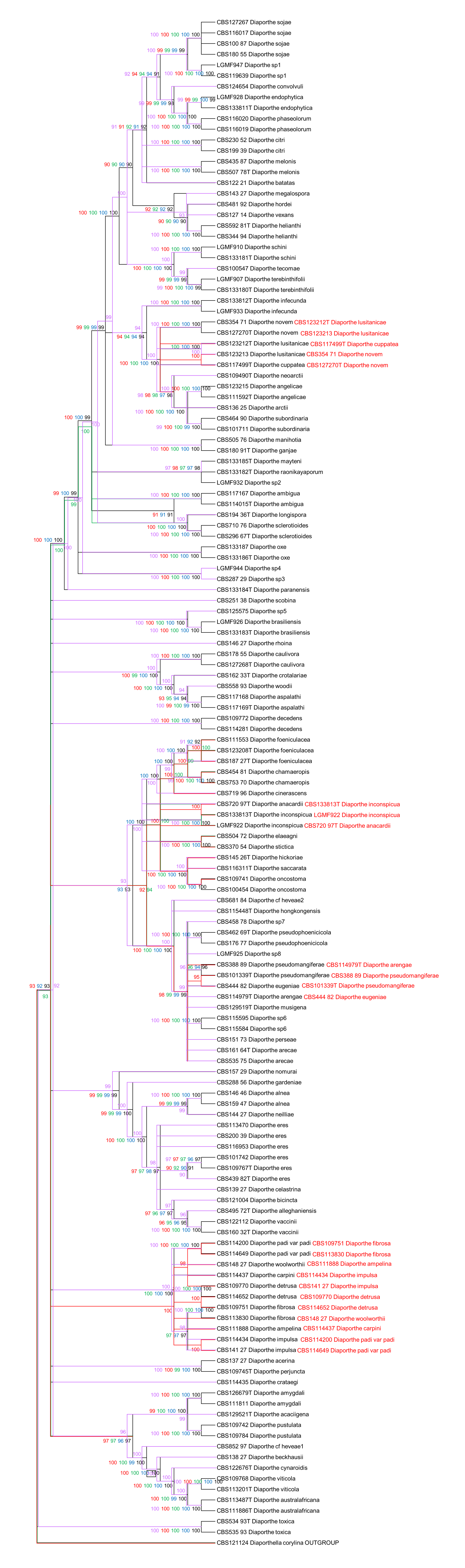
Phylogram for MP tree build using the five loci TEF1-TUB-CAL-HIS-ITS for the 96 Diaporthe species
Phylogram for ML tree build using the five loci TEF1-TUB-CAL-HIS-ITS for the 96 Diaporthe species
Phylogram for MP tree build using the TEF1 locus for the 96 Diaporthe species
Phylogram for ML tree build using the TEF1 locus for the 96 Diaporthe species
Comparison of MP trees generated from the same alignment of 5 genes, when changing the position of the TEF sequences in that alignment
The numbers represent the bootstrap value for the branching in each tree. Red – MP tree with TEF in the first position of the alignment. Mauve – MP tree with TEF in the second position of the alignment. Green – MP tree with TEF in the third position of the alignment. Blue – MP tree with TEF in the fourth position of the alignment. Black – MP tree with TEF in the fifth position of the alignment. The only small effect of shifting the position of TEF is observed in the TEF-ITS-TUB-HIS-CAL (red) and ITS-TEF-TUB-HIS-CAL (mauve) trees. In TEF-ITS-TUB-HIS-CAL case, flipping is observed in a small number of terminal branches. That flipping does not affect the overall topology of the tree, which is identical to that of the remaining trees. These results suggest that changing the order of the genes in the alignment will have a negligible effect on the topology of the phylogenetic trees.
{kind=link}