Proteomics of Nasonia vitripennis and the effects of native Wolbachia infection on N. vitripennis
- Published
- Accepted
- Received
- Academic Editor
- Joseph Gillespie
- Subject Areas
- Entomology, Molecular Biology
- Keywords
- Wolbachia, Immunity, Proteomics, iTRAQ, Nasonia vitripennis
- Copyright
- © 2018 Li et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2018. Proteomics of Nasonia vitripennis and the effects of native Wolbachia infection on N. vitripennis. PeerJ 6:e4905 https://doi.org/10.7717/peerj.4905
Abstract
Background
Nasonia vitripennis, a parasitic wasp, is a good model organism to study developmental and evolutionary genetics and to evaluate the interactions between insect hosts and their symbionts. Wolbachia may be the most prevalent endosymbiont among insect species due to their special ability to improve the fitness of the infected hosts. Transinfection of bacteria or fungi could substantially alter the expression of host immune system components. However, few studies have focused on the effects of native Wolbachia infection. Accordingly, in this study, we evaluated the proteomics of N. vitripennis following Wolbachia infection.
Methods
We studied the proteomics of N. vitripennis following native Wolbachia infection and in antibiotic-treated Wolbachia-free samples using isobaric tags for relative and absolute quantification-liquid chromatography tandem mass spectrometry, accompanying with some ecological experiments.
Results
In total, 3,096 proteins were found to be associated with a wide range of biological processes, molecular functions, and cellular components. Interestingly, there were few significant changes in immune or reproductive proteins between samples with and without Wolbachia infection. Differentially expressed proteins were involved in the binding process, catalytic activity, and the metabolic process, as confirmed by quantitative reverse transcription polymerase chain reaction.
Discussion
Invasion of any pathogen or bacterium within a short time can cause an immunoreaction in the host. Our results implied that during the long process of coexistence, the immune system of the host was not as sensitive as when the symbiont initially infected the host, implying that the organisms had gradually adjusted to cohabitation.
Introduction
Nasonia is the second genus of Hymenoptera to have been subjected to whole-genome sequencing, assembly, and annotation, after Apis mellifera (Werren et al., 2010). Nasonia vitripennis (Hymenoptera: Pteromalidae), a parasitic wasp, lay their eggs into the pupa of many different flies; the eggs then develop into adults in pupa, and during this process, the wasps act as natural enemies to flies (Dong et al., 2009). N. vitripennis is becoming a model organism for developmental and evolutionary genetics (Shuker, Lynch & Peire Morais, 2003; Werren & Loehlin, 2009b) and for the study of interactions between insect hosts and symbionts owing to its overall ease of laboratory use, short generation time (roughly two weeks), tolerance for inbreeding, and straightforward rearing (Li et al., 2017).
The close relationship between N. vitripennis and the maternally inherited endosymbiont Wolbachia has been extensively studied. Wolbachia, an intracellular gram-negative bacterium, naturally infects up to 40% of arthropod species (Werren, Baldo & Clark, 2008; Zug & Hammerstein, 2012). Because Wolbachia is transmitted through the female germline, it has evolved a number of reproductive strategies to favor the Wolbachia-infected females (Sullivan, 2016), such as cytoplasmic incompatibility (CI), male-killing, induction of parthenogenesis, feminization, and speciation (Werren, 1997; Werren, Baldo & Clark, 2008). Among these manipulations, the most common effect is CI (Tram & Sullivan, 2002), whereby infected females mated with infected or uninfected males produce viable embryos, while uninfected females mated with infected males produce inviable embryos (Sullivan, 2016). The reproductive effect of Wolbachia on N. vitripennis is just CI (Breeuwer & Werren, 1993) and can be influenced by temperature, bacterial density, and the bacteriophage WO (Bordenstein & Bordenstein, 2011; Breeuwer & Werren, 1993). Although there are tight associations between Wolbachia and insect hosts, they do not evolve simultaneously due to Wolbachia horizontal transmission, even between phylogenetically distant species (Wang et al., 2016; Zug, Koehncke & Hammerstein, 2012).
