
Spectrometric and computational studies of the binding of HIV-1 integrase inhibitors to viral DNA extremities
- Published
- Accepted
- Received
- Academic Editor
- Jan Jensen
- Subject Areas
- Theoretical and Computational Chemistry, Biophysical Chemistry, Physical Chemistry (other)
- Keywords
- HIV-1 Integrase, Halobenzene inhibitors, Viral DNA, Spectrometric studies, Computational studies
- Copyright
- © 2019 El Khoury et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Physical Chemistry) and either DOI or URL of the article must be cited.
- Cite this article
- 2019. Spectrometric and computational studies of the binding of HIV-1 integrase inhibitors to viral DNA extremities. PeerJ Physical Chemistry 1:e6 https://doi.org/10.7717/peerj-pchem.6
Abstract
Three integrase strand transfer inhibitors are in intensive clinical use, raltegravir (RAL), elvitegravir (EVG) and dolutegravir (DTG). The onset of integrase resistance mutations limits their therapeutic efficiency. As put forth earlier, the drug affinity for the intasome could be improved by targeting preferentially the retroviral nucleobases, which are little, if at all, mutation-prone. We report experimental results of anisotropy fluorescence titrations of viral DNA by these three drugs. These show the DTG > EVG > RAL ranking of their inhibitory activities of the intasome to correspond to that of their free energies of binding, ∆Gs, to retroviral DNA, and that such a ranking is only governed by the binding enthalpies, ∆H, the entropy undergoing marginal variations. We sought whether this ranking might be reproduced through quantum chemistry (QC) Density Functional Theory calculations of intermolecular interaction energies between simplified models consisting of sole halobenzene ring and the highly conserved retroviral nucleobases G4 and C16. These calculations showed that binding of EVG has a small preference over DTG, while RAL ranked third. This indicates that additional interactions of the diketoacid parts of the drugs with DNA could be necessary to further enable preferential binding of DTG. The corresponding ∆Etot values computed with a polarizable molecular mechanics/dynamics procedure, Sum of Interactions Between Fragments Ab initio computed (SIBFA), showed good correlations with this ∆E(QC) ranking. These validations are an important step toward the use of polarizable molecular dynamics simulations on DTG or EVG derivatives in their complexes with the complete intasome, an application now motivated and enabled by the advent of currently developed and improved massively parallel software.
Introduction
HIV-1 Integrase is a key element in viral replication. In addition to its essential role in viral DNA (vDNA) integration into host genomic DNA, it is involved, directly or indirectly, in reverse transcription (Li et al., 2011), nuclear import (Engelman et al., 1995; Ikeda et al., 2004) and HIV-1 particle maturation (Balakrishnan et al., 2013; Fontana et al., 2015). As in addition integrase has no counterpart in human cells, it could represent a privileged target for the design of potent antiretroviral drugs.
The integration step is carried by a multimer of integrase proteins assembled on vDNA ends, referred to as the intasome. In a first step, denoted as 3′-processing, a 3′GT dinucleotide is removed from each end of the long terminal repeats (LTRs) of vDNA. This occurs in the cytoplasm within a multi-component pre-integration complex which gathers the vDNA and several viral and cellular proteins. DNA strand transfer occurs in a second step, after such a complex is chaperoned into the nucleus and results in integration of vDNA as a provirus into the host genome. This requires cutting of two phosphodiester bonds five base pairs apart on opposite strands of the host DNA and is done by free 3′-OH groups that were liberated following LTR processing (Bushman & Craigie, 1991; Pommier, Johnson & Marchand, 2005; Lesbats, Engelman & Cherepanov, 2016). This mechanism was illustrated in two review papers, in Fig. 2 by Pommier, Johnson & Marchand (2005) and Fig. 1 by Lesbats, Engelman & Cherepanov (2016).
Integrase strand transfer inhibitors (INSTIs) proved to be much more effective than processing inhibitors and enabled the development of a successful class of antiretroviral drugs (Zhao et al., 2017). Three inhibitors inspired by the original diketo acids have been successively approved and are commonly used in HIV-1 treatment, namely raltegravir (RAL; MK-0518), elvitegravir (EVG; GS-9137) (Zhao et al., 2017; Grinsztejn et al., 2007; Koelsch & Cooper, 2009) and dolutegravir (DTG; S/GSK1349572) (Grinsztejn et al., 2007; Koelsch & Cooper, 2009; Métifiot, Marchand & Pommier, 2013; Ballantyne & Perry, 2013). The latter is a second generation INSTI aimed at maintaining an effective efficacy to integrase variants resistant to RAL and EVG (Anstett et al., 2017; Wainberg & Han, 2015). A general review of integrase inhibitors was published by Pommier, Johnson & Marchand (2005) encompassing a list of diketoacid inhibitors prior to RAL, EVG and DTG (Fig. 4, Pommier, Johnson & Marchand, 2005).
As was the case for protease and reverse transcriptase inhibitors (Rhee et al., 2010; Tantillo et al., 1994), integrase inhibitors can generate several resistance mutations. These were recently reported following RAL, EVG and DTG treatment (Métifiot et al., 2010; Marinello et al., 2008; Hazuda et al., 2000; Wittkop et al., 2009). They affect, not only residues in direct interactions with INSTIs but also “outer-shell” residues indirectly bound (Anstett et al., 2017; Blanco et al., 2011; Chen et al., 2013). Among these are double mutations such as Q148R/H coupled to G140S/A that produce important synergetic effects on the efficacy of RAL, EVG and even DTG (Goethals et al., 2010; Naeger et al., 2016).
RAL, EVG and DTG (Fig. 1) selectively bind at the interface of integrase and the vDNA ends within the intasome and have in common two distinct structural motives: (a) a large centralized pharmacophore with both keto oxygen and a coplanar neighboring oxygen coordinating the two catalytic Mg(II) cations, structural water molecules, and, either directly or through water, integrase residues; and (b) a halobenzyl group targeting the highly conserved 5′CpA 3′/5′TpG 3′ step on the vDNA ends (Zhao et al., 2017). However, the surface and oxygen arrangement of the central pharmacophore differ among the three INSTIs, as well as the nature and position of the halogenation of their terminal aromatic ring: a single F is attached in para to RAL’s halobenzene ring, whereas EVG has an F in ortho and a Cl in meta, and DTG has two F atoms in ortho and para.
