Compacting and correcting Trinity and Oases RNA-Seq de novo assemblies
- Published
- Accepted
- Subject Areas
- Bioinformatics, Genomics, Computational Science
- Keywords
- RNA-Seq, de novo assembly, compaction, correction, quality assessment
- Copyright
- © 2016 Cabau et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Preprints) and either DOI or URL of the article must be cited.
- Cite this article
- 2016. Compacting and correcting Trinity and Oases RNA-Seq de novo assemblies. PeerJ Preprints 4:e2284v1 https://doi.org/10.7287/peerj.preprints.2284v1
Abstract
Background
De novo transcriptome assembly of short reads is now a common step in expression analysis of organisms lacking a reference genome sequence. Several software packages are available to perform this task. Even if their results are of good quality it is still possible to improve them in several ways including redundancy reduction or error correction. Trinity and Oases are two commonly used de novo transcriptome assemblers. The contig sets they produce are of good quality. Still, their compaction (number of contigs needed to represent the transcriptome) and their quality (chimera and nucleotide error rates) can be improved.
Results
We built a de novo RNA-Seq Assembly Pipeline (DRAP) which wraps these two assemblers (Trinity and Oases) in order to improve their results regarding the above-mentioned criteria. DRAP reduces from 1,3 to 15 fold the number of resulting contigs of the assemblies depending on the read set and the assembler used. This article presents seven assembly comparisons showing in some cases drastic improvements when using DRAP. DRAP does not significantly impair assembly quality metrics such are read realignment rate or protein reconstruction counts.
Conclusion
Transcriptome assembly is a challenging computational task even if good solutions are already available to end-users, these solutions can still be improved while conserving the overall representation and quality of the assembly. The de novo RNA-Seq Assembly Pipeline (DRAP) is an ease to use software package to produce compact and corrected transcript set. DRAP is free, open-source and available at http://www.sigenae.org/drap .
Author Comment
This is a submission to PeerJ for review.
Supplemental Information
Steps in runDRAP workflow
This workflow is used to produce an assembly from one sample/tissue/development stage. It take as input R1 from single-end sequencing or R1 and R2 from paired-end sequencing and eventually a reference proteins set from closest species with known proteins.
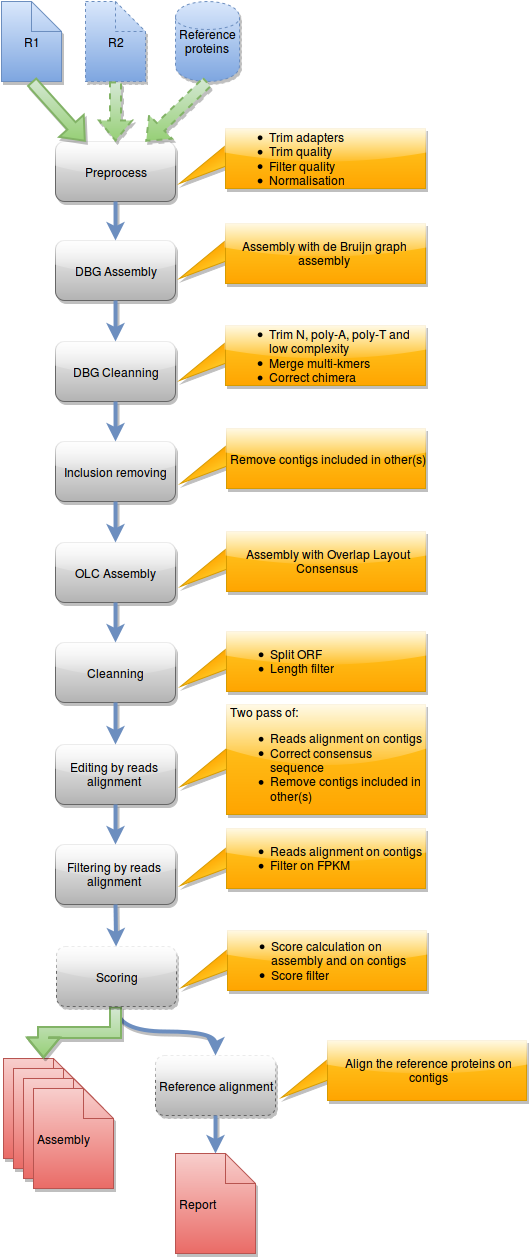{kind=link}
Steps in runMeta workflow
This workflow is used to produce a merged assembly from several samples/tissues/development stage outputted by runDRAP. Inputs are runDRAP output folders and eventually a reference protein set.
