A nested phylogenetic reconstruction approach provides scalable resolution in the eukaryotic Tree Of Life
- Published
- Accepted
- Subject Areas
- Bioinformatics, Computational Biology, Evolutionary Studies, Genomics
- Keywords
- Tree of life, Genomics, Phylogeny, Species tree, Phylogenomics
- Copyright
- © 2014 Huerta-Cepas et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ PrePrints) and either DOI or URL of the article must be cited.
- Cite this article
- 2014. A nested phylogenetic reconstruction approach provides scalable resolution in the eukaryotic Tree Of Life. PeerJ PrePrints 2:e223v1 https://doi.org/10.7287/peerj.preprints.223v1
Abstract
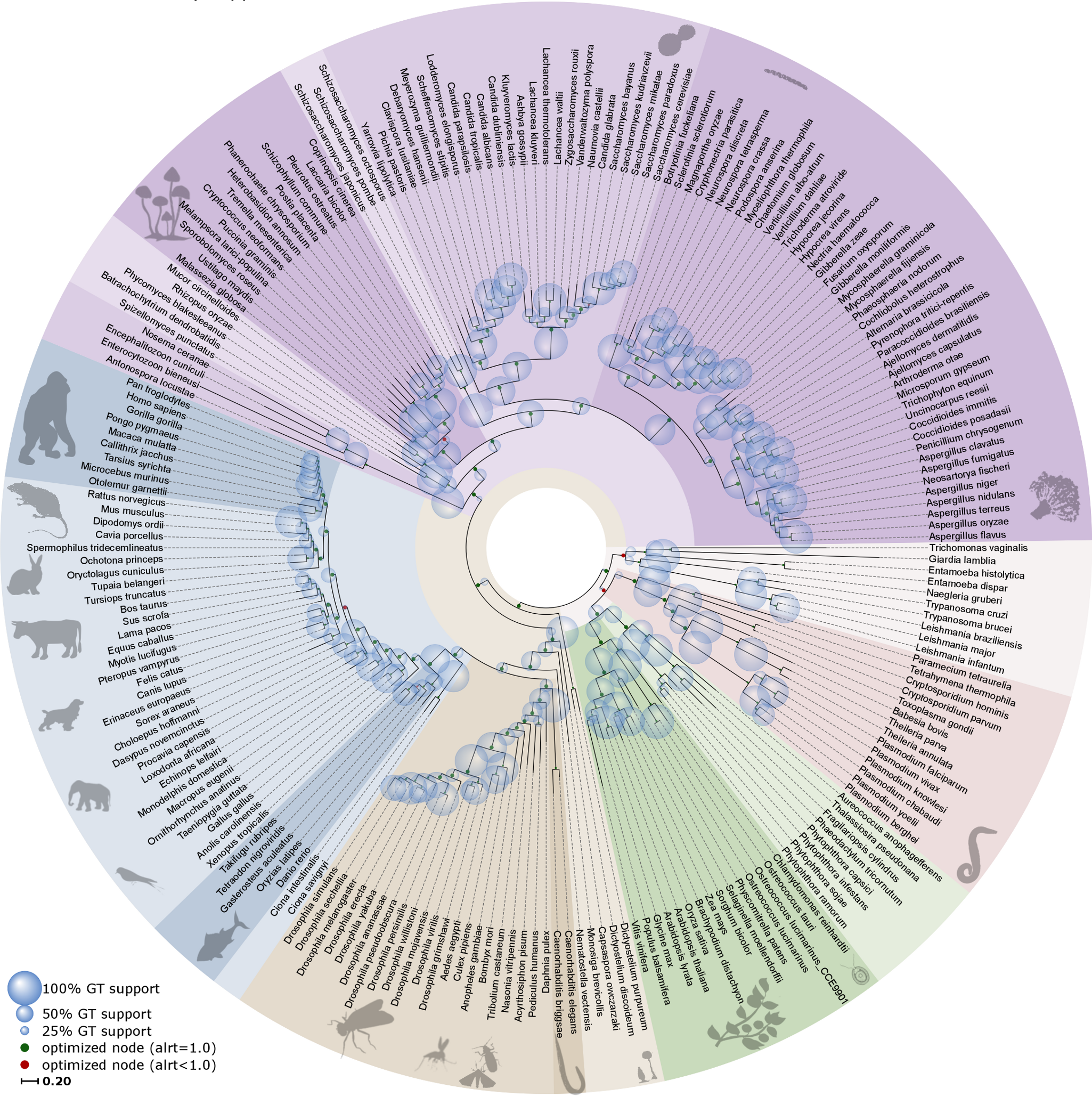
Assembling the Tree Of Life (TOL) faces the pressing challenge of incorporating a rapidly growing number of sequenced genomes. This problem is exacerbated by the fact that different sets of genes are informative at different evolutionary scales. Here, we present a novel phylogenetic approach ( N ested P hylogenetic R econstruction) in which each tree node is optimized based on the genes shared at that taxonomic level. We apply such procedure to reconstruct a 216-species eukaryotic TOL and compare it with a standard concatenation-based approach. The resulting topology is highly accurate, and reveals general trends such as the relationship between branch lengths and genome content in eukaryotes. The approach lends itself to continuous update, and we show this by adding 29 and 173 newly-sequenced species in two consecutive steps. The proposed approach, which has been implemented in a fully-automated pipeline, enables the reconstruction and continuous update of highly-resolved phylogenies of sequenced organisms.