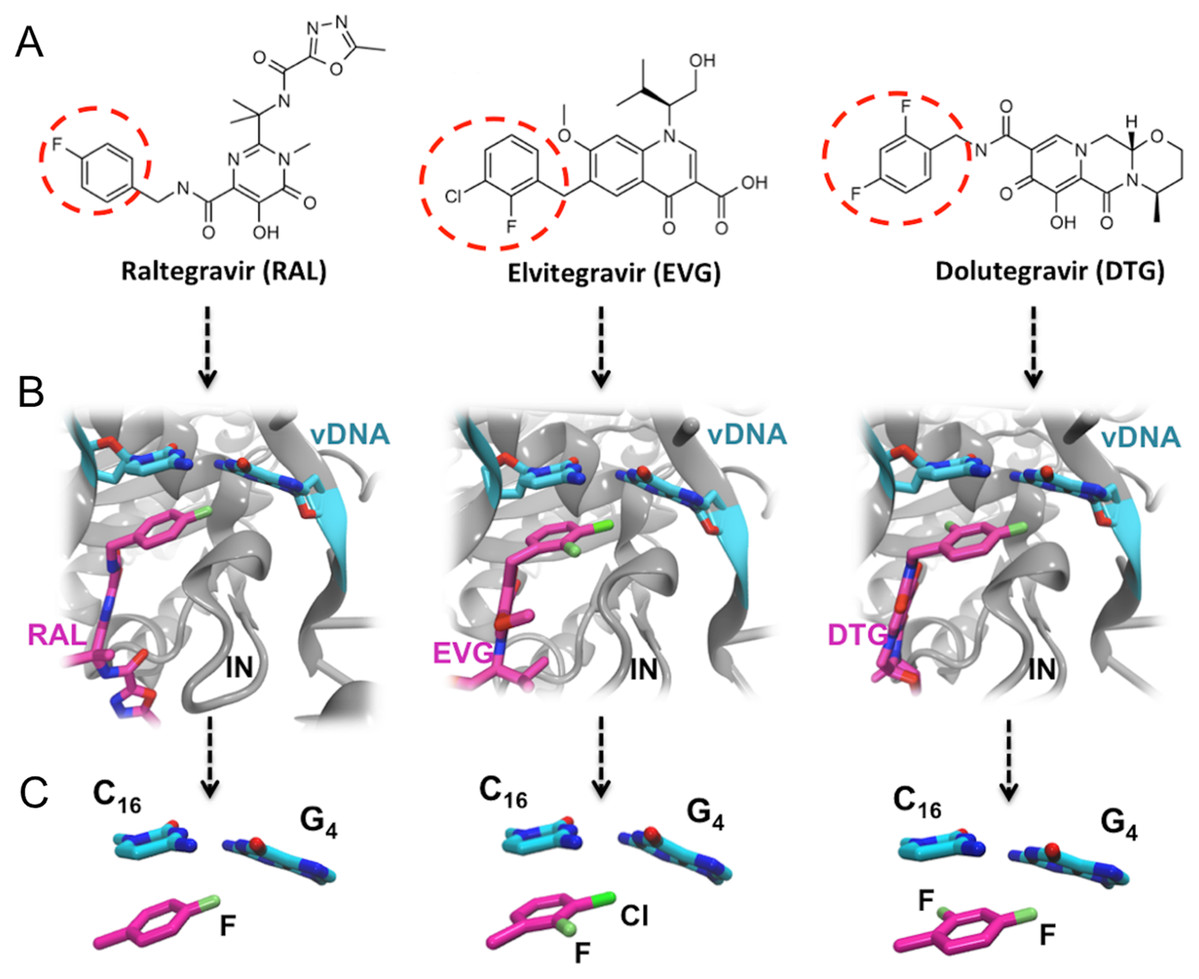
Figure 1: FDA approved Integrase strand transfer inhibitors INSTIs: raltegravir, elvitegravir and dolutegravir.
(A) 2D structures of the inhibitors. The red dashed circle indicates the halobenzyl moiety. (B) High-resolution X-ray 3D structure of each inhibitor in complex with the Integrase (IN) and the viral DNA (vDNA). (C) Close-up on the interactions involving the halobenzene, cytosine 16 and guanine 4.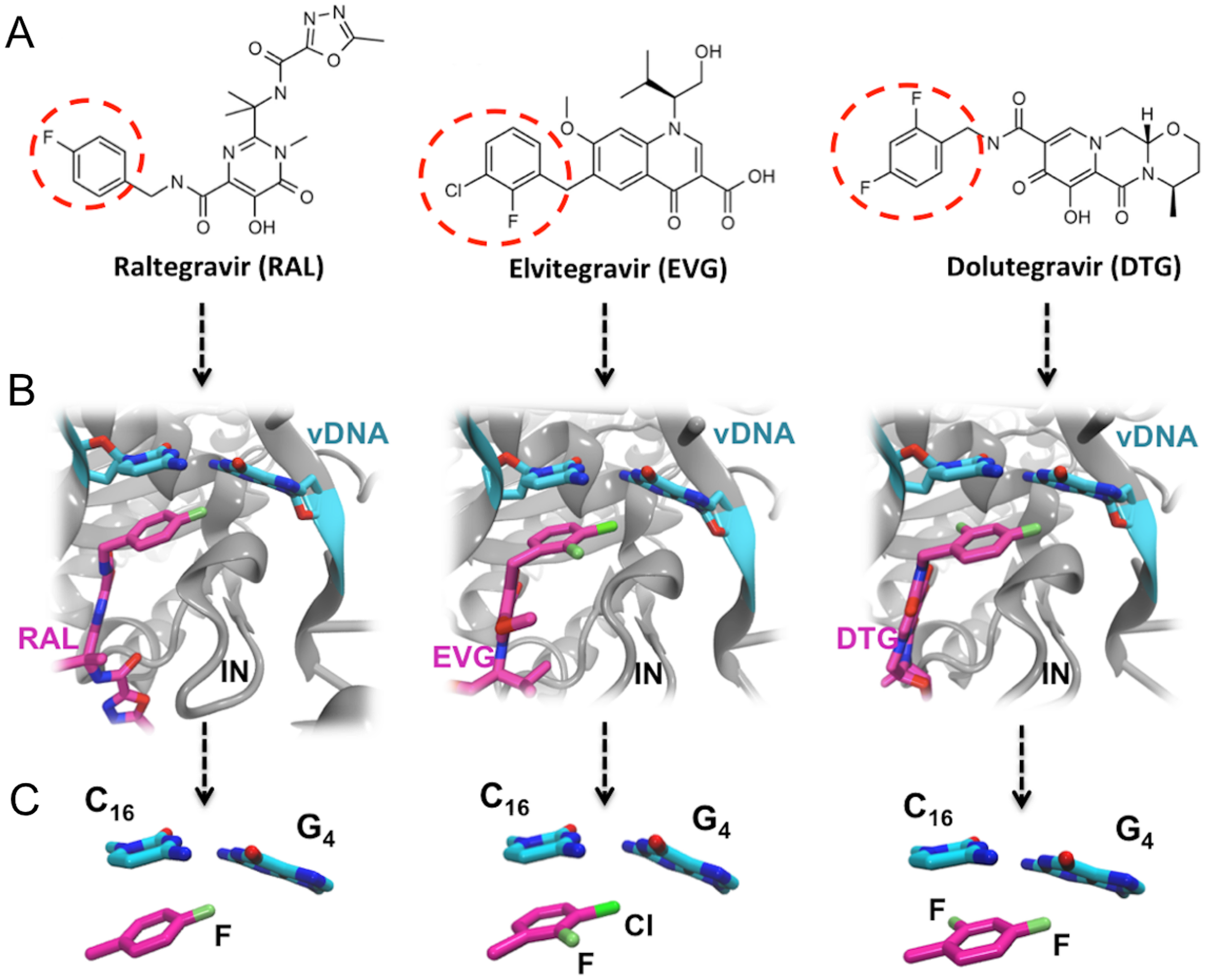{kind=link}
Thus within the intasome, the binding of all three drugs targets both vDNA and the viral protein. In addition to the well-known established interactions with the catalytic and non-catalytic site of integrase, following the 3′ processing reaction, the X-ray structures of integrase-DNA-inhibitor complexes (Fig. 1B) show that the halogenated benzene ring stacks over the C base (C16) upstream, while the C-X bond points toward the center of the G base (G4) downstream of the second strand (Hare et al., 2010a, 2010b, 2011).
