Comprehensive study of autoignition characteristics of propane
- Published
- Accepted
- Received
- Academic Editor
- James Jennings
- Subject Areas
- Theoretical and Computational Chemistry, Physical Organic Chemistry, Kinetics and Reactions
- Keywords
- Propane, Combustion, Ignition delay, Chemical kinetics, Oxidation
- Copyright
- © 2023 Farhan
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ Physical Chemistry) and either DOI or URL of the article must be cited.
- Cite this article
- 2023. Comprehensive study of autoignition characteristics of propane. PeerJ Physical Chemistry 5:e29 https://doi.org/10.7717/peerj-pchem.29
Abstract
Ignition delay times (IDT) for stoichiometric propane (C3H8) diluted with nitrogen were measured in a shock tube facility under reflected shock wave conditions at pressures ranging from 1 to 10 atm and temperatures between 850 and 1500 K. The experiments were limited to a maximum pressure of 10 atm due to the facility’s constraints. In addition, numerical simulations were conducted using several detailed kinetic mechanisms at pressures from 1 to 30 atm and three equivalence ratios (φ = 0.5, 1, and 2) to provide comparative insights. The results indicated that IDT decreases as pressure increases, with a more significant reduction observed between 1 and 10 atm compared to 10 to 30 atm. While most models exhibited similar trends and minimal discrepancies, the GRI Mech 3.0 mechanism demonstrated a slower prediction of ignition delay times at temperatures below 1250 K. In contrast, the POLIMI model exhibited a relatively faster prediction at temperatures above 1250 K, with the deviation between the two models becoming more pronounced as pressure increased. A comparative analysis revealed that the experimental predictions of propane autoignition behavior were in good agreement with the results obtained using the ARAMCO 3.0 mechanism. To further understand the chemistry governing the autoignition process of C3H8, a sensitivity analysis was performed for a stoichiometric mixture at three distinct temperatures (850 K, 1200 K, and 1550 K).
Introduction
Fossil fuels play a crucial role in energy and transportation systems, contributing significantly to the technological development and economic growth of a country (Ritchie & Roser, 2019). Consequently, it is essential to enhance combustion efficiency and reduce the concentration of environmentally harmful substances in combustion products. This concern has motivated the combustion community to extensively examine the physicochemical processes involved in the ignition and combustion of various fuel-air mixtures, ultimately improving system performance and environmental quality. Ignition delay time (IDT) is a critical factor influencing the overall performance of an engine (Maroteaux, Vaglieco & Mancaruso, 2018). Longer IDTs require more fuel for ignition, while shorter IDTs necessitate less. Engine knocking occurs due to very short IDTs, causing autoignition before the flame front reaches the location of autoignition, potentially resulting in engine failure. IDT also helps to better understand the kinetic behavior of the oxidation process and provides meaningful contributions to the kinetics mechanism of the involved mixture (Petrukhin, Grishin & Sergeev, 2016; Shao, Davidson & Hanson, 2018). Since the 1960s, the ignition delay parameter has been extensively studied, with significant research conducted in this area, particularly over the last two decades, due to increasing global demand for fossil fuels and the subsequent environmental degradation.
In the homological series, propane (C3H8) holds a pivotal position as a saturated hydrocarbon, displaying thermochemical and combustion characteristics of larger hydrocarbons more accurately than other small hydrocarbons (Dagaut et al., 1987). Due to its peculiar behavior, propane is a valuable fuel in research aimed at characterizing the oxidation kinetics of light to heavy hydrocarbons. Additionally, propane is considered a highly efficient and clean-burning fuel source due to its lower carbon content (El-Mahallawy & Habik, 2002). Greenhouse gas emissions from propane and methane fuel are lower than those from conventional fuels, extending engine life (Smith, 2017). Consequently, combustion modeling of both fuels is a critical research area.
Substantial research has been conducted on methane gas in various proportions due to its widespread commercial applications. However, investigation into propane oxidation and combustion commenced in the 1970s, stemming from its distinctive behavior and potential use as an alternative fuel on a commercial scale. Numerous researchers have thoroughly examined propane oxidation, chemical kinetics, and ignition characteristics, developing reaction kinetic mechanisms and experimental studies (Lin & Chiu, 2017). Cathonnet (1994) provided an overview of the development of chemical kinetics of hydrocarbons over the past 25 years and predictions for the next 25 years. During this period, the primary focus was on high-temperature combustion, with most mechanisms containing fewer than 100 species in their kinetic models. In the late 1990s, research emphasis shifted towards high-speed and low-temperature combustion (Jachimowski, 1984; Westbrook & Pitz, 1984; Dagaut, Cathonnet & Boettner, 1992; Leung & Lindstedt, 1995).
Although Pease & Munro (1934) conducted the earliest work on propane, studying its negative temperature coefficient (NTC) behavior as a unique property (Pease, 1938), earnest research on propane combustion for its potential application as a substitute fuel began in the 1980s (Dagaut et al., 1987; Jachimowski, 1984; Westbrook & Pitz, 1984; Dagaut, Cathonnet & Boettner, 1992; Burcat et al., 1971; Mclain & Jachimowski, 1977; Westbrook, Pitz & Urtiew, 1983; Sloane, 1992). Advances in theory and kinetics illuminated the critical role of reaction classes in ignition phenomena. Liu et al. (2020) investigated the explosion phenomenon of hydrogen, methane, ethane, and propane, analyzing numerical and experimental data to comprehend reaction pathways and ignition limits under varying temperature and pressure conditions. Ignition delay time (IDT) has been extensively explored to understand the kinetic behavior of the oxidation process and provide significant insights for modifying the combustion kinetics of the mixing mechanism (Petrukhin, Grishin & Sergeev, 2016; Lakshminarayanan & Aghav, 2010).
Numerous researchers have conducted in-depth investigations into propane oxidation using a diverse array of experimental facilities, including rapid compression machines (RCM) (Gallagher et al., 2008; Samimi-Abianeh et al., 2019; Burnett & Wooldridge, 2021), tubular flow reactors (TFR) (Hoffman et al., 1991; Beerer & McDonell, 2011; Sabia et al., 2014), jet-stirred reactors (JSR) (Dagaut et al., 1987), and shock tubes (ST) (Lamoureux, Paillard & Vaslier, 2002; Lam et al., 2011; Agafonov & Tereza, 2015; Reynier, 2016). These studies have primarily focused on examining sensitive and essential species and their reactions, which serve as the driving force behind the combustion process under specific conditions. Nevertheless, numerous questions remain unanswered. In particular, understanding the kinetic chemistry of hydrocarbon fuels continues to be a key objective for refining kinetic models, as current models lack the accuracy required to describe the critical phenomena involved in the ignition process (Miller et al., 2021; Lei et al., 2022). The kinetics and properties of pure propane are characterized using well-established mechanisms, such as ARAMCO, GRI Mech, and San Diego Mech. Augmented experimental research in this area is currently in progress (Burnett et al., 2022; Burnett & Wooldridge, 2021; Molana, Piehl & Samimi-Abianeh, 2020; Ramalingam, Fenard & Heufer, 2020). The aim of this work is to offer a comparative analysis of propane’s autoignition characteristics by employing a range of currently available mechanisms under various pressure and mixture conditions to comprehensively understand propane’s combustion behavior.
Chemical Kinetics and Autoignition of Propane
Chemical kinetics
Yeong & Su (2001) provided an in-depth analysis of propane’s chemical kinetics in high-temperature regimes, utilizing sensitivity analysis to quantify the critical reactions. Upon evaluating the literature, the crucial reactions involved in propane combustion have been compiled in Table 1. These reactions contribute significantly to enhancing the propane ignition process.
The decomposition of C3H8 into C2H5 and CH3 serves as a potent initiation reaction in high-temperature regimes due to the weak C-C bond. Following this reaction, the H-abstraction process generates propyl radicals (C3H7). The literature indicates that Reaction 5 (R5) is a critical chain-branching reaction governing the overall oxidation process and ignition delay of fuels. However, the ignition delay time increases with the rising concentration of C3H8. Reactions 3 (R3) and 4 (R4) decelerate the overall ignition rate since C3H8 can compete with O2 in reactions involving H atoms. This phenomenon has also been observed in the oxidation mechanisms of methane and ethane. O and OH chain carriers produced through R5 react with the fuel C3H8, creating propyl radicals. Notably, typical propyl radicals further decompose into methyl and ethylene radicals. Conversely, isopropyl radicals generate propene, which subsequently reacts with H and OH to form allyl radicals (C3H5 − A). A key aspect of this process is that propyl radicals disintegrate following the β-scission rule, signifying that the breaking bond will be removed from the radical site.