In addition to reproductive modifications, the global spread of Wolbachia has also been attributed to increased host fecundity and protection against pathogens (Hedges et al., 2008; Teixeira, Ferreira & Ashburner, 2008; Zug & Hammerstein, 2015a). Wolbachia confers resistance to various pathogens, e.g., by priming the innate immune system in Aedes aegypti (Bian et al., 2010; Moreira et al., 2009). Insects rely on the innate immune system to mount defense responses against pathogenic invasions (Rodrigues et al., 2010). The insect innate immune system consists of humoral immune and cellular immune responses (Govind, 2008; Rolff & Siva-Jothy, 2003). Humoral immune responses involve immune-related molecules, such as antimicrobial peptides, lysozyme, or phenoloxidase (Urbanski et al., 2014), whereas the main cellular immune responses involve pathogen phagocytosis, nodulation, and encapsulation (Lanz-Mendoza et al., 1996).
Studies have shown that invasion of any pathogen or bacterium within a short time can cause an immunoreaction in the host. The genome-wide analysis of the interaction between Wolbachia and its Drosophila host showed involvement of an antimicrobial humoral response and negative regulation of cell proliferation in the host (Wong et al., 2011). In silkworms, after transinfection with Bacillus subtilis (a gram-negative bacteria) for 24 h, 2,436 genes showed more than two-fold changes in expression (Huang et al., 2009), and the systemic immune response was triggered via the Toll-like receptor pathway, resulting in changes in the expression of antimicrobial peptide genes, such as Attacin, Lebocin, Enbocin, Gloverin, and Moricin (Huang et al., 2009). Such transinfections usually induce a higher bacterial density and cause other alterations to host physiology (McMeniman et al., 2009; Pan et al., 2012).
Compared with transinfection, native or natural infection by Wolbachia has not been extensively studied, except for a few studies in mosquitoes (Hughes et al., 2011b; Joubert & O’Neill, 2017; McMeniman et al., 2009). Stable infection of Wolbachia induced the up- or downregulation of 257 transcripts, with no changes in Toll and immune deficiency (IMD) pathway genes in Aedes fluviatilis (Caragata et al., 2017). Hughes et al. (2011a) found that the immune response in Anopheles after somatic infection is dynamic, as it was first induced and then suppressed as the infection progressed. However, the effects on immune-related genes in the host following long-term coevolution with Wolbachia are still unclear.
Accordingly, in this study, we used native Wolbachia-infected and antibiotic-treated Wolbachia-free N. vitripennis to explore how Wolbachia affects host biology at the protein level by combining isobaric tags for relative and absolute quantification-liquid chromatography tandem mass spectrometry (iTRAQ-LC-MS/MS) and some ecological experiments. Overall, 3,096 N. vitripennis proteins were found to be associated with various biological processes (BPs), molecular functions (MFs), and cellular components (CCs). However, few changes were observed in immune or reproductive proteins, suggesting that during coevolution, the impacts of long-term infection of Wolbachia gradually declined.
Materials and Methods
Nasonia materials
Two Nasonia lines were used in our experiments, native Wolbachia-infected N. vitripennis and Wolbachia-free N. vitripennis; both lines had been grown in our laboratory since 2011. The Wolbachia-infected line was naturally infected with single Supergroup A Wolbachia strain, as confirmed by Wolbachia surface protein (WSP). The Wolbachia-free N. vitripennis line was treated with rifampin to remove native Wolbachia at the beginning, and repeated polymerase chain reaction (PCR) with primers targeting the WSP gene was used to confirm the effects of removal. Wolbachia-free organisms used in our study were reared after 30 generations to reduce the effects of antibiotics. Two lines were maintained using standard insectary conditions at 28 °C for light and 25 °C for dark (L:D = 16:8 h) with 60% ± 10% relative humidity (Werren & Loehlin, 2009c). In our experiment, N. vitripennis parasitised Wolbachia-free Sarcophaga marshalli pupa, where the eggs, larvae, and pupae of the wasp were developed (Werren & Loehlin, 2009c), and the adults were reared with 10% honey water.