On another note, it was reported that, compared with RAL and EVG, DTG displays a more potent in vitro anti-HIV activity and a distinct resistance profile (Anstett et al., 2017; Wainberg & Han, 2015; Vandekerckhove, 2010; Osterholzer & Goldman, 2014). Furthermore, we have ourselves reported that an increase in the drug-vDNA complex stability correlates with an increase in drug activity and a decrease in viral resistance (Anstett et al., 2017; Hare et al., 2010b, 2011), highlighting the important contribution of the vDNA end recognition for the binding affinity of INSTIs to the intasome (Hobaika et al., 2010; El Khoury et al., 2017; Ammar et al., 2012, 2016). In this connection, we have shown a narrow correlation between the strongest DTG-DNA affinity and DTG’s highest barrier to resistance (El Khoury et al., 2017).
Maximizing shape complementarity at the integrase-vDNA-inhibitor surface could serve as a guiding principle for the development of new INSTIs (Zhao et al., 2017; Hare et al., 2010a; Hobaika et al., 2009, 2010). An important feature of second generation INSTIs consists into their increased contacts not only with protein active and non-active site, but also with processed vDNA, right before the strand transfer step. This is supported by the fact that the sequence of the nucleic bases at the ends of the LTR represents stringent requirements concerning retroviral integration and that there is no evidence of mutations in the LTRs that could lead to resistance to INSTIs (Hobaika et al., 2009; LaFemina, Callahan & Cordingley, 1991; Sherman, Dickson & Fyfe, 1992; Zargarian et al., 2003).
Could attempts to design novel INSTIs with enhanced affinities focus on the vDNA, which would render the new inhibitors much less sensitive to mutations occurring on integrase?
As a continuation of our previous studies, we experimentally measure the ΔG values for the binding of RAL, EVG and DTG to vDNA. This is done by fluorescence anisotropy experiments at three different temperatures. We also analyze, by both ab initio quantum chemistry (QC) and polarizable molecular mechanics/dynamics, the intermolecular interaction energies of their halobenzyl rings with G4 and C16. The individual energy contributions of ΔE(QC) are also compared to their Sum of Interactions Between Fragments Ab initio computed (SIBFA) counterparts. Such analyses and validations of inhibitor interactions within the core of vDNA binding site constitute a necessary step toward long-duration polarizable molecular dynamics of drug-intasome complexes.
An outstanding feature of the CX ring in halobenzenes, discovered on the basis of QC (Clark et al., 2007; Politzer, Murray & Clark, 2010) is the existence of a zone of electron depletion along the extension of the bond with a magnitude increasing along the series F > Cl > Br > I. This “sigma-hole” goes along with a zone of electronic build-up on a cone around the halogen. It has been earlier shown that atom-centered point charges used in “classical” force-fields cannot account for the impact of the sigma-hole on the Coulomb electrostatic contribution EC: this could be only partly remedied upon resorting to an additional fictitious atom prolonging the CX bond with a partial charge and a distance to the X bearer that have to be fit on the basis of QC calculations (Ibrahim, 2011; Jorgensen & Schyman, 2012; Kolar & Hobza, 2012). On the other hand, anisotropic potentials such as SIBFA, with distributed atomic multipoles up to quadrupoles, were shown to closely account for the impact of the sigma-hole along the F, Cl and Br series on the magnitude of EC both along and around the CX- bond without extra calibration effort (El Hage et al., 2013). Along these lines, recent work showed that another possibility, resorting to a distributed charge model to reproduce the local quadrupole around the CX could also enable to account for the impact of the sigma-hole on EC (Devereux et al., 2014; El Hage et al., 2016). An additional incentive to validate SIBFA in the present work is the perspective of its applications to the entirety of the inhibitor-intasome complex, in which the presence of two Mg(II) cations close to one another and of structured water molecules render it preferable to resort to polarizable potentials than to “classical,” non-polarizable ones.
At this stage we do not intend to compute true ΔGs to compare the binding affinities of the three inhibitors to vDNA and a fortiori to the intasome. These would only be meaningful at the outcome of long-duration Molecular Dynamics, and such an outcome could be very sensitive to the accuracy of the intermolecular potential. As a first step toward this evaluation, we focus here on the sole halobenzene-G4/C16 interactions expressed in terms of actual enthalpies of binding and evaluate: (a) how well the ranking of ΔE(QC) intermolecular interaction energies in the ternary complexes compares to the experimental binding enthalpy ranking; (b) how well the magnitudes of ΔEtot(SIBFA) compare to those of ΔE(QC) in the three complexes and if they have the same ranking.
Materials and Methods
DNA sample and inhibitors
The oligonucleotide LTR32 (Fig. 2) was purchased from Eurogentec (Belgium). It was designed to adopt a folded double-stranded hairpin structure even under the low concentrations (10−9 to 10−5 M) used in fluorescence anisotropy experiments. RAL, EVG and DTG were purchased from AdooQ and Medchemexpress, respectively and their structures are represented in Fig. 1A.