Understanding propane oxidation at low temperatures is essential for comprehending the negative temperature coefficient (NTC) phenomena. Numerous studies have been conducted on propane ignition, with ongoing efforts to present more comprehensive research. The latest study in this regard is by Burnett (2022), while other important studies include (Tereza et al., 2023; Yu, Liu & Ma, 2020; Bai et al., 2019; Merchant et al., 2015; Titova, Kuleshov & Starik, 2011; Cord et al., 2012; de Vries et al., 2009). According to Merchant et al. (2015), Reactions 6 (R6) and 7 (R7) serve as two initiation reactions in low-temperature propane oxidation. The resulting normal propyl radicals further oxidize to peroxy radicals, which undergo internal isomerization to form QOOH radicals and are subsequently oxidized to the O2O2QOOH radical. Following this, a decomposition chain commences via the H-abstraction class, producing three reactive OH radicals. In contrast, isopropyl radicals undergo H-abstraction similar to high-temperature regimes. Titova, Kuleshov & Starik (2011) also presented a detailed mechanism of propane and elucidated the oxidation process at temperatures below 1,000 K.
| Reaction No. | Reactions |
|---|---|
| R1 | C3H8 (+M) →C2H5 + CH3 (+M) |
| R2 | C2H5 (+M) →C2H4 + H (+M) |
| R3 | C3H8 + H →NC3H7 + H2 |
| R4 | C3H8 + H →IC3H7 + H2 |
| R5 | H + O2→ O + OH |
| R6 | C3H8 + OH →NC3H7 + H2O |
| R7 | C3H8 + OH →IC3H7 + H2O |
| R8 | C3H8 + O →C3H7 + OH |
| R9 | C3H8 + OH →C3H7 + H2O |
| R10 | NC3H7→CH3 + C2H4 |
| R11 | IC3H7→ H + C3H6 |
| R12 | C3H6 + H →C3H5-A + H2 |
| R13 | C3H6 + OH →C3H5-A + H2O |
Ignition delay time
Understanding the kinetic behavior of the oxidation process and providing practical suggestions for modifications in the combustion kinetics of the reaction mechanism necessitates the consideration of ignition delay time (IDT), a critical physicochemical characteristic of the combustible fuel-air mixture employed in combustion design (Petrukhin, Grishin & Sergeev, 2016; Shao, Davidson & Hanson, 2018). Furthermore, combustion timing significantly influences various aspects of engine performance, including power output, combustion efficiency, emissions, and peak cylinder pressure (Maroteaux, Vaglieco & Mancaruso, 2018). As previously discussed, considerable progress has been made over the past two decades in examining the ignition and combustion kinetics of saturated and unsaturated hydrocarbons (Titova, Kuleshov & Starik, 2011). A longer IDT requires more fuel during ignition, while a shorter IDT demands less fuel. An extended IDT enhances fuel vaporization and augments the chemical ignition degree due to a more uniform fuel-air mixture. However, this can result in explosive combustion, which causes knocking phenomena and may lead to combustion chamber failure due to turbulent operation. Conversely, a shorter IDT smoothens engine starting and softens diesel engine running. The optimal ignition delay for gas turbines, diesel, and gasoline engines ranges between 2 and 6 ms, depending on the engine type (Petrukhin, Grishin & Sergeev, 2016; Lakshminarayanan & Aghav, 2010).
Cadman, Thomas & Butler (2000) investigated the IDT behavior of lean propane-air mixtures in a shock tube at low temperatures (835–1400 K) and high pressures (5-39 atm). Zhukov, Sechenov & Starikovskii (2005) also measured the IDT of lean propane-air mixtures using a shock tube over a temperature range of 800–1500 K and a pressure range of 2–500 atm. Based on their findings, they revised the detailed kinetic mechanism to accommodate their experimental observations. Similarly, Herzler, Jerig & Roth (2004) and Petersen et al. (2009) performed experimental analyses on shock tubes at temperatures of 750–1300 K and pressures of 10-30 atm. Their results concurred with Cadman, Thomas & Butler (2000)’s measurements. It was observed that experimental IDT at lower temperatures (i.e., <1000 K) began to deviate significantly from numerical simulations using detailed kinetic models and became nearly temperature-independent (Agafonov & Tereza, 2015). Kochar et al. (2011) noted that IDT behavior could not be experimentally reproduced with similar modeling of IDT using a detailed propane oxidation mechanism due to entropy losses. The negative temperature coefficient (NTC) behavior was observed experimentally at low temperatures. Consequently, interest in propane ignition at high pressures and low temperatures has grown in the past decade, owing to the importance of confirming accurate chemical kinetic processes under such conditions (Lam et al., 2011). Studies in shock tubes at higher temperatures and pressures have demonstrated variations in the temperature dependence of autoignition time for propane/oxygen mixtures transitioning from intermediate to high temperatures (Sabia et al., 2014).
Materials and Methods
All experiments in this study were conducted at Xi’an Jiaotong University using a preheated stainless steel shock tube. The shock tube comprises a 6.0 m driver section with a 75 mm (approximately 2.95 in.) internal diameter and a 7.6 m driven section with a 150 mm (approximately 5.91 in.) internal diameter. A double diaphragm segment separates both sections. The inner surface was polished to Ra = 0.4 µm to minimize shock wave bifurcation caused by wall friction and to mitigate fuel corrosion. A schematic of the shock tube is depicted in Fig. 1, and further details of the facility can be found in Sun et al. (2020)’s paper.
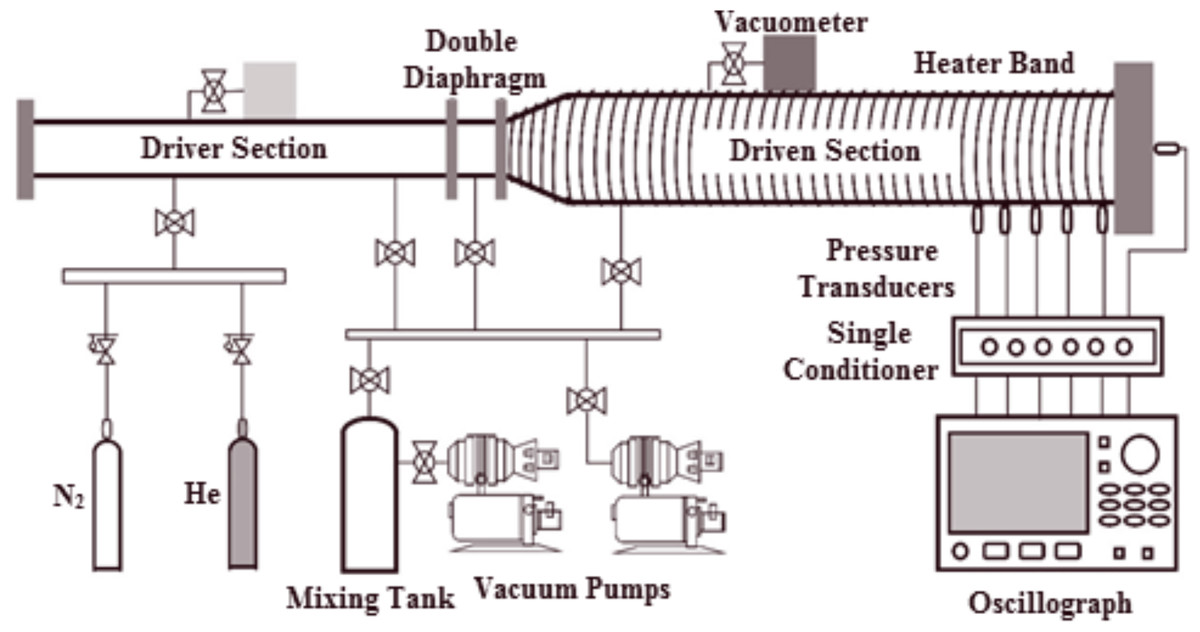
Figure 1: Detailed diagram of stainless steel shock tube apparatus.