Supplemental Information
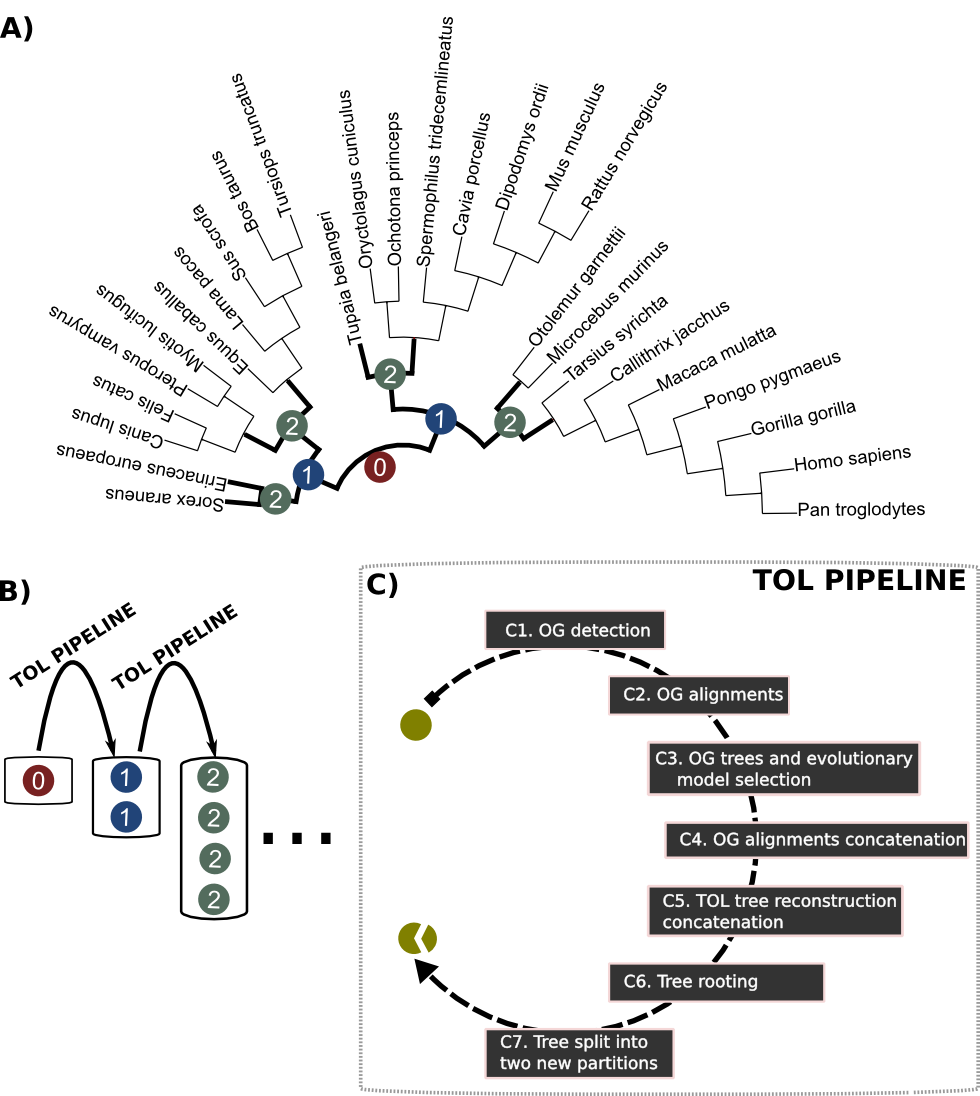
Schematic representation of the nested phylogenetic reconstruction approach.