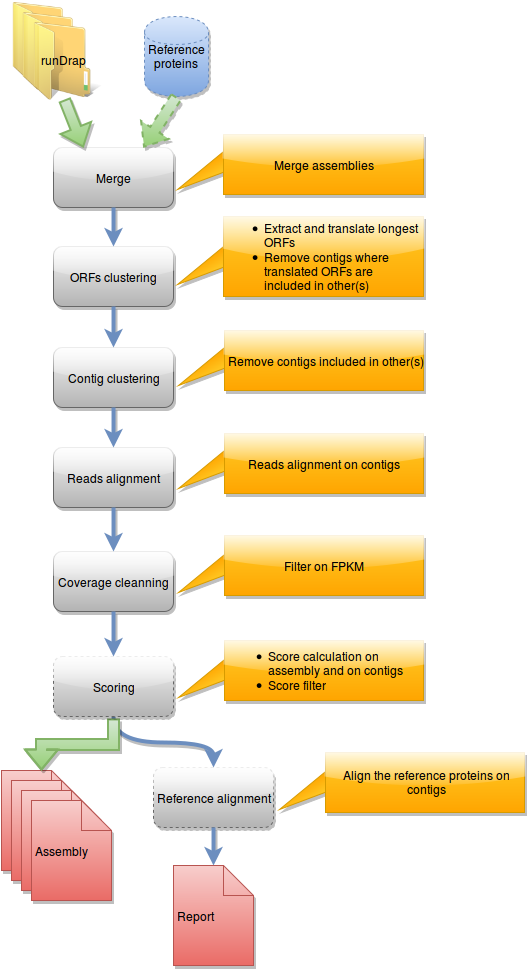{kind=link}
Steps in runAssessment workflow
This workflow is used to evaluate quality for one assembly or for compare several assemblies produced from the same dataset. Inputs are the assembly/ies, R1 and eventually R2, and a reference protein set.
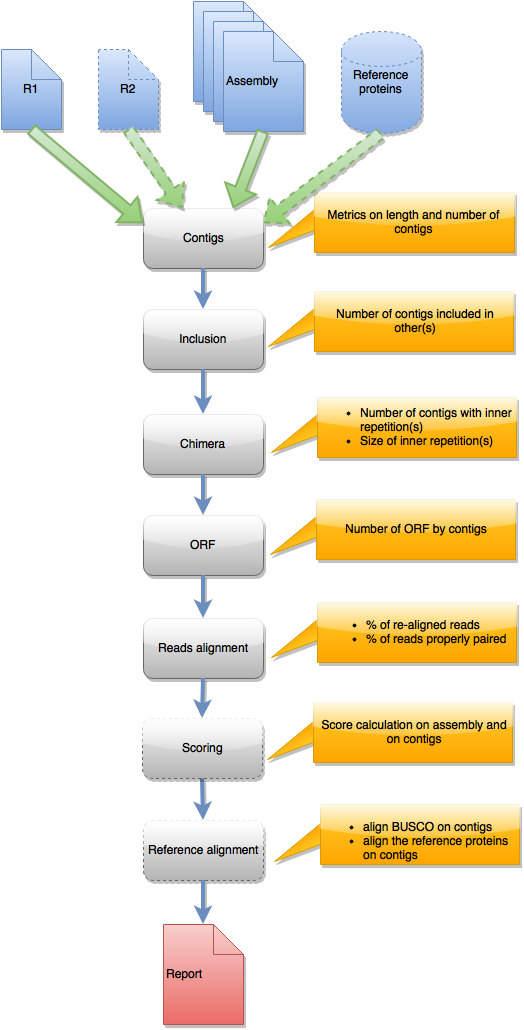{kind=link}