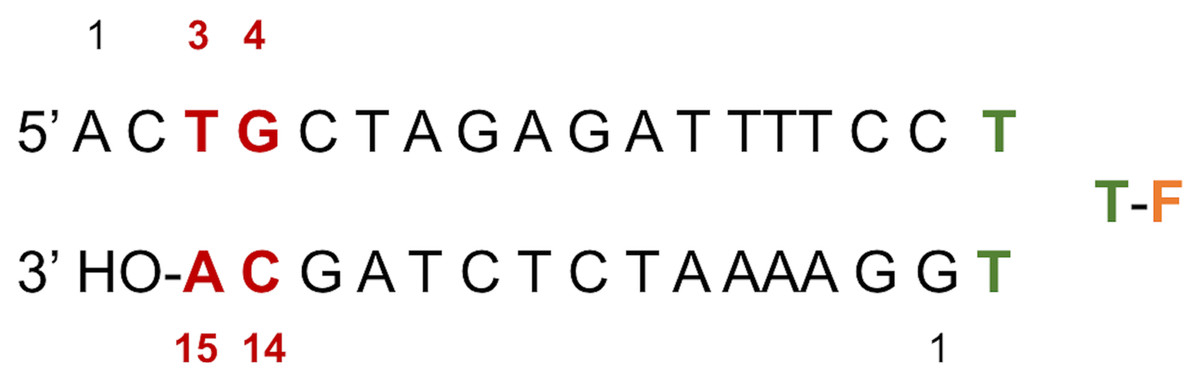
Figure 2: LTR32 is a linear oligonucleotide sequence designed to adopt a double strand hairpin structure in solution upon folding around a loop created by a purposely added thymine triplet (TTT in green) with the sensitive fluorescein reporter (F, in orange) grafted to its central T.
The latter allows fluorescence studies in solution at low concentrations. The stem that reproduces the 3′ processed LTR end comprises a 17-nucleotide strand and a 15-nucleotide strand corresponding to the unreactive strand and the reactive strand, respectively. Their pairing leaves an unpaired dinucleotide 5′ AC 3′ at the 5′ end on the unreactive strand. In each strand the nucleotide numbering goes from the 5′ to the 3′ extremity. The highly conserved doublet of base pairs, here numbered C14-G4 and A15-T3, is colored in red. The base pair C14-G4 is the equivalent of C16-G4 mentioned in the text based on the PDB entries used in the computations.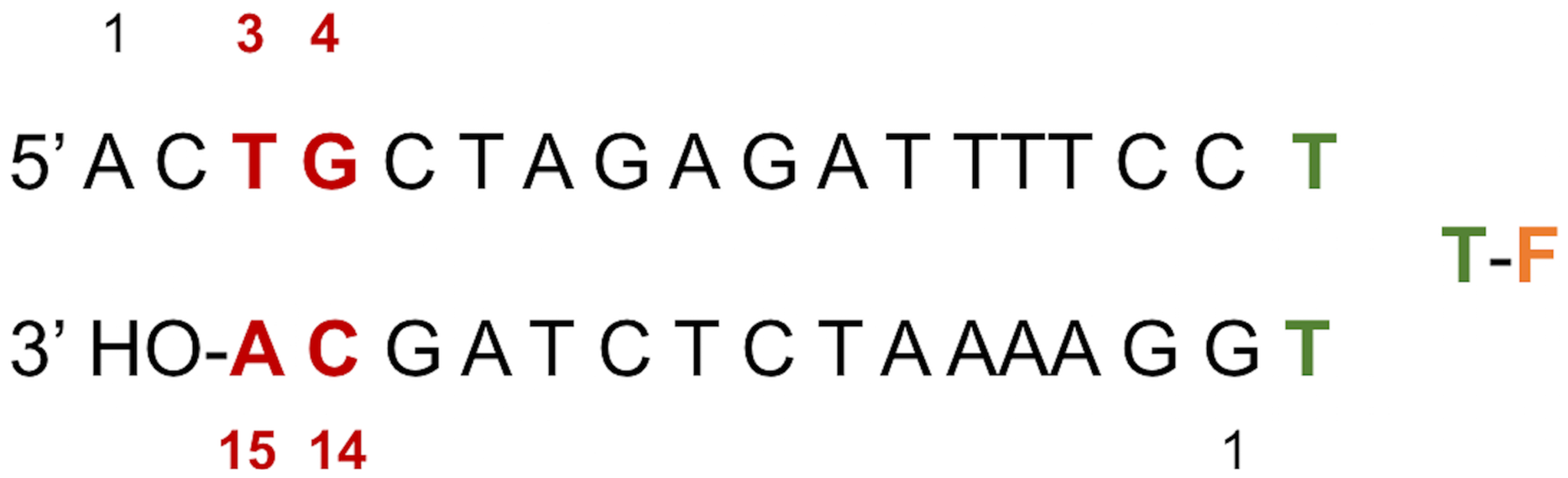{kind=link}
LTR32 is a linear oligonucleotide sequence designed to adopt a double strand hairpin structure in solution upon folding around a loop created by a purposely added thymine triplet (TTT in green) with the sensitive fluorescein reporter (F, in orange) grafted to its central T. The latter allows fluorescence studies in solution at low concentrations. The stem that reproduces the 3′ processed LTR end comprises a 17-nucleotide strand and a 15-nucleotide strand corresponding to the unreactive strand and the reactive strand, respectively. Their pairing leaves an unpaired dinucleotide 5′ AC 3′ at the 5′ end on the unreactive strand. In each strand the nucleotide numbering goes from the 5′ to the 3′ extremity. The highly conserved doublet of base pairs, here numbered C14-G4 and A15-T3, is colored in red.
Fluorescence measurements
Thermodynamic parameters of ligand-processed DNA complexes were identified using fluorescence anisotropy (Heyduk & Lee, 1990; Hill & Royer, 1997) on a Jobin-Yvon Fluoromax II instrument. RAL was purchased from AdooQ and EVG and DTG were purchased from Medchemexpress. The LTR32 oligonucleotide, reproducing the processed vDNA, was purchased from Eurogentec (Belgium). It contains a thymine loop bearing the fluorescein reporter for fluorescence studies. During titrations, labeled DNA (LTR32) was dissolved in phosphate buffer (10 mM, pH 6, I = 0.1) and placed in thermally jacketed quartz cells (one cm) at 5, 15 and 25 °C; increasing concentrations of the inhibitors (RAL, EVG or DTG) were then added. The excitation was recorded at 488 nm and the emission at 516 nm. The equilibrium dissociation constants (Kd) were determined with GraphPAD Prism 5 applying the non-linear regression (curve fit)-Least square procedure. This analysis led to the calculation of binding free energies using the following equation: ΔG = −RT ln(1/kd). In order to transform the obtained ΔG into ΔH and ΔS, we graphed a linear function: ΔG = ΔH – TΔS for each DNA-ligand complex (LTR32-RAL, LTR32-EVG, and LTR32-DTG); ΔG (kcal/mole) is the binding free energy at 5, 15 and 25 °C and T is the respective temperature in Kelvin. The fluorescence anisotropy profiles of RAL, EVG and DTG are displayed in Figs. S1–S3 respectively.
PDB entries
In the used computational approaches, all complexes were extracted from the X-ray structures of the PFV intasome (IN-viral DNA-Mg2+) in complex with EVG (Hare et al., 2010a) (PDB code: 3L2U), DTG (Hare et al., 2011) (PDB code: 3S3M) and RAL (Hare et al., 2010b) (PDB code: 3OYA).