{kind=link}
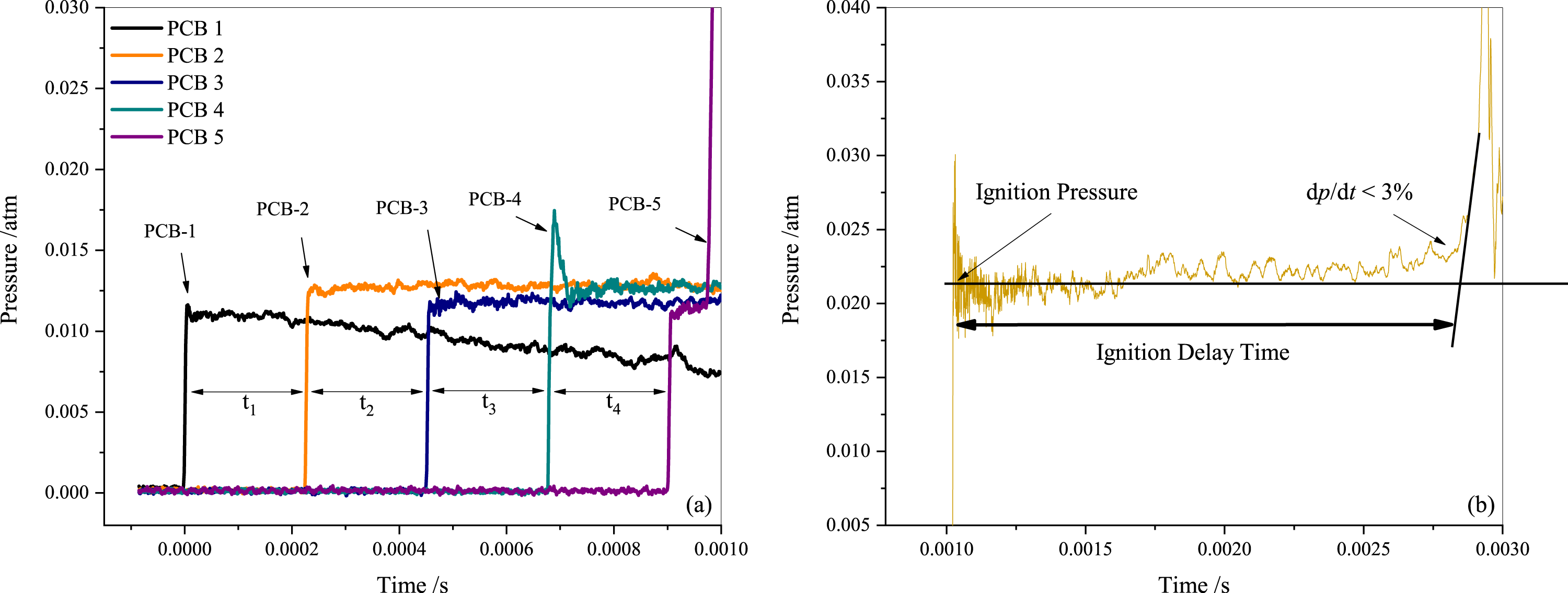
Tailored conditions were employed to achieve a relatively longer test time for lower temperature tests and to attain different pressures. Dalton’s partial pressure law was utilized to generate the test mixtures in a 140 L mixing tank, where they were allowed to rest for over 12 h. A combination of a mechanical vacuum pump (D60C, Leybold) and a roots vacuum pump (ZJP-150; Chengdu Rankuum Machinery Ltd., Chengdu, China) were used to evacuate the shock tube to approximately 1.0 atm before each test, and the leak rate was maintained at less than 0.2 atm/min using a vacuometer (MPG400; Inficon, Bad Ragaz, Switzerland). The incident shock velocity was recorded by five fast-responding piezoelectric pressure transducers (PCB 113B22) that were spaced 200 mm apart and installed in the axial direction near the end face of the low-pressure section. Figure 2A displays the recorded pressure traces measured by these five PCBs (piezoelectric pressure transducers). Measurements of incident excitation and shock wave intervals traveling through two transducers were made using four-time interval counters (FLUKE PM6690). The shock velocity was obtained based on the principle of solid-wall reflection of the shock wave. Finally, the reflected shock temperature (T5) was calculated using the one-dimensional shock theory and the chemical equilibrium software package GASEQ (Morley, 2005). A charge output dynamic pressure sensor measured the reflected shock pressure (PCB 113B03) mounted on the end wall of the shock tube with acceleration compensation. The ignition delay time is defined as the time interval between the incident shock wave arriving at the end wall and the maximum slope of the pressure profile being extrapolated to the baseline. A digital oscilloscope (Yokogawa DL850E) was employed to acquire all the pressure traces. The larger internal diameter of the driven section significantly reduced non-ideal effects, such as flow viscosity, shock acceleration, and partial diaphragm rupture. The results of several experiments indicate that the pressure rise ratio (shock attenuation rate, (dp5/dt)) is within 3 %/ms in this study and has been considered during simulations using the SENKIN/VTIM approach (Chaos & Dryer, 2010). According to Deng et al. (2016), the simulated ignition delay time is defined as the time of maximum dT/dt, as it is close to the experimental definition as illustrated in Fig. 2B. Various well-established mechanisms have been employed to more accurately represent the chemical kinetics of propane, including ARAMCO 2.0, ARAMCO 3.0, GRI Mech 3.0, POLIMI Mech, San Diego, and USC Mech 2.0, for numerical predictions.

Figure 2: Pressure sensor measurement results (A) and definition of the ignition delay period (B).
{kind=link}
Results and Discussion
In this study, the ignition delay times (IDTs) of stoichiometric propane-air mixtures behind a reflected shock wave were investigated at various pressure levels. The ARAMCO 3.0 kinetic mechanism provided a reasonable description of the experimental data pertaining to propane ignition as shown in Fig. 3. The experimental IDT results displayed a variation within 25%, which is consistent with studies reported in the literature (Burke et al., 2014; Ramalingam et al., 2021). Lam et al. (2011) emphasized the importance of accounting for pressure and temperature variations before ignition in shock tubes for accurate interpretation of results. The variations in experimental data may be partially attributed to the absence of such treatment. It was observed that IDT decreased with increasing pressure, with a significant reduction from 1 to 10 atm.
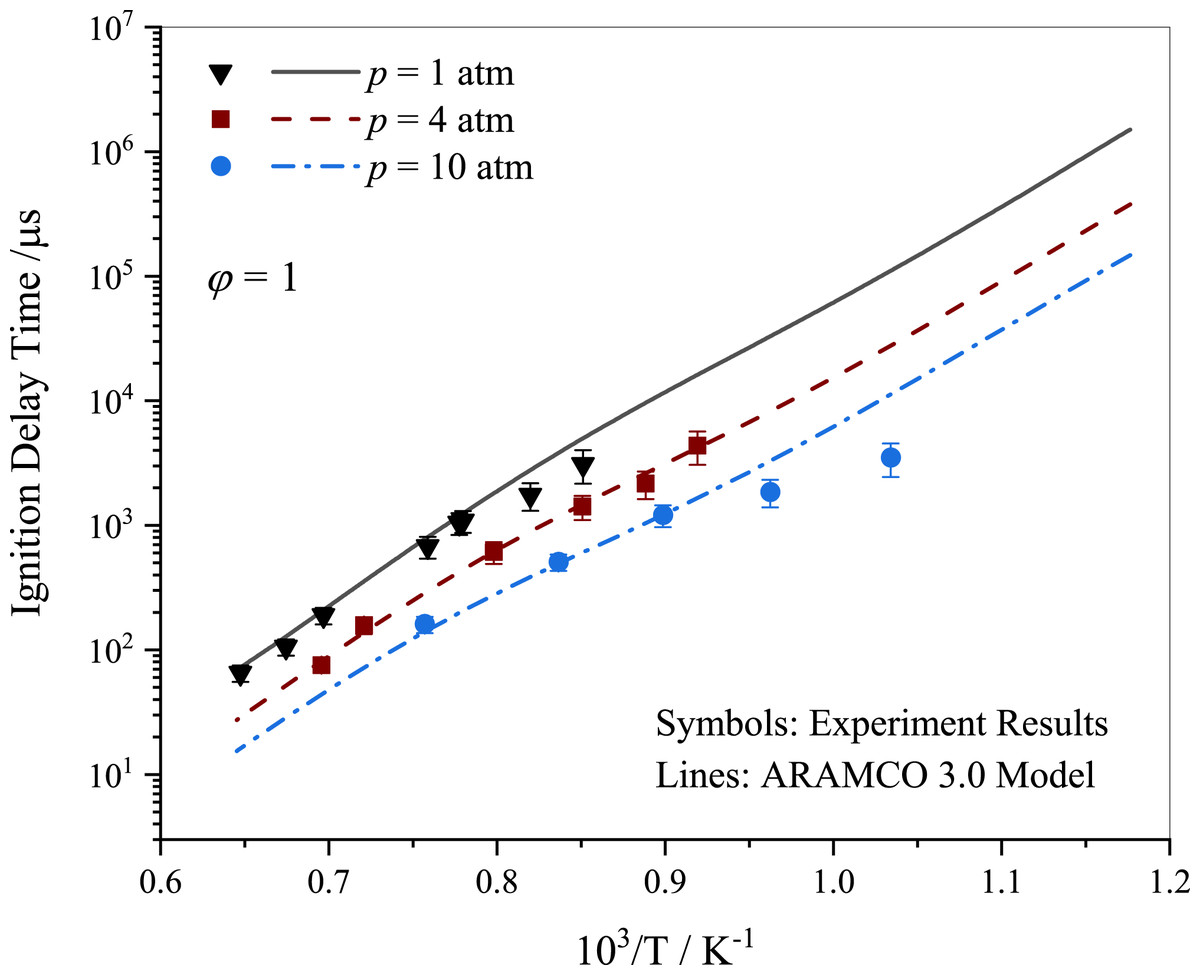
Figure 3: Ignition delay time comparison between the current work measurements and simulations using ARAMCO 3.0 for the stoichiometric propane mixtures.
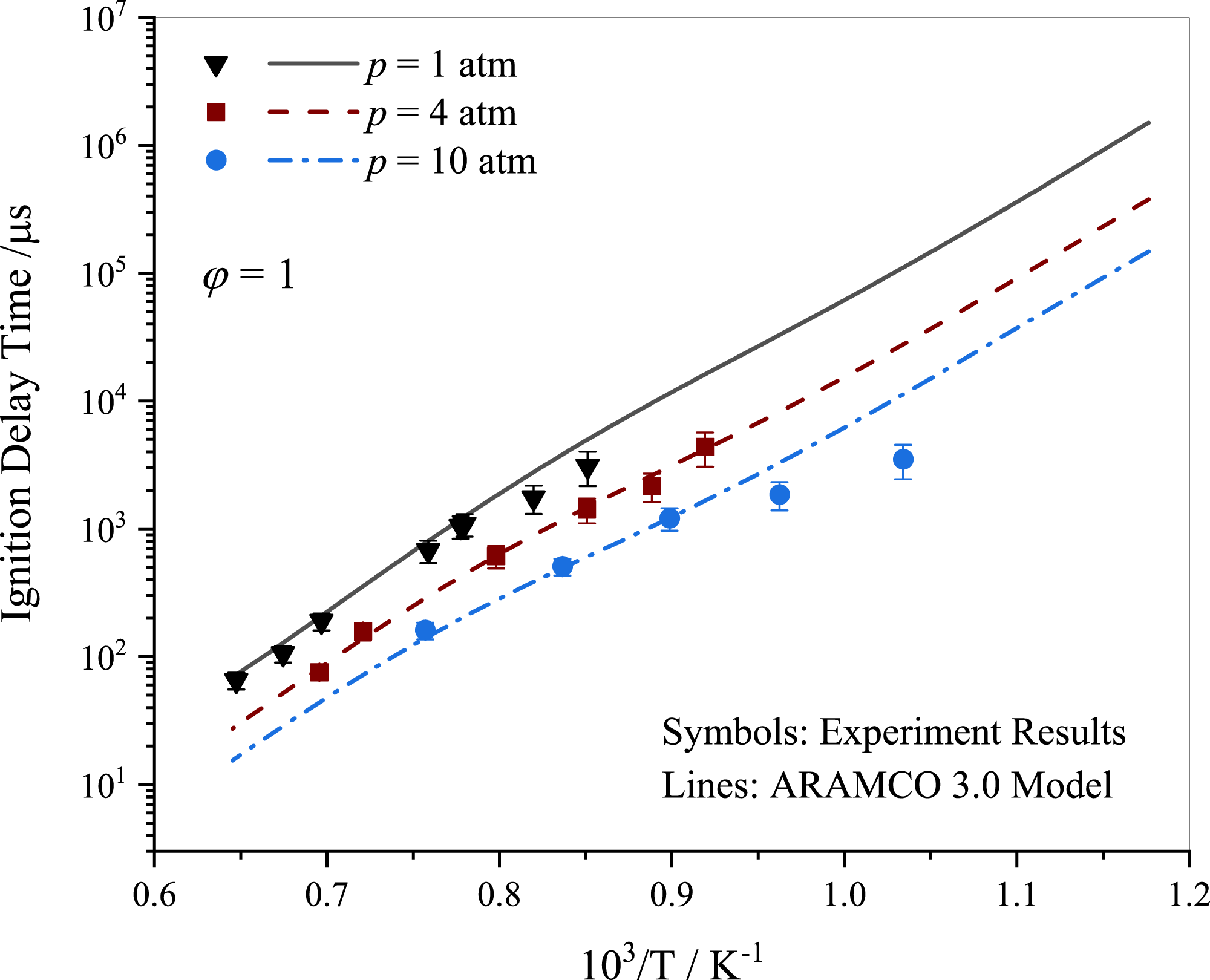{kind=link}
Experiments were conducted up to 10 atm for stoichiometric propane mixtures due to the pressure measurement limitations of the shock tube facility. Numerical simulations were performed for different propane mixture equivalence ratios (ϕ = 0.5 and 2), as shown in Table 2, to understand the variations in IDT.