Schematic representation of the nested phylogenetic reconstruction approach. First, a starting unrooted tree is reconstructed including all species (iteration 0, red node in panel A) and using a Gene Concatenation Methodology (GCM, panel C). GCM includes: C1) searching for groups of one-to-one orthologs (Ortholog Groups, OGs), C2) reconstruction of multiple sequence alignments of each OG, C3) phylogenetic reconstruction for each single OG, C4) concatenation of OG alignments, C5) species tree reconstruction based on the concatenated alignment. Secondly, the first resulting tree is split into two well supported clades, each of them defining a subset of species. GCM is then applied to each of the new sets of organisms, including four extra species as rooting anchors. As a result, two new trees are obtained (iteration 1, blue nodes in panel A). Subsequently, each of the new sub-trees is rooted using their anchor species (C6) and split into its two major clades (C7). The four resulting partitions (iteration 2, green nodes in panel A) are used to continue the same procedure until reaching a given limit for the size (number of species) in the recomputed partitions (panel B). An animation showing how the tree is re-shaped at each iteration can be seen at http://tol.cgenomics.org/TOL_animation.gif .
{kind=link}
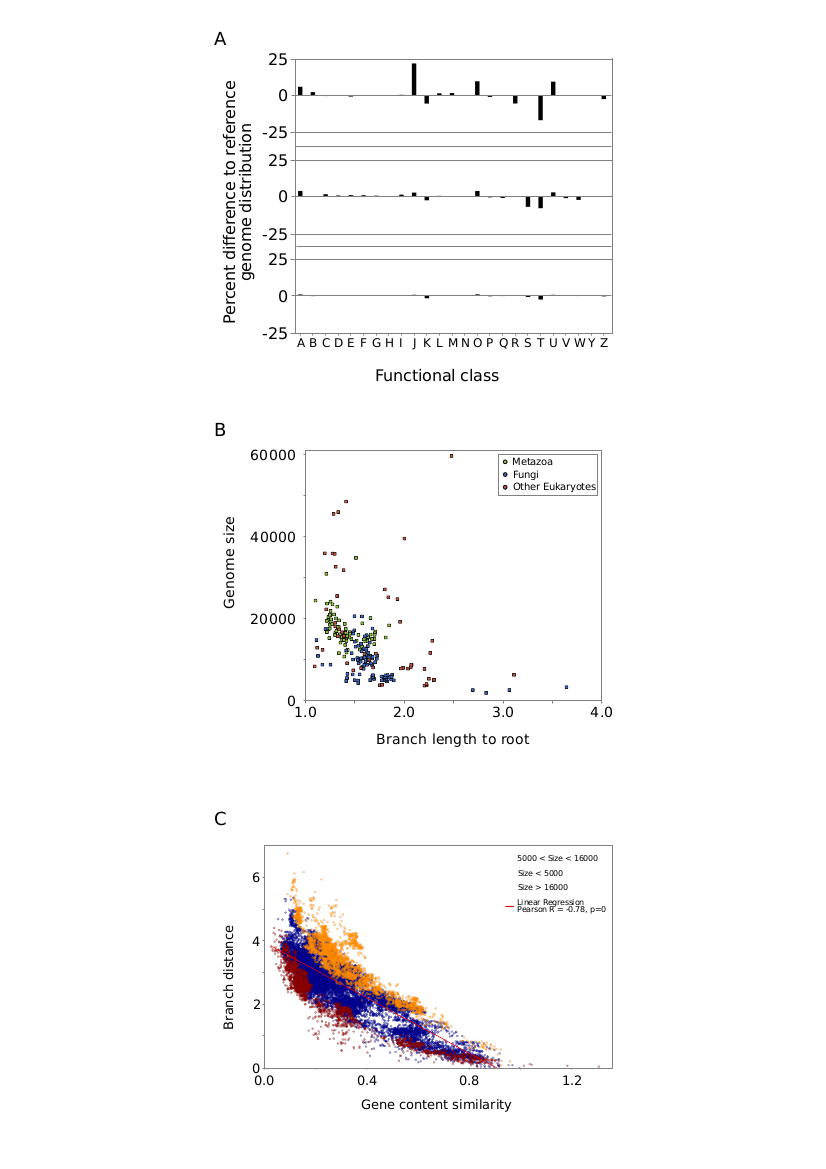
TOL analyses I
TOL analyses I: A-B) Grey lines represent topological distance between reference trees and the TOL (A-Chordates, B-Fungi, see Figure S5). Black line represents the number of protein families used at each iteration. C) Number of NCBI taxonomic groups not recovered at each iteration.
{kind=link}
{kind=link}
{kind=link}
Supplementary data
Supplementary methods, figures and tables