Ab-initio QC computations
The systems were first energy-minimized at the correlated level using the dispersion—corrected B97-D3 functional by Goerigk & Grimme (2011) and the cc-pVTZ basis set (Dunning, 1989; Feller, 1996) with the Gaussian 09 (G09) software (Frisch et al., 2009). The C1’ atoms of G4 and C16 were replaced by an H atom and the H-N9 and H-N1 bonds were set at 1.0 Å, retaining the N in the position it occupies in DNA in the corresponding X-ray structure. During minimization, these two bonds were held in place. This constraint was set to account for the anchoring of G4 and C16 in the DNA backbone. At the outcome of energy-minimization, we could verify that the G4-C16 base-pair intermolecular interaction energies had equal values in the three complexes. This was not the case without base-pair relaxation, the intermolecular G4-C16 interaction energy being three kcal/mole less favorable in the EVG ternary complex than in the RAL and DTG ones. Thus, such a relaxation, even though partial, removes any bias that would disfavor the EVG complex with respect to the two other ones. Similarly, the C atom connecting the halobenzene ring to the diketoacid was replaced by an H atom, the CH bond being set at 1.09 Å, the halobenzene C occupying the same position as in the crystal structure. We found it necessary to also retain the HC bond in place, so that minimization of the halobenzene ring was limited to actual rotations around this bond along with minor intramolecular ring geometry relaxation. It was found that otherwise for the DTG complex a translation of the halobenzene ring could occur with full stacking underneath G4, but this would be prevented in the presence of the anchoring diketoacid entity which would then undergo unfavorable steric interactions with DNA. Figures 3A–3C give representations of the three ternary complexes, with a superimposition of the starting and final structures.
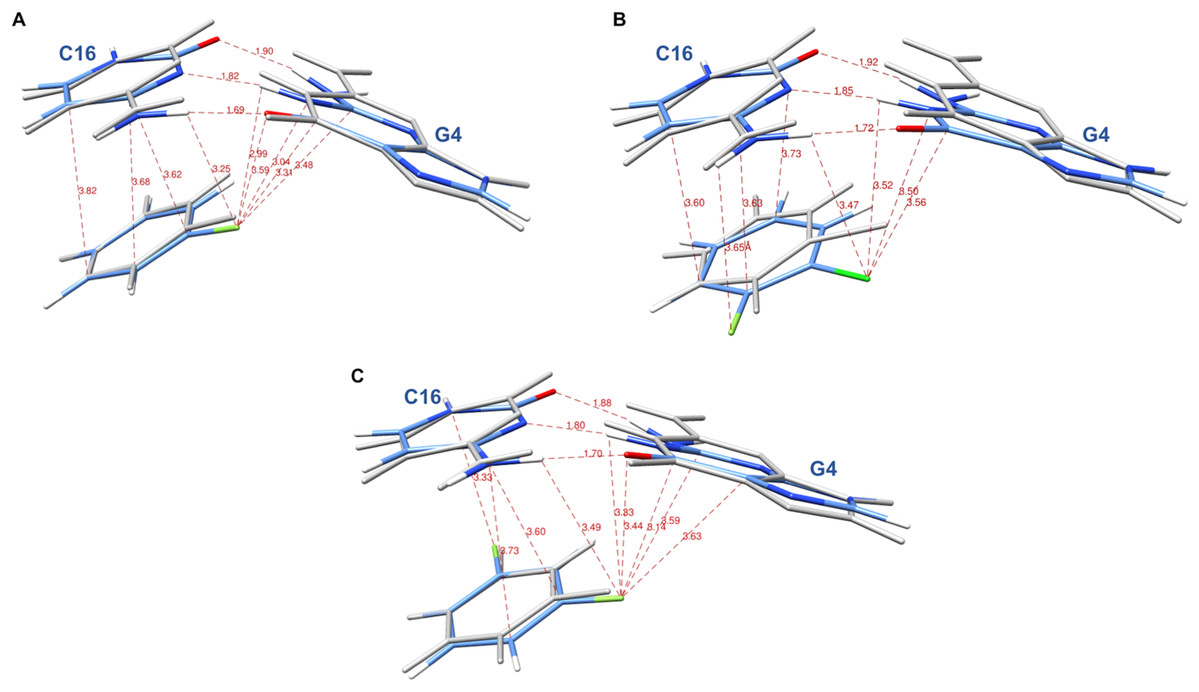
Figure 3: Representation of the complex of G4/C16 with the halobenzene rings of RAL (A), EVG (B) and DTG (C) at the outcome of restrained energy-minimization, with relevant intermolecular distances.
The starting X-ray structures are shown in white.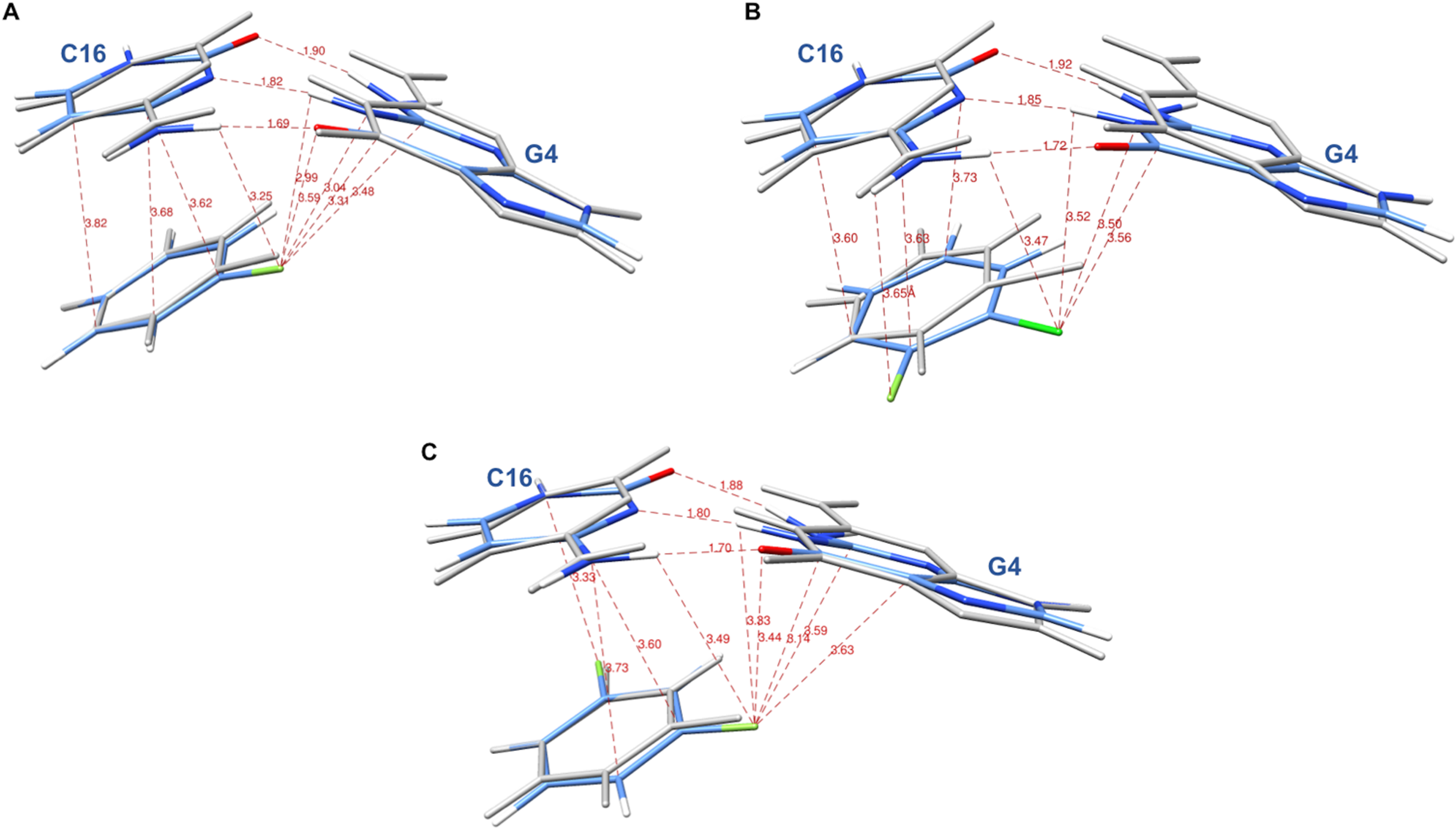{kind=link}
At the outcome of energy-minimization, single-point calculations were performed with the B97D3 functional including the correction for basis set superposition error (BSSE) (Boys & Bernardi, 1970; Simon, Duran & Dannenberg, 1996). They were also done with the B3LYP-D3 (Goerigk & Grimme, 2011), ωB97X-D (Mardirossian & Head-Gordon, 2014) and B97-D functionals (Grimme, 2006). Energy-decomposition analyses (EDA) were performed using the ALMOEDA procedure (Khaliullin et al., 2007; Horn, Mao & Head-Gordon, 2016) with the QChem software (Shao et al., 2015).
SIBFA computations
The intermolecular interaction energy (ΔEtot) is computed as the sum of five contributions: electrostatic multipolar (EMTP), short-range repulsion (Erep), polarization (Epol), charge transfer (ECT) and dispersion (Edisp) (Gresh et al., 2007). EMTP is computed with distributed multipoles derived from the QC molecular orbitals of the individual ligands (Stone, 1981; Stone & Alderton, 1985; Vigné-Maeder & Claverie, 1988), augmented with penetration (Piquemal, Gresh & Giessner-Prettre, 2003). The anisotropic polarizabilities are distributed on the centroids of the localized orbitals using the Garmer and Stevens procedure (Garmer & Stevens, 1989) coded in the GAMESS software (Schmidt et al., 1993). Erep and ECT are computed using representations of the molecular orbitals on the chemical bonds and the lone-pairs. Edisp has an expansion into 1/R6, 1/R8, and 1/R10 along with an exchange—dispersion component (Creuzet, Langlet & Gresh, 1991).
