Characterisation of the human uterine microbiome in non-pregnant women through deep sequencing of the V1-2 region of the 16S rRNA gene
- Published
- Accepted
- Subject Areas
- Microbiology, Molecular Biology, Gynecology and Obstetrics, Women's Health
- Keywords
- uterus, human microbiome, vaginal microbiome, reproduction, Illumina, microbiota, uterine microbiome, 16S ribosomal RNA, endometrium, bacterial vaginosis
- Copyright
- © 2015 Verstraelen et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ PrePrints) and either DOI or URL of the article must be cited.
- Cite this article
- 2015. Characterisation of the human uterine microbiome in non-pregnant women through deep sequencing of the V1-2 region of the 16S rRNA gene. PeerJ PrePrints 3:e1269v2 https://doi.org/10.7287/peerj.preprints.1269v2
Abstract
Background. It is widely assumed that the uterine cavity in non-pregnant women is physiologically sterile, also as a premise to the long-held view that human infants develop in a sterile uterine environment, though likely reflecting under-appraisal of the extent of the human bacterial metacommunity. In an exploratory study, we aimed to investigate the putative presence of a uterine microbiome in a selected series of non-pregnant women through deep sequencing of the V1-2 hypervariable region of the 16S ribosomal RNA (rRNA) gene. Methods. Nineteen women with various reproductive conditions, including subfertility, scheduled for hysteroscopy and not showing uterine anomalies were recruited. Subjects were highly diverse with regard to demographic and medical history and included nulliparous and parous women. Endometrial tissue and mucus harvesting was performed by use of a transcervical device designed to obtain endometrial biopsy, while avoiding cervicovaginal contamination. Bacteria were targeted by use of a barcoded Illumina MiSeq paired-end sequencing method targeting the 16S rRNA gene V1-2 region, yielding an average of 41,194 reads per sample after quality filtering. Taxonomic annotation was pursued by comparison with sequences available through the Ribosomal Database Project and the NCBI database. Results. Out of 183 unique 16S rRNA gene amplicon sequences, 15 phylotypes were present in all samples. In some 90% of the women included, community architecture was fairly similar in as much B. xylanisolvens, B. thetaiotaomicron, B. fragilis and an undetermined Pelomonas taxon constituted over one third of the endometrial bacterial community. On the singular phylotype level, six women showed predominance of L. crispatus or L. iners in the presence of the Bacteroides core. Two endometrial communities were highly dissimilar, largely lacking the Bacteroides core, one dominated by L. crispatus and another consisting of a highly diverse community, including Prevotella spp., Atopobium vaginae, and Mobiluncus curtisii. Discussion. Our findings are, albeit not necessarily generalizable, consistent with the presence of a unique microbiota dominated by Bacteroides residing on the endometrium of the human non-pregnant uterus. The transcervical sampling approach may be influenced to an unknown extent by endocervical microbiota, which remain uncharacterised, and therefore warrants further validation. Nonetheless, consistent with our understanding of the human microbiome, the uterine microbiota are likely to have a previously unrecognized role in uterine physiology and human reproduction. Further study is therefore warranted to document community ecology and dynamics of the uterine microbiota, as well as the role of the uterine microbiome in health and disease.
Author Comment
Following peer review, we have substantially changed our manuscript.
Supplemental Information
Supplementary Table 1. Nucleotide sequences of all 183 phylotypes determined using Illumina-based amplicon deep-sequencing and their phylogenetic assignment
Phylotype number is a unique number to designate each annotated operational taxonomic unit in this study. Annotation refers to the highest taxonomic level to which an phylotype could be annotated by comparison with sequences available through the Ribosomal Database Project and the NCBI database.
