Tiling the genome into consistently named subsequences enables precision medicine and machine learning with millions of complex individual data-sets
- Published
- Accepted
- Subject Areas
- Genomics, Medical Genetics
- Keywords
- clinical screening, human genetics, machine learning, precision medicine
- Copyright
- © 2015 Guthrie et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ PrePrints) and either DOI or URL of the article must be cited.
- Cite this article
- 2015. Tiling the genome into consistently named subsequences enables precision medicine and machine learning with millions of complex individual data-sets. PeerJ PrePrints 3:e1426v1 https://doi.org/10.7287/peerj.preprints.1426v1
Abstract
The scientific and medical community is reaching an era of inexpensive whole genome sequencing, opening the possibility of precision medicine for millions of individuals. Here we present tiling: a flexible representation of whole genome sequences that supports simple and consistent names, annotation, queries, machine learning, and clinical screening. We partitioned the genome into 10,655,006 tiles: overlapping, variable-length sequences that begin and end with unique 24-base tags. We tiled and annotated 680 public whole genome sequences from the 1000 Genomes Project Consortium (1KG) and Harvard Personal Genome Project (PGP) using ClinVar database information. These genomes cover 14.13 billion tile sequences (4.087 trillion high quality bases and 0.4321 trillion low quality bases) and 251 phenotypes spanning ICD-9 code ranges 140-289, 320-629, and 680-759. We used these data to build a Global Alliance for Genomics and Health Beacon and graph database. We performed principal component analysis (PCA) on the 680 public whole genomes, and by projecting the tiled genomes onto their first two principal components, we replicated the 1KG principle component separation by population ethnicity codes. Interestingly, we found the PGP self reported ethnicities cluster consistently with 1KG ethnicity codes. We built a set of support-vector ABO blood-type classifiers using 75 PGP participants who had both a whole genome sequence and a self-reported blood type. Our classifier predicts A antigen presence to within 1% of the current state-of-the art for in silico A antigen prediction. Finally, we found six PGP participants with previously undiscovered pathogenic BRCA variants, and using our tiling, gave them simple, consistent names, which can be easily and independently re-derived. Given the near-future requirements of genomics research and precision medicine, we propose the adoption of tiling and invite all interested individuals and groups to view, rerun, copy, and modify these analyses at https://curover.se/su92l- j7d0g-swtofxa2rct8495
Author Comment
As millions of people get their genome sequenced, physicians and researchers as well as the individuals themselves will want to ask questions of these data. To ask questions we need a consistent naming scheme for parts of the genome. To address this we invented tiling – a technique that divides the genome into about 10 million overlapping, variable-length sequences, or “tiles”, each with a unique 24-base tag at each end. We use examples from public data to show tiling supports simple and consistent names, annotation, queries, machine learning, and clinical screening. In particular, we found six PGP participants with previously undiscovered pathogenic BRCA variants, and using our tiling, gave them simple, consistent names, which can be easily and independently re-derived. Someday soon the general public will have a chance to know the tiles in their own genomes and physicians may use the information to realize precision medicine.
Supplemental Information
S2 Text: Upper-bounding Estimates for Storing 1 Million Tiled Genomes
S4 Text: Clinical Analysis of PGP Participants with BRCA frameshifts
S6 Text: Methods for the VCF-based Clinical Analysis of the BRCA region
Table S1: Fisher’s exact test for Hashimoto’s thyroiditis
The Fisher’s exact test statistic value is 0.012545, which is significant at p < 0.05. The total number of PGP participants included is the number of participants from our cohort of 178 who completed the “Endocrine, Metabolic, Nutritional, and Immunity Survey” (last updated in 2012, downloaded on 2015, February 18).
Table S2: Fisher’s exact test for breast cancer for all participants reporting to be female
The Fisher’s exact test statistic value is 0.172704, which is not significant at p < 0.05. The total number of PGP participants included is the number of participants from our 178 who completed the “Cancer Survey” (last updated in 2012, downloaded on 2015, February 18) and who reported to be female with no conflicting reports of other genders. The power associated with this study is 48%, if we assume the probability of developing breast cancer when a participant has a deleterious BRCA mutation is 57% [69] and the probability of developing breast cancer when a participant has functional BRCA proteins is 12% [70]. To obtain a power greater than 95%, we would require a population size of 181 females.