| Composition | XC3H8 (%) | XO2 (%) | XN2 (%) |
|---|---|---|---|
| ϕ = 0.5 | 2.06 | 20.57 | 77.37 |
| ϕ = 1 | 4.03 | 20.15 | 75.82 |
| ϕ = 2 | 7.75 | 19.37 | 72.88 |
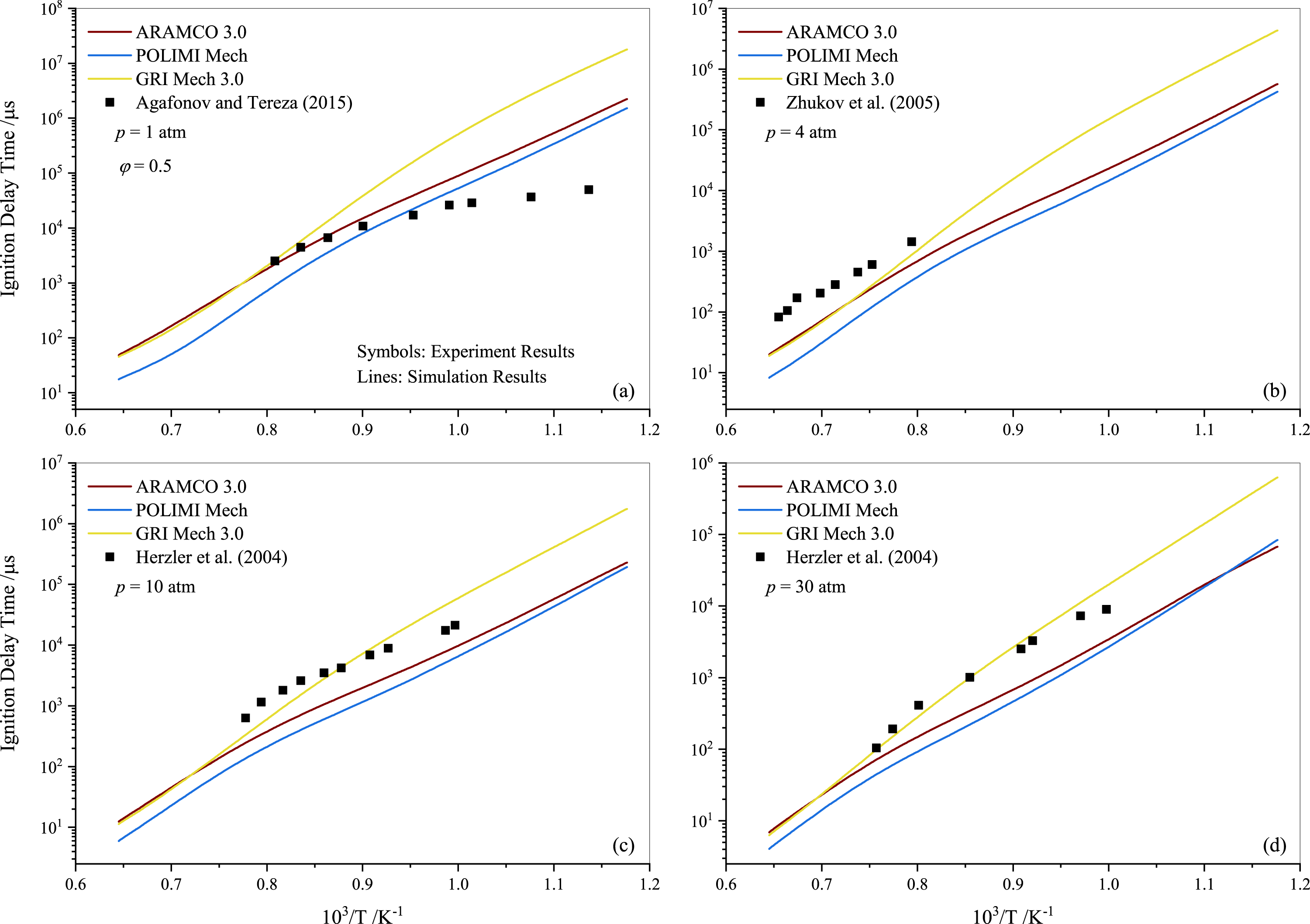
Tereza et al. (2023) argued that the validation of most kinetic models is often described by the ignition delay. In Fig. 4, a comparison between the experimental results for stoichiometric propane mixtures and several well-established mechanisms, including ARAMCO 2.0, ARAMCO 3.0, GRI Mech 3.0, POLIMI Mech, San Diego, and USC Mech 2.0, is presented. The experimental data from Horning, Davidson & Hanson (2002); Brown & Thomas (1999), and Tereza et al. (2023) are used to compare with the results of the present study in Figs. 4A, 4B, and 4D, respectively. It is evident that the experimental data from the current work align more closely with model predictions than the literature data. The most apparent cause for these disparities is the variation in mixture compositions, which significantly influences the results. For instance, Horning, Davidson & Hanson (2002) used argon gas as a diluent instead of nitrogen, which considerably impacts the ignition delay behavior (Würmel et al., 2007). Additionally, Brown & Thomas (1999) utilized both argon and nitrogen as diluents in their experiments and observed minor differences; however, their shock tube facility differed from the one used in this study. The GRI Mech 3.0 mechanism exhibited slower prediction of ignition delay times at temperatures below 1250 K, deviating more significantly with increasing pressures. In contrast, the POLIMI mechanism (Faravelli, Frassoldati & Ranzi, 2003; Frassoldati, Faravelli & Ranzi, 2003) over-predicted the IDT for pure propane mixtures. The underperformance of the GRI model is unsurprising given its age, while research on the POLIMI model and the ARAMCO mechanism is ongoing. Ramalingam, Fenard & Heufer (2020) measured the IDT of propane at high pressures (30 and 50 atm) using a rapid compression machine and compared different mechanisms with their experimental data. They concluded that the ARAMCO 3.0 mechanism qualitatively captured the IDT data better than the remaining mechanisms.
