Transcriptome analysis of immature xylem in Chinese fir at different developmental phases
- Published
- Accepted
- Subject Areas
- Evolutionary Studies, Genomics, Molecular Biology, Plant Science, Soil Science
- Keywords
- Transcriptome, Chinese fir, RNA-Seq, wood formation, xylem
- Copyright
- © 2016 Zhang et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Preprints) and either DOI or URL of the article must be cited.
- Cite this article
- 2016. Transcriptome analysis of immature xylem in Chinese fir at different developmental phases. PeerJ Preprints 4:e2032v1 https://doi.org/10.7287/peerj.preprints.2032v1
Abstract
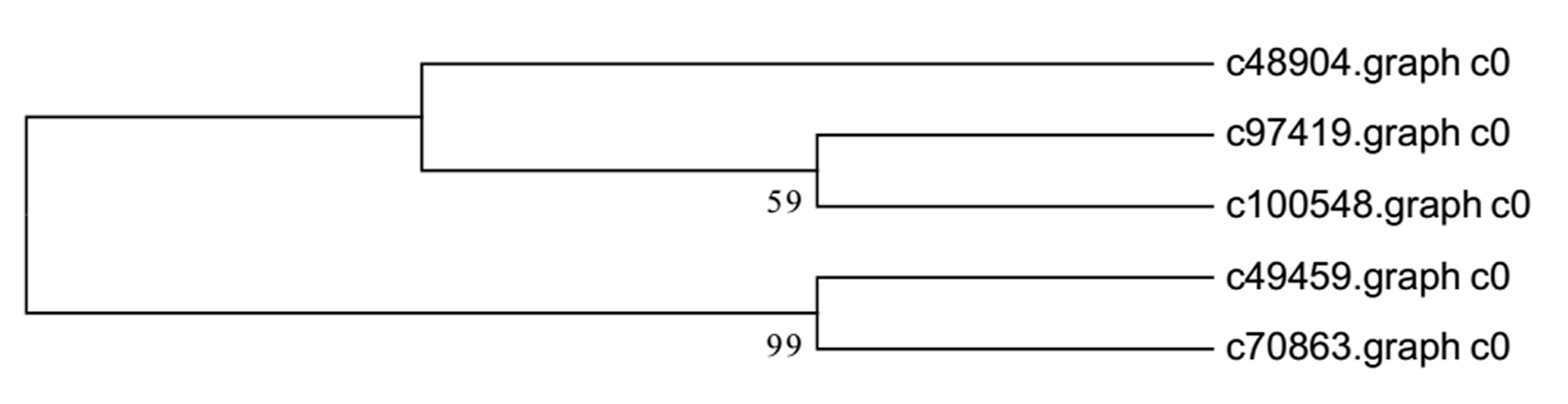
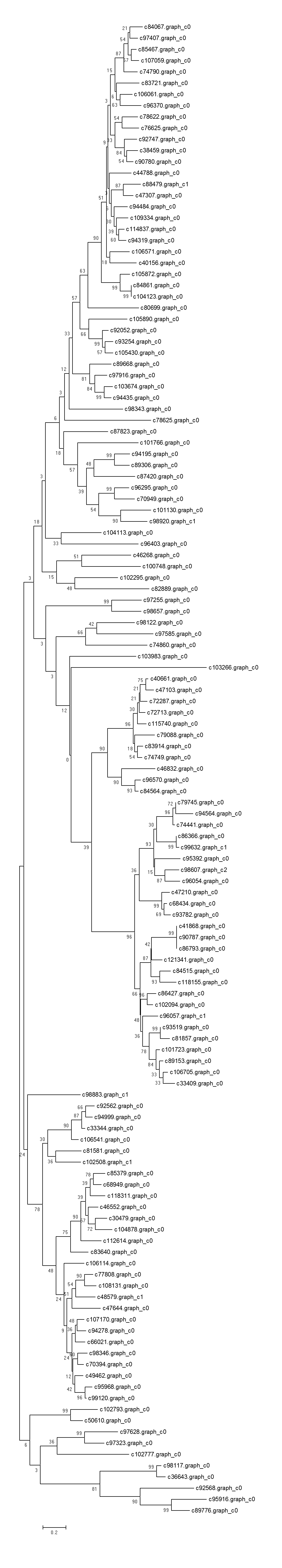
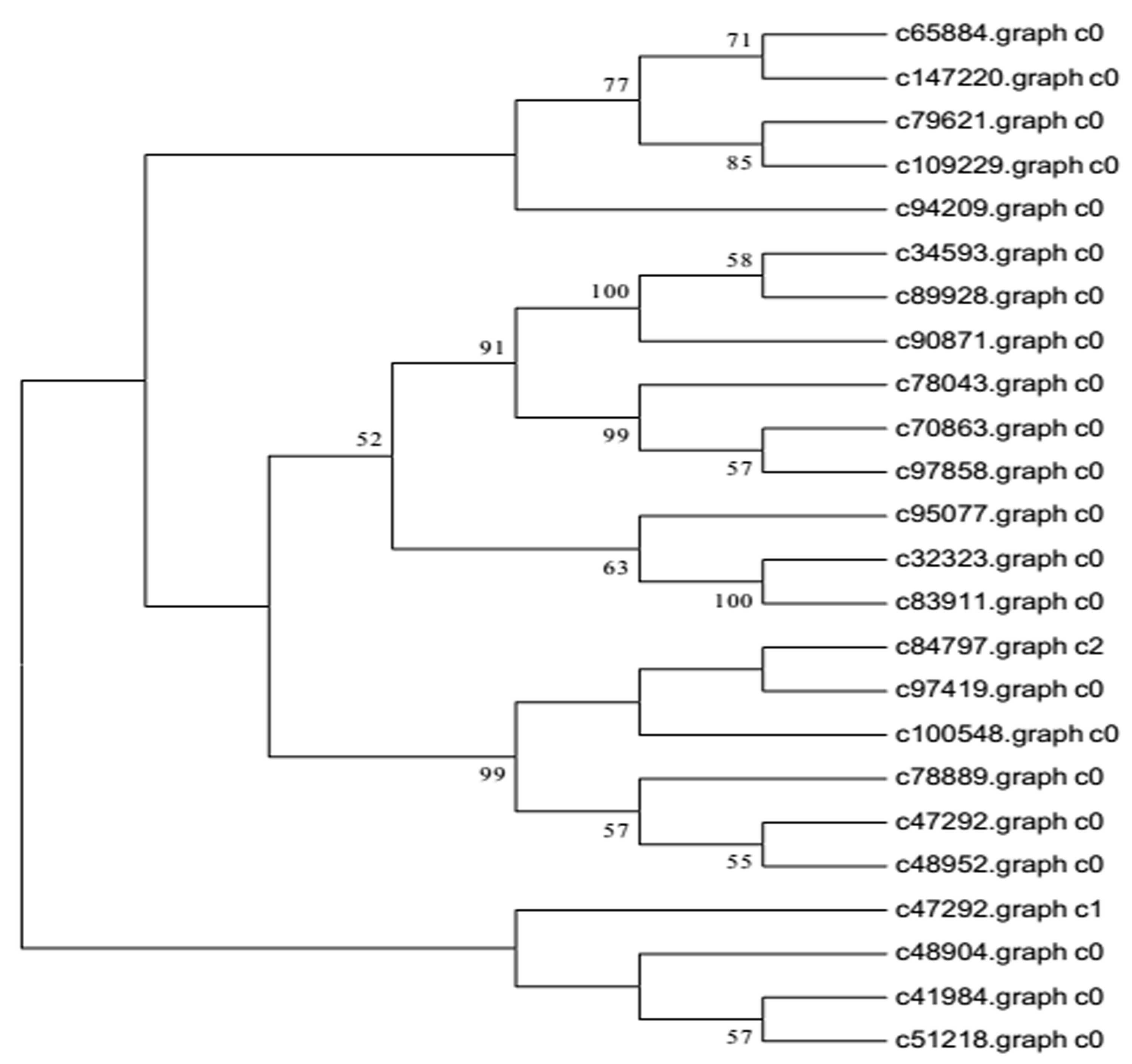
Background: Chinese fir [Cunninghamia lanceolata (Lamb .) Hook.], is one of the most important native tree species for timber production in southern China. An understanding of overall fast growing stage, stem growth stage and senescence stage cambium transcriptome variation is lacking. We used transcriptome sequencing to identify the repertoire of genes expressed during development of xylem tissue in Chinese fir, aiming to delineate the molecular mechanisms of wood formation. Results: We carried out transcriptome sequencing at three different cultivation ages (7Y, 15Y and 21Y) generating 68.71 million reads (13.88 Gbp). A total of 140,486 unigenes with a mean size of 568.64 base pairs (bp) were obtained via de novo assembly. Of these, 27,427 unigenes (19.52%) were further annotated by comparison to public protein databases. A total of 5,331 (3.79%) unigenes were mapped into 118 pathways by searching against the Kyoto Encyclopedia of Genes and Genomes Pathway database (KEGG). Differentially expressed genes (DEG) analysis identified 3, 16 and 5,899 DEGs from the comparison of 7Y vs. 15Y, 7Y vs. 21Y and 15Y vs. 21Y, respectively, in the immature xylem tissues, including 2,638 significantly up-regulated and 3,280 significantly down-regulated genes. Besides, five NAC transcription factors, 190 MYB transcription factors, and 34 WRKY transcription factors were identified respectively from Chinese fir transcriptome. Conclusion: Our results revealed the active transcriptional pathways and identified the DEGs at different cultivation phases of Chinese fir wood formation. This transcriptome dataset will aid in understanding and carrying out future studies on the molecular basis of Chinese fir wood formation and contribute to future artificial production and applications.
Author Comment
This is a submission to PeerJ for review.
{kind=link}
{kind=link}
{kind=link}