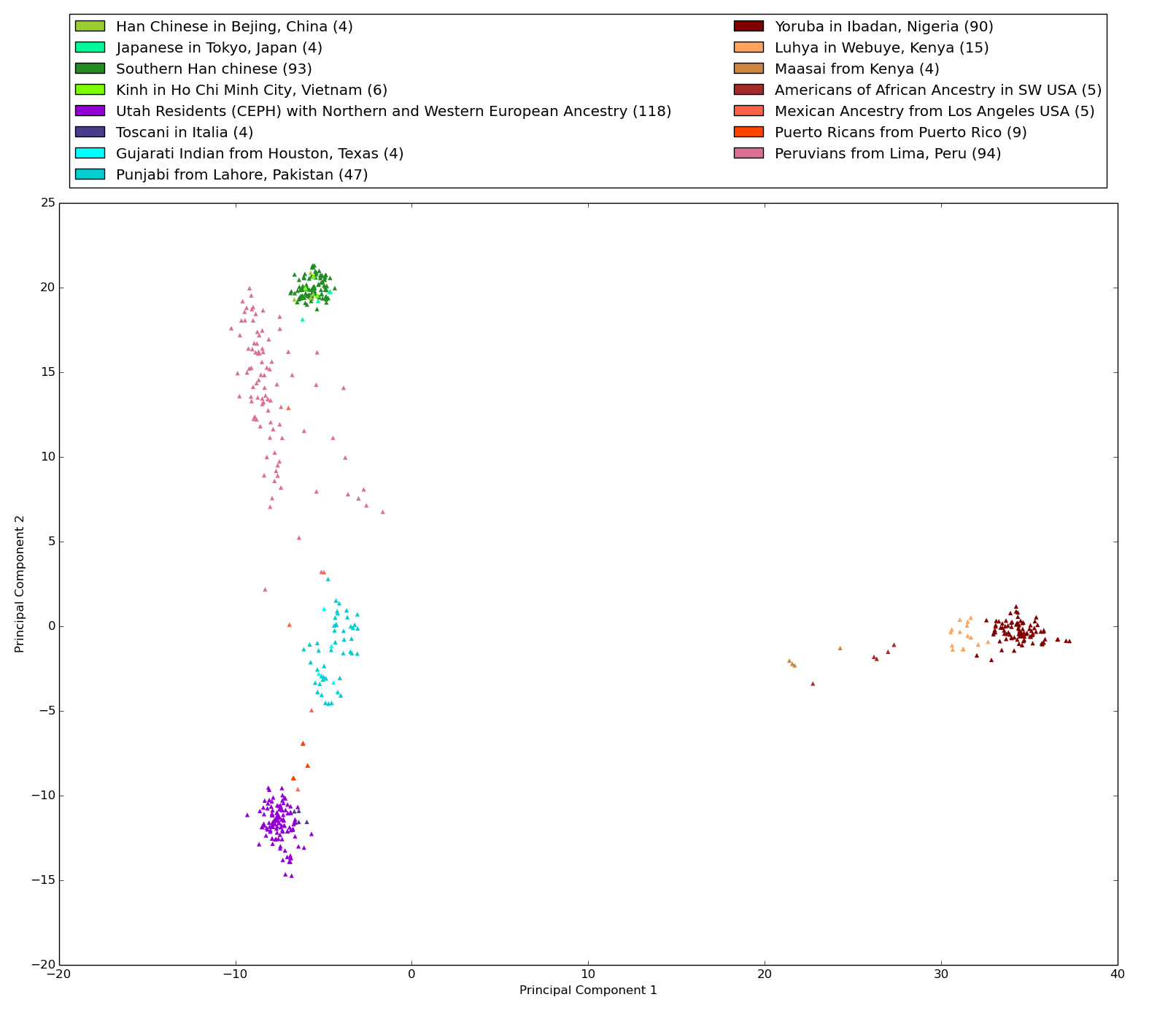
Figure S1: Projection of 502 1000 Genomes Project whole genome sequences along 489 their first two principal components
This projection replicates the 1000 Genomes Project PCA projection, which used single nucleotide variants. Only well sequenced positions in autosomal chromosomes were used in our PCA: 29,366 positions out of 20,160,996 (10,080,498 per phase) (0.146% of the genome). As expected, the first principal component separates the participants of African descent from the other participants. The second principal component separates the participants of European and Asian descent. The African super population is colored with brown shades, the Ad Mixed American super population is colored with red shades, the East Asian super population is colored with green shades, the South Asian super population is colored in light blue shades, and the European super population is colored in purple shades. The number in parenthesis in the legend is the number of whole genome sequences labeled with this ethnicity.