Results
We have previously reported that the strong INSTIs bind tightly to the ends of the 3′-processed vDNA, at the LTR ends (El Khoury et al., 2017); thus, justifying their function as a blocker of the strand transfer step (Hare et al., 2010a). Table 1 reports the Kd values for the binding of RAL, EVG and DTG to the LTR32 oligonucleotide, as determined by fluorescence anisotropy titrations in a phosphate buffer at 5, 15 and 25 °C. The fluorescence anisotropy titration curves of LTR32 for increasing concentrations of drugs are reported in Figs. S1–S3. Those of LTR32 by DTG and EVG at 5 °C already reported in El Khoury et al. (2017), are added to this study for completeness. The DTG > EVG > RAL Kd ranking of inhibitor-vDNA binding affinities is the same as the one reported for their binding to the complete intasome (Hightower et al., 2011).
| Temperature | RAL | EVG | DTG |
|---|---|---|---|
| 278 | 5.90E−09 | 9.50E−11 | 1.63E−12 |
| 288 | 7.69E−09 | 1.44E−10 | 2.74E−12 |
| 298 | 1.26E−08 | 2.65E−10 | 6.07E−12 |
Note:
Temperatures in Kelvin, Kd values in molar.
We have further unraveled the enthalpy and entropy components of the free energies of LTR32-INSTI complexation. The ΔG, ΔH and ΔS values are reported in Tables 2A–2C. These results show the affinity ranking of the three inhibitors for end vDNA to be dominated by the enthalpy component. This finding is fully consistent with the microcalorimetry study reported by Chaires (2006) which covered 26 DNA ligands and led to the conclusion that formation of DNA-intercalator complexes is enthalpy-driven, while that of DNA-groove binder complexes is entropy-driven. RAL-LTR32, EVG-LTR32 and DTG-LTR32 interactions are characterized by a mean ΔH/ΔG ratio in the 1.3–1.4 range. This is within the 0.83–1.97 ΔH/ΔG ratio range, considered as a signature of an enthalpy-dominated interaction.
| Temperature | ΔG | ΔH | TΔS |
|---|---|---|---|
| (A) Thermodynamic contributions of LT32-raltegravir interactions. | |||
| 278 | −10.07 | −14.16 | 4.09 |
| 288 | −10.29 | −14.53 | 4.24 |
| 298 | −10.37 | −14.75 | 4.39 |
| (B) Thermodynamic contributions of LT32-elvitegravir interactions. | |||
| 278 | −12.27 | −16.36 | 4.09 |
| 288 | −12.48 | −16.71 | 4.23 |
| 298 | −12.56 | −16.94 | 4.38 |
| (C) Thermodynamic contributions of LT32-dolutegravir interactions. | |||
| 278 | −14.43 | −18.50 | 4.07 |
| 288 | −14.66 | −18.88 | 4.22 |
| 298 | −14.72 | −19.08 | 4.37 |
Note:
Temperatures in Kelvin, energies in kcal/mole.
The above experimental results are an incentive for SIBFA polarizable molecular dynamics simulations of complexes of various halogenated drugs with retroviral DNAs, which should benefit from the massively parallel Tinker-HP software, co-developed in one of our Laboratories (Lagardère et al., 2018). We deemed it necessary, however, to perform a prior validation in addressing the question: to which extent would the binding of the drug halobenzyl rings to G4 and C16 be accountable for the DTG > EVG > RAL ranking, and how well could the outcome from high-level QC computations be accounted for by the SIBFA polarizable molecular mechanics procedure?