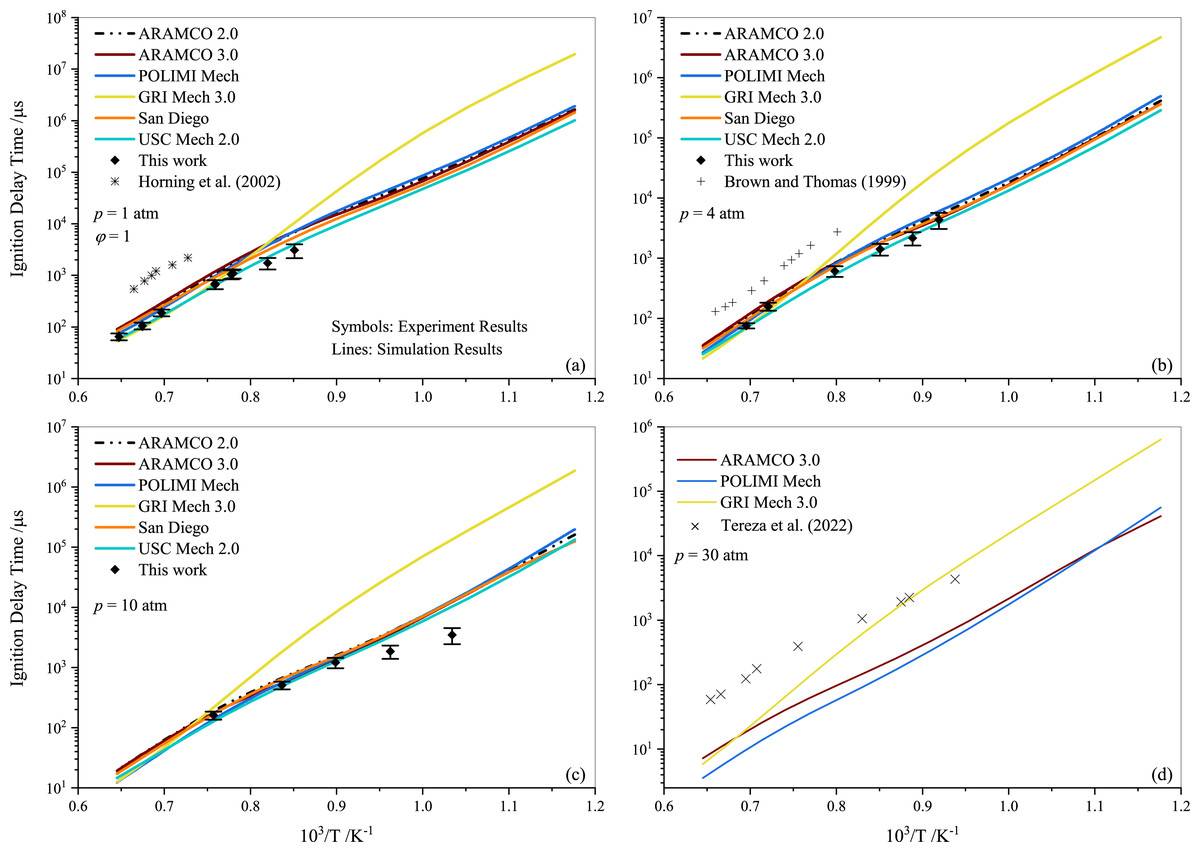
Figure 4: Ignition delay time comparison between the measurements and simulations for the stoichiometric propane-air mixture (ϕ= 1) at pressures of 1.0 atm (A), 4.0 atm (B) 10 atm (C) and 30 atm (D).
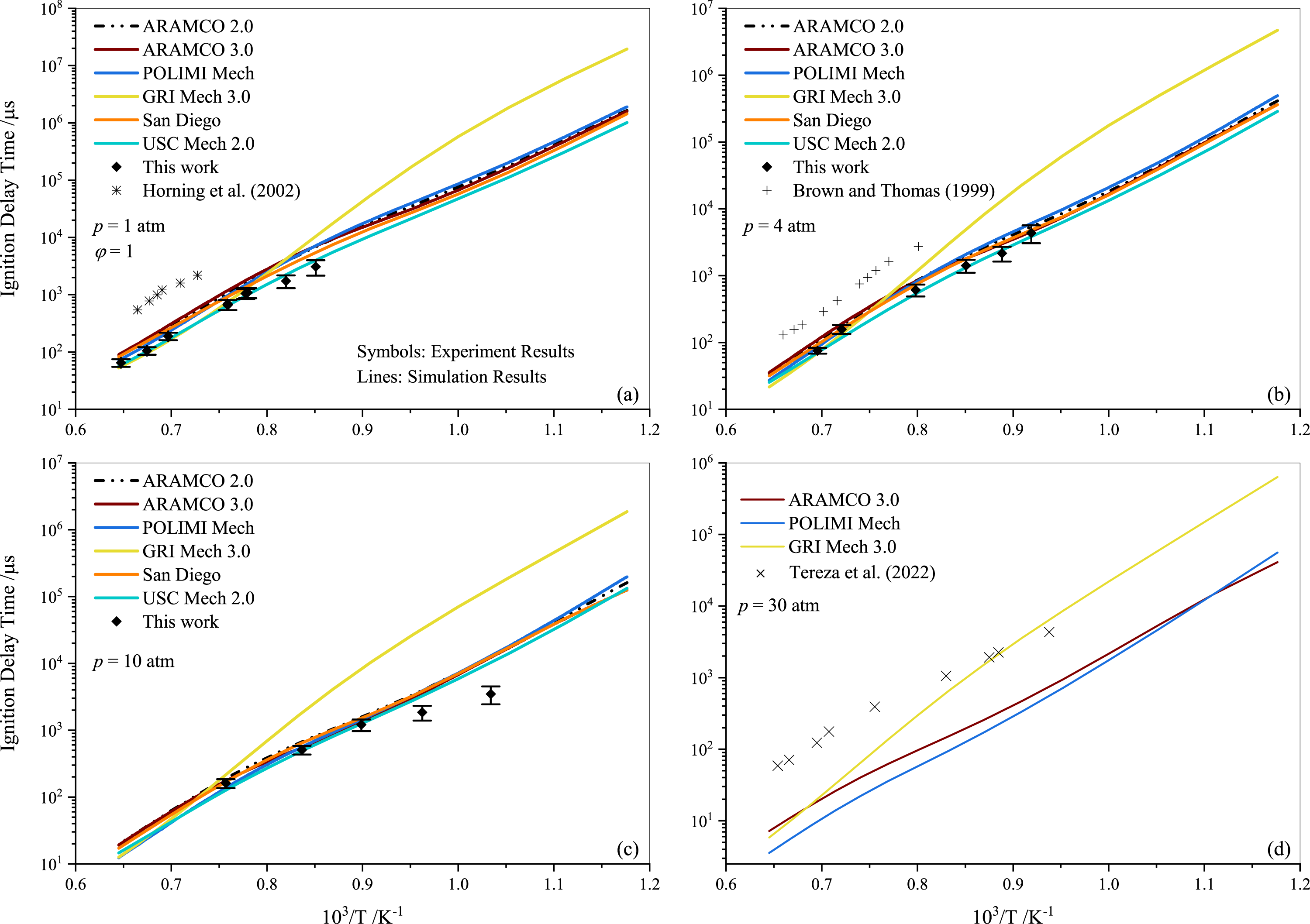{kind=link}
It is worth noting that both ARAMCO 2.0 and ARAMCO 3.0 produced similar results, although ARAMCO 3.0 incorporated the isopropanol sub-model. This suggests that the influence of the isopropanol sub-model on intermediate to high-temperature chemistry may be negligible. At 10 atm, a considerable difference between experimental and numerical results was observed for the lower temperature regime. Burnett & Wooldridge (2021) identified sources of scatter in experimental facilities due to complex ignition behavior during propane ignition under low-temperature combustion conditions. This implies that model refinement and further experiments are required to understand the deviation. Continued research into C1- C3 mixtures’ ignition will enable significant advances in the design and improvement of kinetic mechanisms that can accurately explain ignition delays in propane-air mixtures.
Following the experimental and numerical results for stoichiometric propane auto-ignition, numerical simulations were also conducted for other equivalence ratios using the selected three mechanisms (ARAMCO 3.0, POLIMI, and GRI Mech 3.0), given their widespread applicability. Figures 5 and 6 illustrate that the IDT behavior in fuel-lean and fuel-rich mixtures is somewhat similar to stoichiometric conditions, respectively. The experimental data from Agafonov & Tereza (2015); Zhukov, Sechenov & Starikovskii (2005) and Herzler, Jerig & Roth (2004) were used in Figs. 5A, 5B, and 5C & 5D, respectively. The comparison indicates that the experimental literature data exhibit a non-linear discrepancy with all model predictions. Furthermore, Fig. 7 presents the effect of the equivalence ratio on the IDT of pure propane. It was observed that IDT decreased with increasing propane concentration over the air (i.e., increasing equivalence ratio), but the difference was marginal in terms of IDT improvement. This comparison of equivalence ratios serves as a reference, considering that the equivalence ratio plays a critical role in the overall combustion process and engine performance.
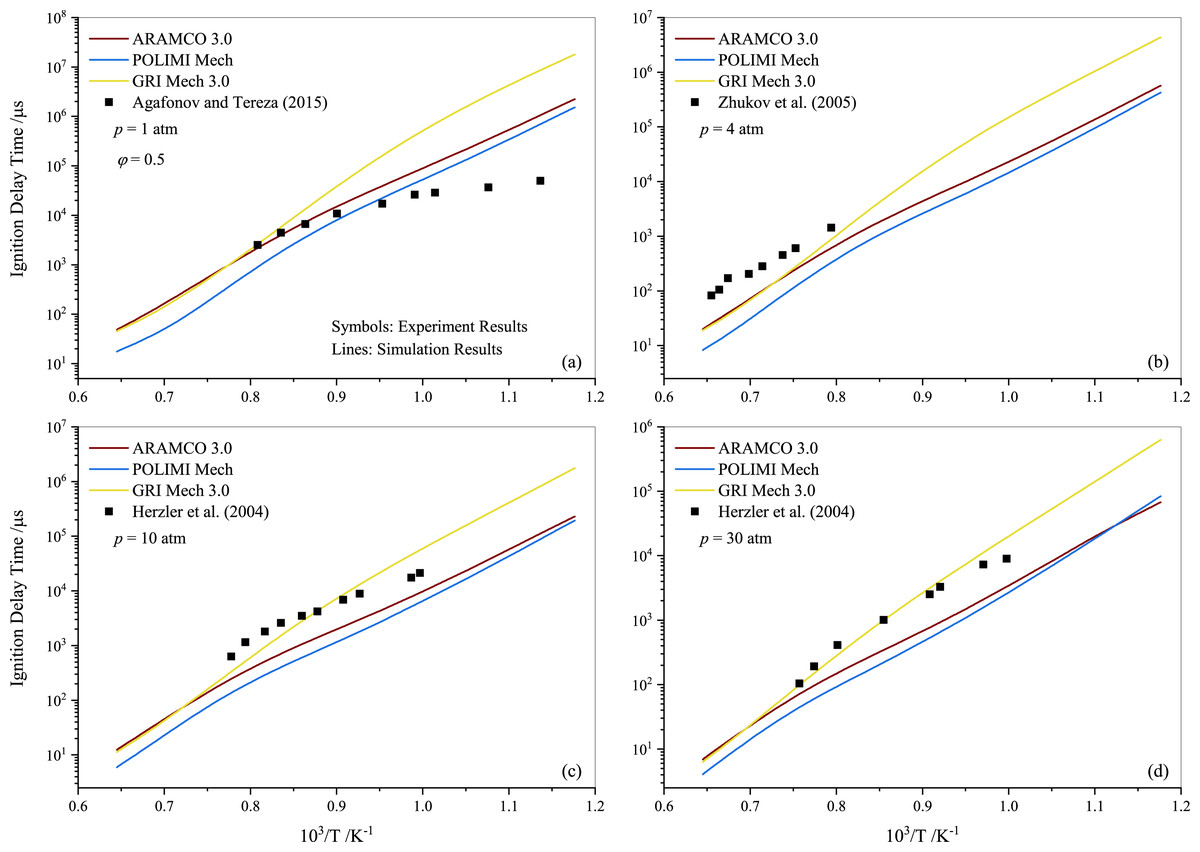
Figure 5: Numerically computed ignition delay time of lean-propane-air mixture ( ϕ= 0.5) at pressures of 1.0 atm (A), 4.0 atm (B) 10 atm (C) and 30 atm (D).