{kind=link}
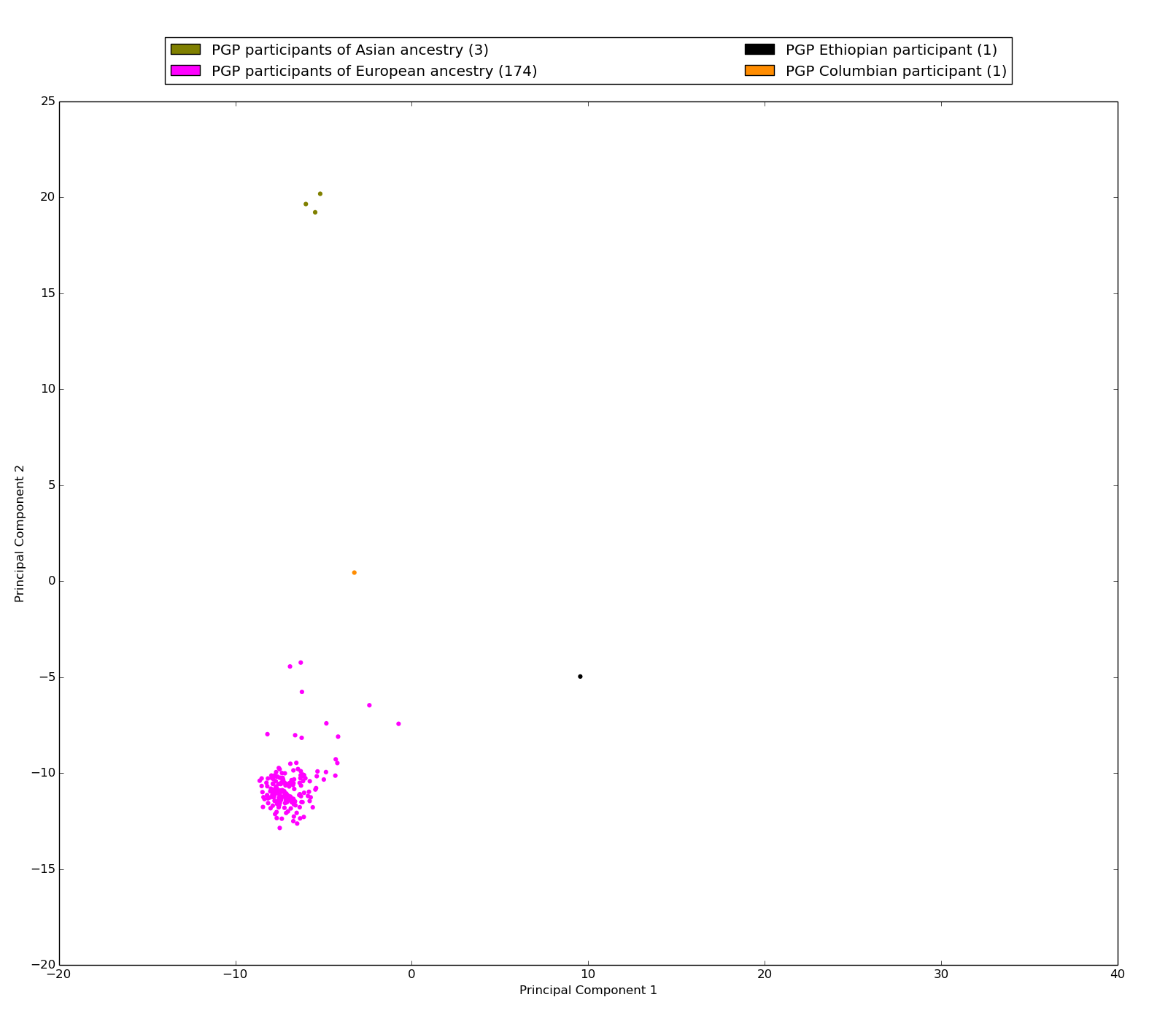
Figure S2: Projection of 178 PGP whole genome sequences along their first two principal components
Only well sequenced positions in autosomal chromosomes were used in PCA: 29,366 positions out of 20,160,996 (10,080,498 per phase) (0.146% of the genome). As expected, the first principal component separates the participants of African descent from the other participants. The second principal component separates the participants of European and Asian descent. The African super population is colored with brown shades, the Ad Mixed American super population is colored with red shades, the East Asian super population is colored with green shades, the South Asian super population is colored in light blue shades, and the European super population is colored in purple shades. The number in parenthesis in the legend is the number of whole genome sequences (callsets) with this ethnicity. Note the large proportion of participants with european ancestry indicating the ethnic homogeneity of the PGP.
{kind=link}