The considered complexes have small sizes and, for the present purposes, energy-minimizations bore on the sole halobenzyl ring. An evaluation of the SIBFA accuracy is nevertheless mandatory, as there would be little hope that inconsistencies between the SIBFA and QC results at this early stage could be obliterated or restored by subsequent large-scale molecular dynamics simulations on the entire drug-IN-vDNA complex. It also is in line with our previous analyses on the binding of a series of mono- and poly-halogenated rings to G4/C16 and the sensitivity of ΔE and its individual contributions to diverse chemical substitutions (El Hage et al., 2014, 2015).
Table 3A lists the ΔE(QC) and ΔEtot(SIBFA) values, and Table 3B reports the ALMOEDA contributions to ΔE(B97-D) and ΔE(ωB97X-D) and to ΔEtot(SIBFA). The chloro-fluorobenzene ring of EVG is found to be favored over the difluoro ring of DTG in their ternary complexes, by a small margin. It amounts to 1 and 1.6 kcal/mole out of 40 with the B97-D3 BSSE-uncorrected and corrected functional, respectively, and is reduced to 0.4 and 1.0 kcal/mol with the B3LYP-D3 BSSE-uncorrected and corrected functionals. The ALMOEDA differences are smaller, namely 0.1 (B97D) and 0.7 kcal/mole (ωB97X-D). Such differences could simply reflect an inherent preference for the G4/C16 base pair of the chloro-fluorobenzene ring rather than the difluorobenzene ring. The DTG ring is on the other hand, consistently favored over the RAL one. The differences are more stable, amounting to 0.8–1 and kcal/mole with all four functionals. They may simply reflect the gain in energy brought about by the second fluorine substituent.
| (A) | |||||
|---|---|---|---|---|---|
| Procedure | B97D3 | B3LYPD3 | ωB97D | B97D | SIBFA |
| RAL complex | |||||
| ΔEtot | −41.4a | −42.7a | −38.5c | −36.2c | −38.6 |
| −37.2b | −38.7b | ||||
| EVG complex | |||||
| ΔEtot | −43.3a | −44.1a | −39.9c | −37.2c | −42.0 |
| −39.6b | −40.5b | ||||
| DTG complex | |||||
| ΔEtot | −42.3a | −43.7a | −39.2c | −37.1c | −39.3 |
| −38.0b | −39.5b | ||||
| (B) | ||||
|---|---|---|---|---|
| B97D | ωB97X-D | HF | SIBFA | |
| RAL complex | ||||
| EMTP | −47.5 | |||
| Erep | 50.9 | |||
| E1 | −6.1 | −11.3 | 3.3 | 3.5 |
| Epol | −13.0 | −13.1 | −13.7 | −11.5 |
| Ect | −17.1 | −14.1 | −9.3 | −6.8 |
| ΔE | −19.7 | −14.8 | ||
| Edisp | −23.8 | |||
| ΔEtot | −36.1 | −38.5 | −38.5 | |
| EVG complex | ||||
| EMTP | −46.3 | |||
| Erep | 46.4 | |||
| E1 | −8.7 | −14.1 | 3.9 | 0.1 |
| Epol | −12.3 | −12.5 | −13.0 | −11.2 |
| Ect | −16.2 | −13.3 | −8.6 | −6.5 |
| ΔE | −17.8 | −17.5 | ||
| Edisp | −24.5 | |||
| ΔEtot | −37.1 | −39.9 | −42.1 | |
| DTG complex | ||||
| EMTP | −47.7 | |||
| Erep | 52.1 | |||
| E1 | −6.3 | −11.3 | 4.0 | 4.4 |
| Epol | −13.2 | −13.3 | −13.8 | −11.8 |
| Ect | −17.5 | −14.6 | −9.7 | −7.2 |
| ΔE | −19.7 | −14.6 | ||
| Edisp | −24.8 | |||
| ΔEtot | −37.1 | −39.2 | −39.3 | |
We have computed the zero-point vibration energies (ZPE) in the three minimized complexes. For each ligand, the differences of ZPE between the ternary complex and the three separate monomers were the same, 2.2 kcal/mole. ZPE is thus not expected to impact the ΔE(QC) ranking.
For consistency, in each complex, the present SIBFA calculations were done with the same internal geometries of the three monomers as the B97-D3 minimized ones. ΔEtot(SIBFA) reproduces the QC ranking. It has values consistently intermediate between the BSSE-uncorrected and corrected ΔE(B97-D3) ones, and this is also the case with respect to ΔE(B3LYP-D3) values concerning the difluoro DTG and monofluoro rings of RAL. ΔEtot(SIBFA) appears to overestimate the EVG vs DTG ring preference, namely 2.5 kcal/mole, while such a difference does not exceed 1.6 kcal/mole at the DFT level.
Table 3B lists the values of the ALMOEDA energy contributions to ΔE(B97D) and ΔE(ωB97X-D) along with the SIBFA ones. ΔE(ωB97X-D) has 2.1–2.8 kcal/mole larger magnitudes than ΔE(B97D). While both functionals have in each complex very similar values of Epol, they differ significantly regarding E1 and Ect. E1 is app. Five kcal/mole larger with the ωB97X-D than the B97-D functional, while Ect is smaller by three kcal/mole. ΔEtot(SIBFA) is very close to ΔE(ωB97X-D) for the two fluorinated derivatives but larger by 2.2 kcal/mole for the fluoro-chlorobenzene ring of EVG. The comparison should not be carried out further at this stage, since (a) the present SIBFA calculations resort to HF-derived multipoles and polarizabilities, Edisp being a post-HF correction; (b) all DFT-ALMOEDA contributions embody a van der Waals kernel encompassing correlation/dispersion effects, and there is not explicit dispersion contribution.
Consideration of the HF results for both mono- and difluoro rings of RAL and DTG shows unusually large differences between the magnitudes of ΔE(HF) and ΔE(SIBFA) (without the Edisp contribution), as they differ by five kcal/mole out of 19. While the ALMOEDA and SIBFA E1 values are close for both rings, both Epol and Ect(SIBFA) are underestimated by 2–2.5 kcal/mole each. Thus the much closer agreements between ΔEtot(SIBFA) and ΔE(ωB97X-D) found with these two rings (<0.3 kcal/mole out of 39) stem from a compensation of errors, Edisp(SIBFA) having a five kcal/mole larger magnitude than the stabilization energy brought upon passing from the HF to the ωB97X-D level. On the other hand, ΔE(SIBFA) has an only 0.3 kcal/mole out of 18 smaller magnitude than ΔE(HF) for the fluoro-chlorobenzene ring of EVG, but this stems from a four kcal/mole less repulsive value of E1(SIBFA) than E1(HF), compensating for the four kcal/mole undererestimation of Epol + Ect. Such analyses show the continuous need to check for a term-to-term identification of the SIBFA and QC contributions. Improvements along these lines are being carried out presently, but with correlated multipoles and polarizabilities and on the basis of correlated EDA approaches. Each ligand of interest is probed in a diversity of positions by cationic and dipolar probes (El Khoury et al., 2017; Kwapien et al., 2017) and using an automated procedure which, for each individual contribution, optimizes the relevant parameters by least-squares fit minimizing the error with respect to its QC counterpart (M. Devillers, et al., 2019, unpublished data).