{kind=link}
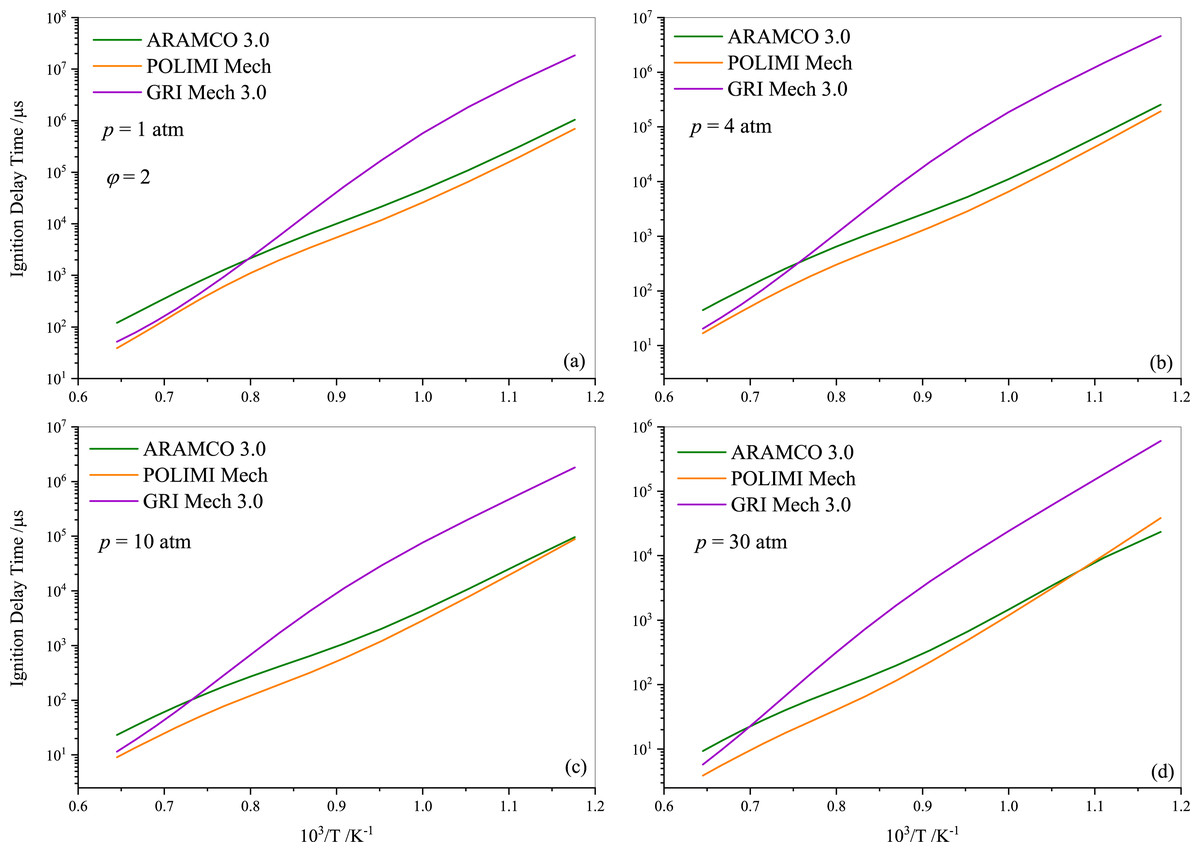
Figure 6: Numerically computed ignition delay time of rich-propane-air mixture ( ϕ= 2) at pressures of 1.0 atm (A), 4.0 atm (B) 10 atm (C) and 30 atm (D).
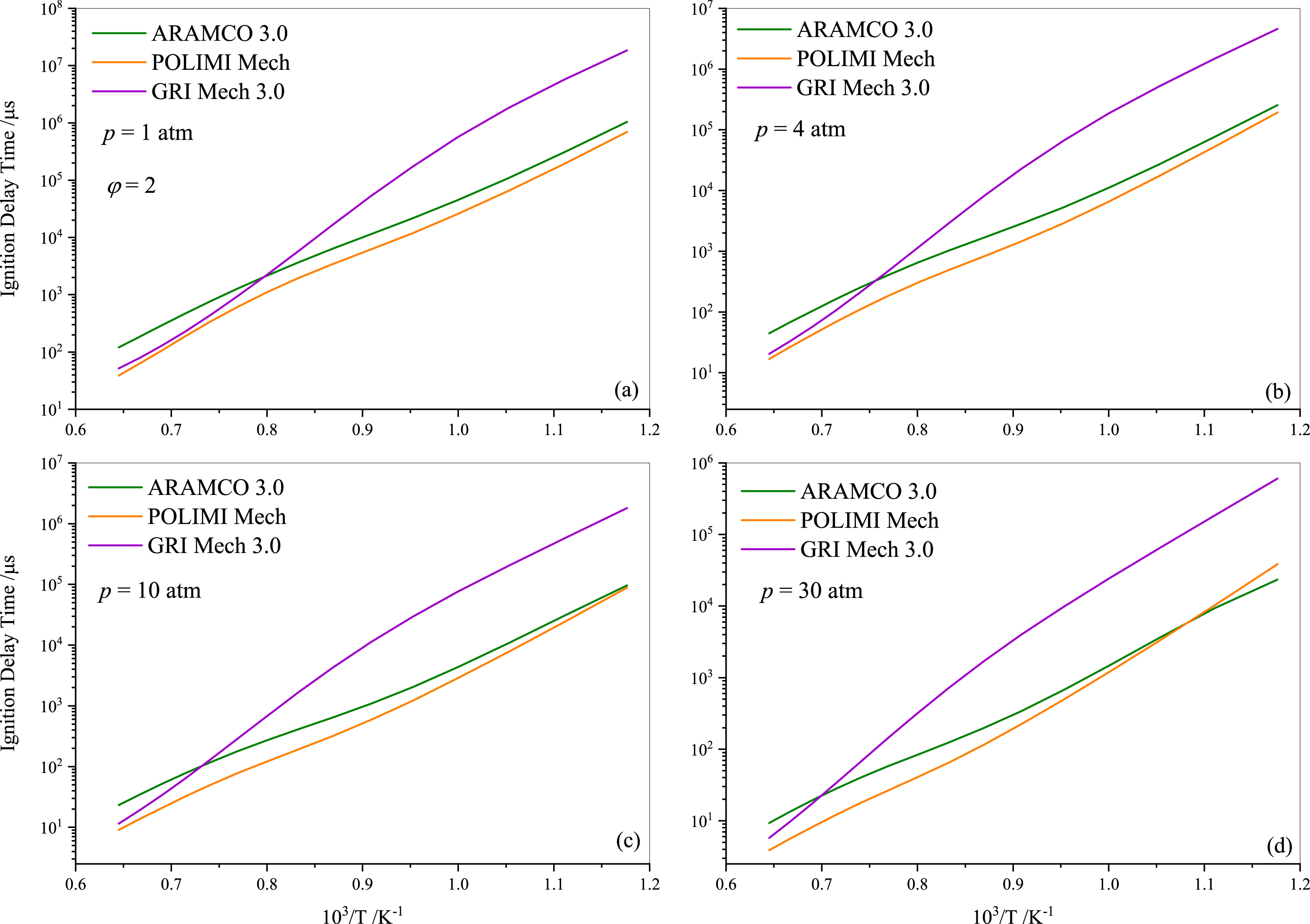{kind=link}

Figure 7: Ignition delay time comparison between the current work measurements and simulations using ARAMCO 3.0 for different propane-air mixtures at pressures of 1.0 atm (A), 4.0 atm (B) 10 atm (C) and 30 atm (D).
{kind=link}
In conclusion, this study offers valuable insights into the ignition delay times of propane-air mixtures under various pressure levels and equivalence ratios. The ARAMCO 3.0 kinetic mechanism demonstrated its ability to provide a reasonable description of the experimental data. The discrepancies between the experimental results and model predictions highlight the need for further research to refine the models and improve the understanding of complex ignition behavior in propane combustion. By examining the effects of equivalence ratios, this study contributes to a better understanding of the role these ratios play in the combustion process and engine performance, which can ultimately lead to the development of more efficient and cleaner engine technologies.