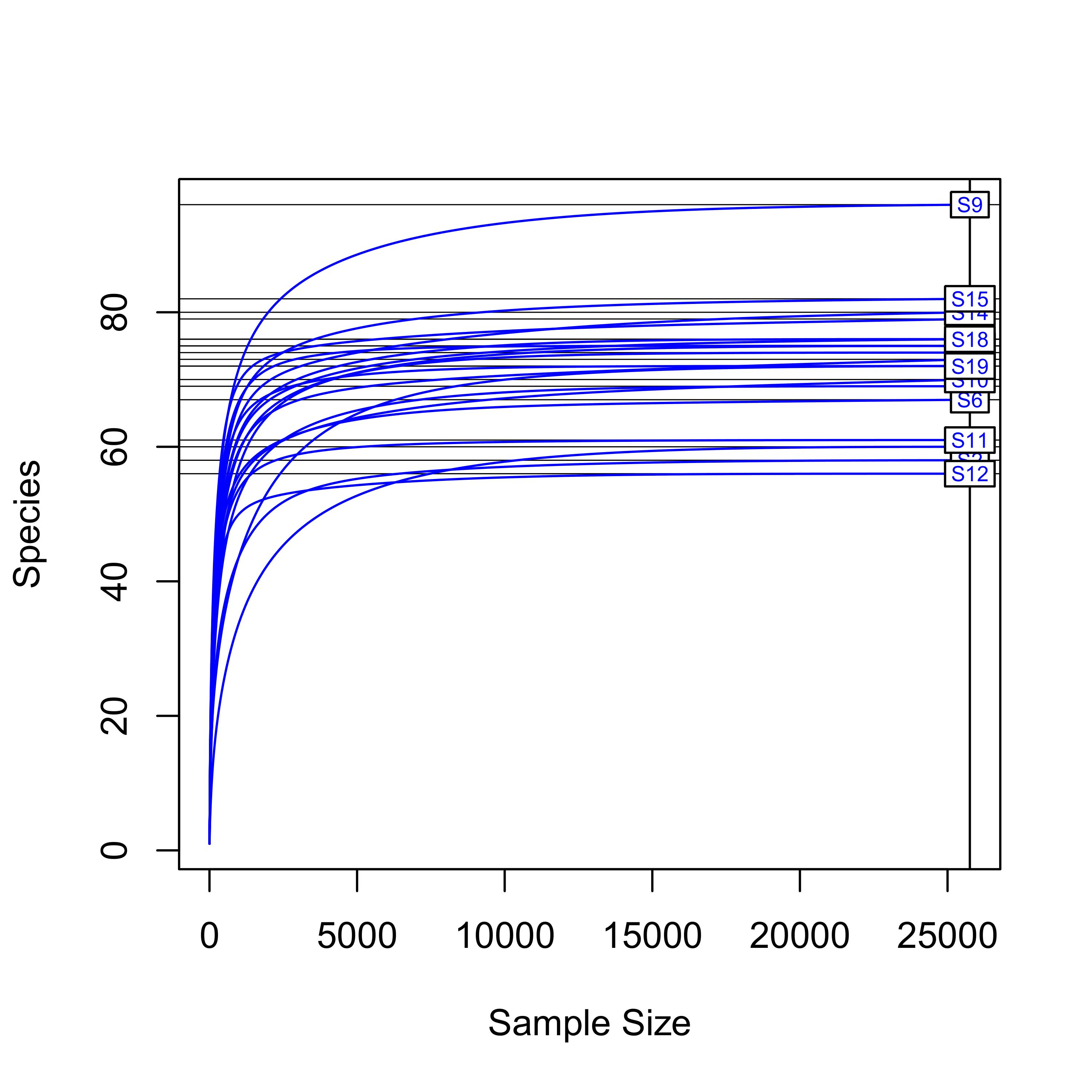
Figure S1. Rarefaction curves illustrating sampling depth
Phylotype number is a unique number used in this study corresponding to defined amplicon sequence as listed in Supplementary Table 1. For each endometrial community (subsequent columns) the relative abundances of those phylotypes is listed as a percentage.
Supplementary Figure 1. Rarefaction curves
Rarefaction curves were generated using the vegan package from the R program and show that saturation was reached at >15,000 reads per sample, and hence sufficient for all samples.
{kind=link}