We have further considered whether the EVG ring preference over the DTG ring could be possibly biased by their initial “anchorings” by their diketoacid connector preventing the displacement of the HC halobenzene connecting bond. For that purpose, we have permuted the DTG and EVG ring positions in the ternary complexes. That is, the HC connecting bond of the DTG ring was superimposed over that of the EVG ring in the EVG complex and its plane then superimposed over the EVG one. We proceeded in a similar fashion to superimpose the EVG ring over the DTG one in the ternary DTG complex. We have also superimposed the RAL ring over the DTG and EVG ones in their ternary complexes. These three “chimeric” complexes were then energy-minimized with the B97-D3 functional, the HN9, HN1, and HC connecting bonds remaining unrelaxed. The results are reported in Table 4. On the one hand, in each binding site, RAL has a consistently lesser binding affinity than EVG and DTG. On the other hand, in the EVG binding site, the DTG-EVG difference is reduced to 0.2 kcal/mole. Conversely, it increased to 2.5 kcal/mole in the DTG binding site.
| RAL site | EVG site | DTG site | |
|---|---|---|---|
| ΔE(B97D3)-RAL | −37.2 | −38.3 | −37.7 |
| ΔE(B97D3)-EVG | −39.6 | −40.5 | |
| ΔE(B97D3)-DTG | −39.4 | −38.0 |
This indicates that regardless of the additional interactions involving the diketoacid anchor and DNA, a modulation of the ring affinities can depend on the location of the anchor in the DNA grooves. Thus ΔE(QC) for EVG binding appears more favored in the DTG site (−40.5 kcal/mole) than in its own site (−39.6 kcal/mole). Everything else being equal, this could imply that connecting the diketoacid ring of DTG to the fluoro-chlorobenzene ring of EVG rather than difluorobenzene could improve the drug binding affinity, albeit by a modest amount (one kcal/mole out of 40). A similar inversion occurs in the case of DTG, with ΔE(QC) values of −39.4 in the EVG site and −38.0 in its own site, so that the difluorobenzene ring could better contribute to the biding affinity if grafted to the diketoacid of EVG rather than that of DTG.
Discussion
ΔEtot(SIBFA) had values and trends consistent with the ΔE(QC) ones. However, in the case of the fluoro-chorinated ring of EVG, ΔEtot(SIBFA) presently overestimates ΔE(QC) by amounts in the two out 40 kcal/mole range. SIBFA is being presently refined using correlated rather than HF-derived multipoles, and its calibration done on the basis of correlated energy decomposition analyses. These should further narrow down the errors with respect to ΔE(QC) with the appropriate functional, in terms of both the total energy and its individual contributions.
Conclusions and Perspectives
This study focused on three INSTIs successively used in anti-HIV-1 therapy, RAL, EVG, and DTG. We carried out measurements of their free energies of binding, ΔG, to the vDNA end in solution, and unraveled their enthalpy and entropy components in solution. We found that the ΔG ranking DTG > EVG > RAL parallels that inferred for the intasome (Hightower et al., 2011). The ΔG ranking is also paralleled by the ΔH one, a signature for intercalation-driven binding (Chaires, 2006).
We have considered three model ternary complexes, involving G4, C16 and the halobenzene ring of DTG, EVG, and RAL. These were extracted from the corresponding X-ray crystal structures and energy-minimized them by DFT/B97-D3 computations, freezing the HN9, HN1, and HC bonds connecting G4, C16, and the ring to their DNA and diketoacid anchors. The mono-fluorobenzene ring of RAL had the least favorable ΔE(QC) values, whatever the DFT functional. However, the chloro-fluorobenzene ring of EVG had more favorable ΔE(QC) values than the difluorobenzene of DTG, the energy difference being in the range 0.1–1.6 kcal/mole out of 40. Such a preference could plainly indicate that fluoro-chlorobenzene interacts more favorably with the G4/C16 base pairs than difluorobenzene, on account of its larger sigma-hole as well as more favorable van der Waals dispersion effects. But this leaves open the contribution of the dikeoacid connector in the relative affinities. On the one hand, it contributes interactions of its own with the backbone. But it also anchors the halobenzene ring with respect to the G4/C16 base-pair. Along these lines, it was found that grafting the fluoro-chlorobenzene ring of EVG on the diketoacid arm of DTG resulted, in the DTG binding site, into a one kcal/mole ΔE(QC) gain. A similar gain was observed in the case of the difluorobenzene ring of DTG, if transposed into the EVG binding site.
As put forth in El Hage et al. (2015), it is possible to leverage the “Janus-like” properties of the CX bond (X=F, Cl, Br), electron-deficient along the bond and electron-rich in a cone around it, to target respectively and simultaneously electron-rich and electron-deficient sites of the nucleotide bases. Polarizable molecular mechanics is responsive to the electronic changes brought about by substitutions as these impact the magnitude of both QC-derived distributed multipoles and polarizabilities used to compute the EMTP* and Epol contributions. These should enable to fine-tune and further evolve the affinity of halobenzenes for targeted HIV-1 DNA bases.
Along these lines, several novel compounds were recently designed and endowed with significantly more favorable ΔE(QC) and ΔE(SIBFA) values than the difluoro benzene ring of DTG and the fluoro-chlorine ring of EVG. They will be reported in a forthcoming paper (P. El Darazi, et al., 2019, unpublished data).
In addition to handling the halobenzene interactions (El Hage et al., 2013, 2014), a further asset of SIBFA and related polarizable potentials (Bell et al., 2016) is the reliable handling of poly-ligated complexes of divalent cations (El Khoury et al., 2017; Kwapien et al., 2017) and interactions involving “discrete” structural waters (De Courcy et al., 2010; Gresh et al., 2011). Such structural motives are also encountered in the intasome-INSTI complexes.
Grounded on these validations, we plan to undertake long-duration polarizable molecular dynamics simulations on a diversity of INSTI complexes with intasome, resorting to the massively parallel computer code Tinker-HP. These will enable to quantify the extent to which the halobenzene-G4/C16 interactions are modulated by the conformational flexibilities of each partner within the complex, by the electrostatic potentials and fields exerted by the neighboring INT residues and vDNA bases, and possibly as well by the two neighboring divalent Mg(II) cations and by the structural waters.