Sensitivity analysis
Sensitivity reaction pathway analysis is an invaluable method for comprehending the underlying chemical mechanisms and rate-limiting reactions implicated in the oxidation of a fuel, such as propane, under diverse conditions. In this study, a sensitivity analysis using Cantera (Goodwin David, Moffat Harry & Speth Raymond, 2018) was conducted to explore the kinetic chemistry governing propane oxidation in ARAMCO 3.0 at 1 and 4 atm, with temperatures ranging from 850 to 1550 K.
At lower temperatures, propane oxidation is dominated by low-temperature chemistry, which involves intricate reaction pathways that lead to the formation of alkyl radicals and oxygenated intermediates, such as peroxy radicals, aldehydes, and ketones. The branching reactions and isomerization processes of these intermediates play a crucial role in the system’s overall reactivity. The results indicate that at T = 850 K (Fig. 8), propane dehydrogenates to form isopropyl and normal-propyl by reacting with OH and HO2 radicals. The ensuing C3 radicals are predominantly dehydrogenated and react with other HO2 radicals. Sabia et al. (2014) suggested that methyl radicals generate OH, H, and HO2 radicals at intermediate temperatures. The branching reactions remain connected to the formation and degradation of H2O2. However, the production of HO2 radicals is generally limited to lower temperature regime. Key reactions to consider in this temperature regime include:
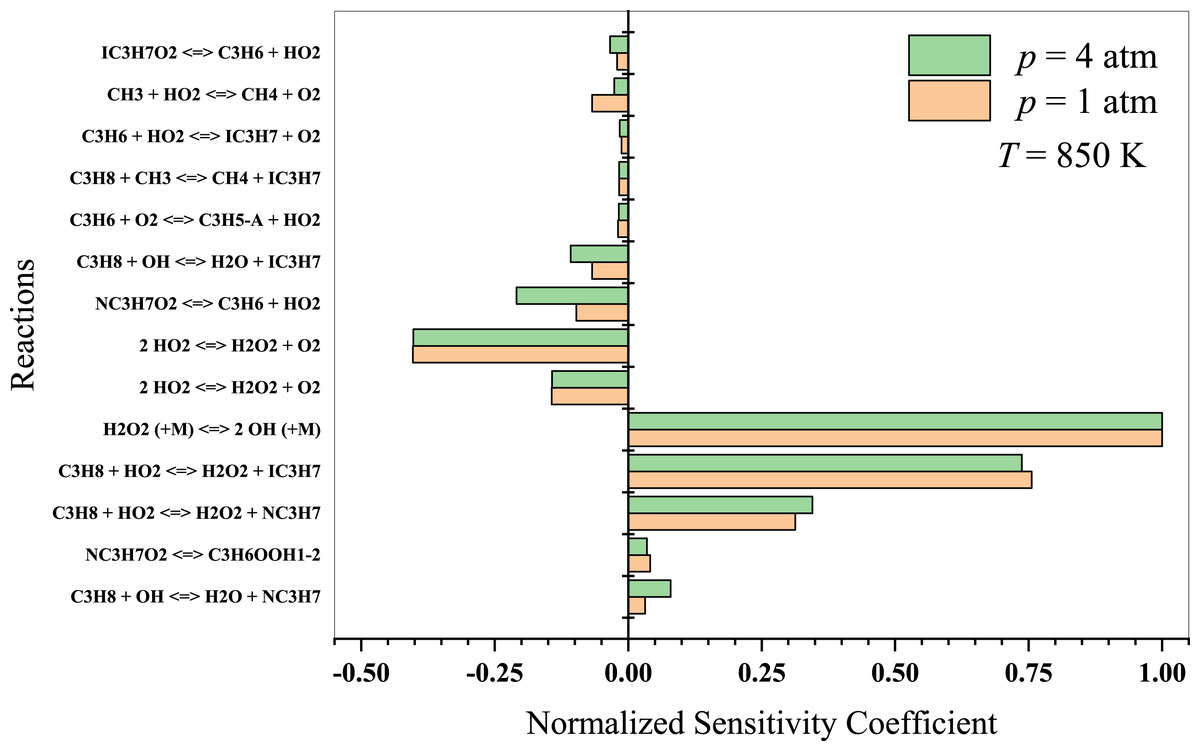
Figure 8: Sensitivity analysis of stoichiometric propane-air mixture at Temperature - 850 K.
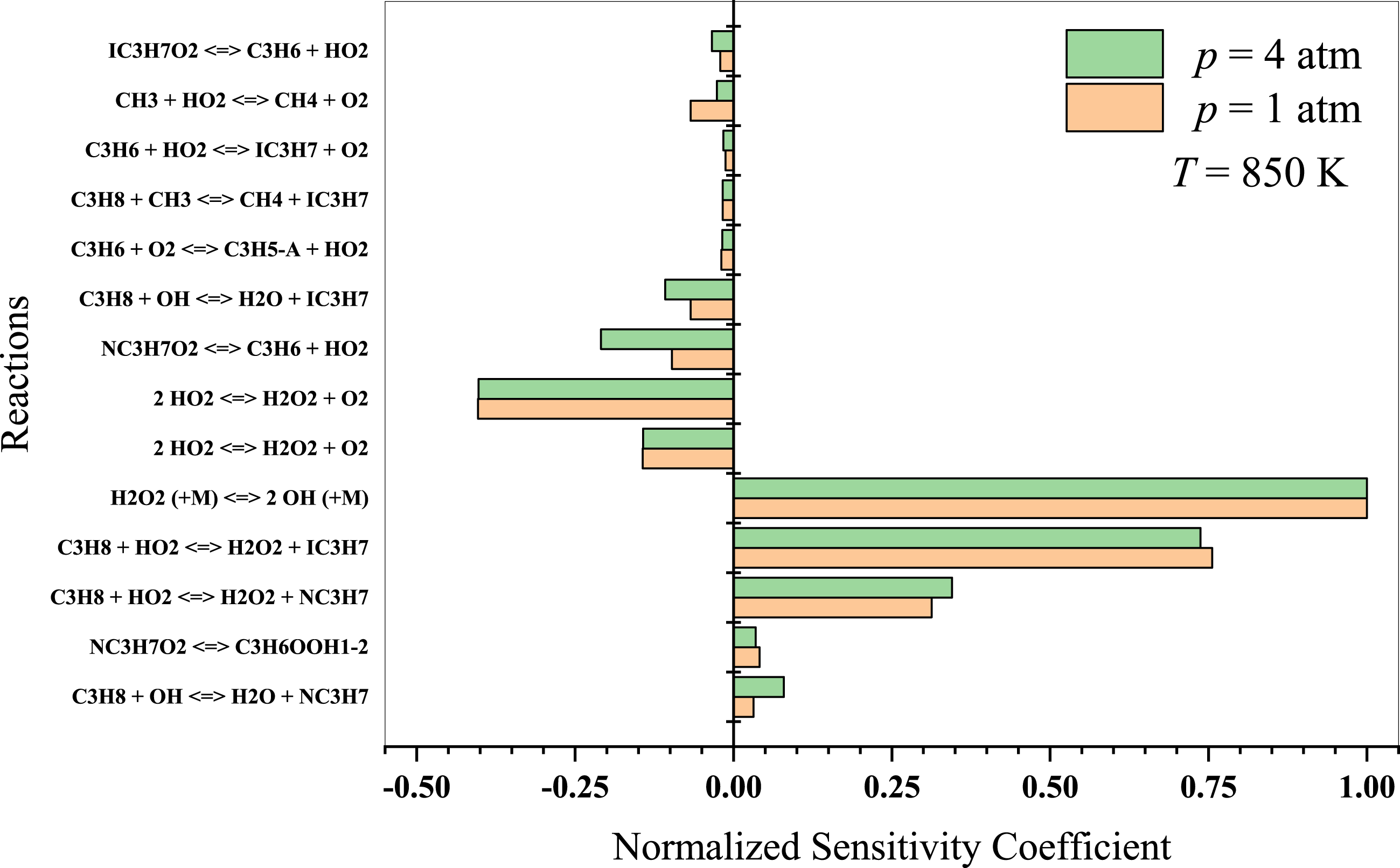{kind=link}
The initial propane H-abstraction by molecular oxygen (O2) or hydroxyl radical (OH) to form alkyl radicals and the subsequent formation of peroxy radicals.
Isomerization and branching reactions involving peroxy radicals, which can lead to chain-branching and the formation of cyclic ethers or hydroperoxides.
The decomposition of cyclic ethers and hydroperoxides to form smaller oxygenated species and radicals, which continue to propagate the oxidation process.
At intermediate temperatures, the chemistry transitions from low-temperature to high-temperature oxidation, and the significance of low-temperature pathways decreases. In Fig. 9, it is evident that the reaction CH3 + HO2 → CH4 + O2 inhibits ignition, while CH3 + HO2 → CH3O + OH promotes combustion. In this regime, the following reactions are more significant:
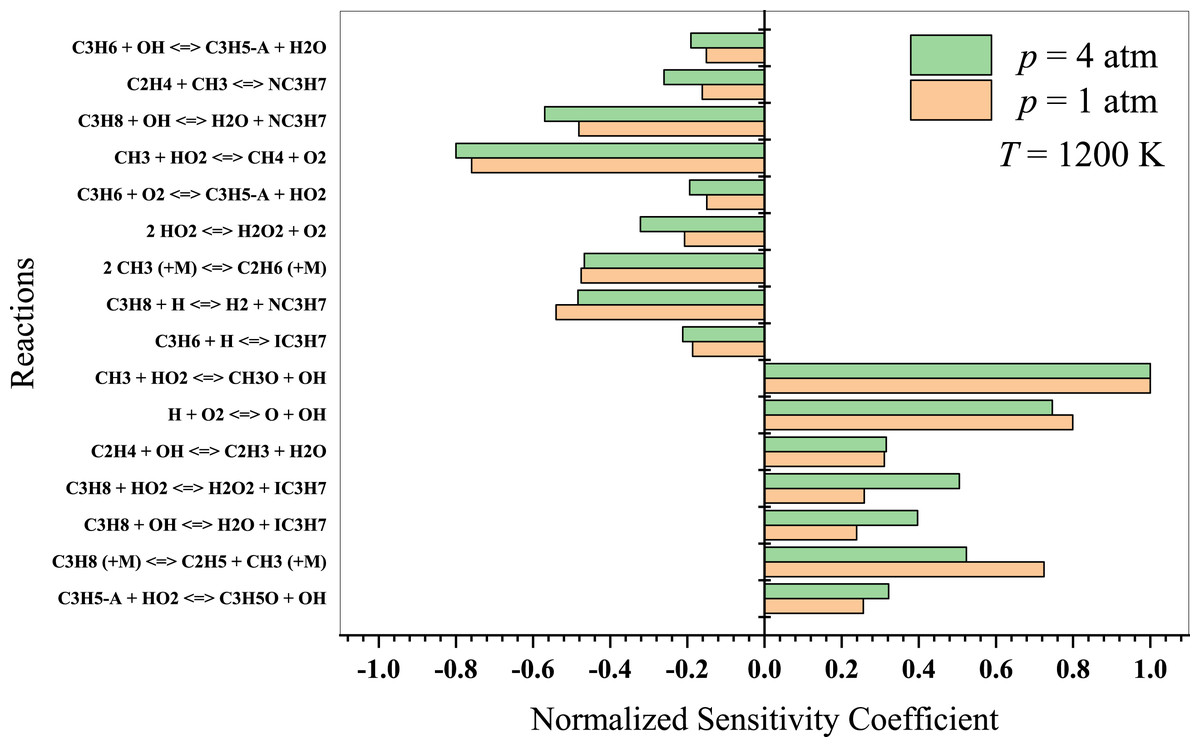
Figure 9: Sensitivity analysis of stoichiometric propane-air mixture at Temperature - 1200 K.
{kind=link}
The reaction of propane with OH to form alkyl radicals and water. The decomposition of alkyl radicals to form smaller hydrocarbon radicals and alkenes.
The oxidation of alkenes and smaller hydrocarbon radicals by O2, OH, and HO2, leading to the formation of more stable products, such as CO, CO2, and H2O.
At high temperatures, propane oxidation is governed by high-temperature chemistry, wherein the formation of oxygenated intermediates becomes less essential. In Fig. 10, the typical high-temperature branching reaction (H + O2 → OH + O) promotes autoignition at temperatures above 1,300 K. It considerably enhances the system’s reactivity, resulting in an extended autoignition latency (Sabia et al., 2014). The authors also suggested that for T >1,000 K, the reaction 2 CH3 + (M) →C2H6 + (M) plays a crucial role in promoting recombination and pyrolytic reactions. The primary reactions to consider in this regime include:
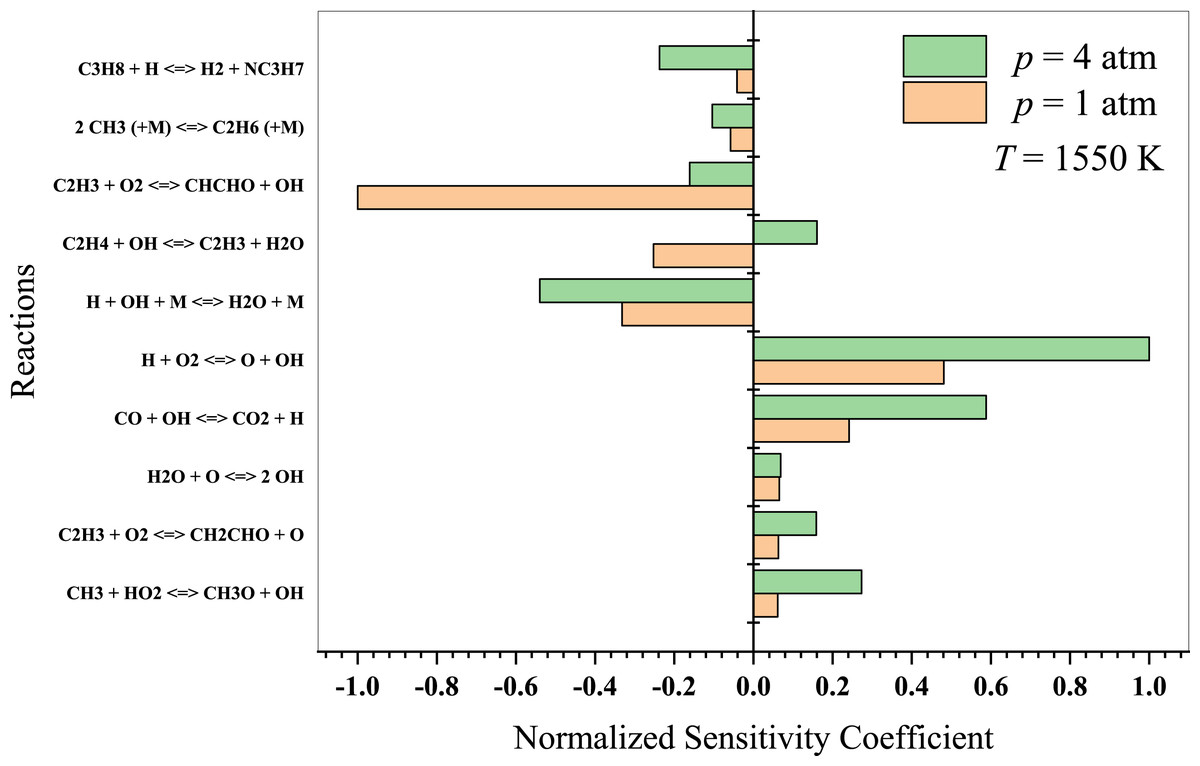
Figure 10: Sensitivity analysis of stoichiometric propane-air mixture at Temperature - 1550 K.
{kind=link}
The direct H-abstraction from propane by O2, OH and atomic oxygen (O), leading to the formation of alkyl radicals and smaller hydrocarbon radicals.
The rapid decomposition of radicals to form stable products, such as CO, CO2, and H2O, via reactions with O2, OH and O.
Hashemi et al. (2019) investigated the high-pressure oxidation of propane, focusing on the reaction of C3H8 with HO2. According to their findings, H-abstraction by HO2 from C3H8 is sensitive in determining ignition delays. At the lower pressure of 10 atm, the reaction CH3 + HO2 → CH4 + O2 inhibits ignition. The sensitivity coefficients of C3H8 + OH →n − C3H7 + H2O and n − C3H7 → C2H4 + CH3 are of great importance at 30 atm, as both promote ignition at 900 K but inhibit it at 1100 K. Nevertheless, high-temperature propane oxidation differs from low-to-intermediate temperature oxidation. As previously discussed, Yeong & Su (2001) presented detailed propane chemistry at high temperatures. Kazakov et al. (2006) argued that varying models can yield different deductions on reaction chemistry due to different rate coefficients and thermochemistry. Consequently, researchers will persist in their work on detailed kinetic modeling to enhance combustion chemistry and draw more reliable conclusions.
Conclusion
The escalating consumption of fossil fuels and the subsequent deterioration of air quality have prompted researchers to explore potential alternative fuels for cleaner combustion and develop effective engine technologies. The ignition delay time (IDT) is a crucial physicochemical characteristic of the combustible fuel-air mixture considered when attempting to understand and enhance an engine’s combustion performance. IDT governs the fuel kinetics during combustion and plays a significant role in shock explosion phenomena, particularly at low temperatures and high pressures (Prince & Williams, 2012; Kuo, 2005). This study aims to provide a comparative analysis of the autoignition characteristics of propane using various available mechanisms at pressures (1-30 atm) for three equivalence ratios ϕ = 0.5, 1, and 2, offering comparative results for better interpretation.
The primary conclusions of this study are as follows:
The results clearly indicate that IDT decreases with increasing pressure. A significant reduction in IDT is observed from 1 to 10 atm, with a more pronounced difference at lower temperature regions. The variation in experimental results falls within the normal range and is comparable to those found in previous literature studies.
Using a variety of kinetic models available in the literature, numerical simulations demonstrate that several kinetic mechanisms yield similar outcomes under the specified conditions of reported experimental data, with the exception of the GRI mechanism. The GRI mechanism, commonly used at the commercial level, does not yield satisfactory results. Conversely, the POLIMI mechanism over-predicts the IDT for pure propane mixtures.
At high pressure and low temperature, a discrepancy exists between experimental and numerical results. Lam et al. (2011) highlighted the importance of considering pressure and temperature variations before ignition in shock tubes when interpreting results. The deviation in experimental data may be partly attributed to the absence of such treatment. However, the experimental results are generally in good agreement with those produced by the ARAMCO 3.0 model, which incorporates the detailed chemical kinetics of propane.
Lastly, a sensitivity analysis was conducted to identify the chemical kinetics governing the autoignition process of propane at different temperature ranges. At lower and intermediate temperatures, the decomposition and branching reactions of the OH and HO2 radicals are of considerable importance. At higher temperatures, the formation of OH radicals through H-abstraction plays a critical role in determining ignition delays.