Ecological experiment
At 10–13 days after N. vitripennis oviposited into Sarcophaga marshalli pupae, the pupae were dissected to obtain virgin adults. The male and female pupa were identified individually and collected separately once upon eclosion. Four crossing types based on different Wolbachia infection statuses were carried out (♀ W+ × ♂ W+, ♀ W+ × ♂ W−, ♀ W− × ♂ W−, ♀ W− × ♂ W+). A virgin female and a virgin male adult were placed in a 7 mL tube, together with one fresh Sarcophaga marshalli pupa for oviposition. The offspring of the crossings were collected and counted, and the percent females (the proportion of females of the total number of individuals) was calculated. The offspring number and percent females based on the number of fly pupae per day were calculated for nine consecutive generations. Moreover, the effects of Wolbachia on longevity of N. vitripennis were studied. Approximately 300 samples were used for life testing. Each experiment was repeated with six groups.
Protein preparation
Wolbachia-infected and Wolbachia-free line were prepared for iTRAQ separately, each line contains three replicates. The female adults were collected after eclosion two to three days, and all adults of a sample were from the same generation. Approximately 200 adult wasps per sample were homogenized, and total proteins were extracted using the phenol extraction procedure. The protein content in the supernatant was quantified using a BCA Protein Assay Kit (Thermo Scientific, Milford, MA, USA).
Proteome analysis by iTRAQ
The proteome was quantified using iTRAQ-LC-MS/MS. Protein abundances that changed more than 1.2-fold were regarded as significantly differentially expressed. The extracted proteins were digested, and peptides were quantified as described by Wisniewski et al. (2009). For peptide labeling, the peptide mixture from each group was labeled with iTRAQ Reagent-8plex multiplex kit (AB SCIEX, Foster City, CA, USA) according to the manufacturer’s instructions. Wolbachia-free and Wolbachia-infected samples were labeled with iTRAQ reagents 113, 115, 117 and 114, 116, 118, respectively. The labeled peptide mixtures were pooled for strong cation-exchange chromatography. Samples were separated by nanoLC-MS/MS (EASY-nLC1000). The mass spectrometry data were analyzed using a Q-Exactive mass spectrometer (Thermo Finnigan, Waltham, MA, USA) in the positive ion mode with a selected mass range of m/z 300–1,800.
Quantitative real-time reverse transcription PCR
Total RNA was isolated using TransZol UP (TRANSGEN BIOTECH, Beijing, China) from Wolbachia-infected and Wolbachia-free N. vitripennis adults (50 individuals each). The first-strand cDNA was reverse-transcribed from 2 μg total RNA using TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (TRANSGEN BIOTECH, Beijing, China) at 42 °C for 15 min according to the manufacturer’s instructions. Specific primers for tested genes were designed on the basis of cDNA sequences from NCBI database by Primer Premier 6 software. All primers used in quantitative PCR (qPCR) are shown in Table S1. qPCR was performed on a real-time PCR machine (Bio-Rad, Hercules, CA, USA) with TransStart Tip Green qPCR Supermix (TRANSGEN BIOTECH, Beijing, China). The amplification reaction conditions were as follows: 95 °C for 3 min; followed by 40 cycles of 95 °C for 10 s, 60 °C for 30 s, and 95 °C for 10 s; and then a melting curve was constructed from 65 to 95 °C. The relative expression of each gene was normalized to the expression of RPL13a and UBC using the 2−ΔΔCT method. Results are expressed as means ± standard deviations. Three biological replicates were used in the experiment.
Data analysis
The raw MS/MS spectra data were searched and identified using Mascot 2.2 and Proteome Discoverer 1.4 (Thermo Fisher Scientific, Waltham, MA, USA). The database used in this study was NCBI_Nasonia_vitripennis_25492_20141104.fasta, loaded by Uniprot in November 2014. Assembled protein identifications were qualitatively analyzed using Proteome Discoverer 1.4 software. All data were reported based on 99% confidence for protein identification, as determined by a false discovery rate (FDR) of less than or equal to 1%. Statistical analysis was conducted using one-way analysis of variance (ANOVA). Results with P values of less than or equal to 0.05 by Tukey’s test were considered significant. Among the statistically significant proteins detected by ANOVA (P < 0.05), proteins showing more than 1.2-fold changes in abundance were regarded as significantly differentially expressed.
Bioinformatics
All proteins that were found in abundance among Wolbachia-infected and Wolbachia-free group were further analyzed for functional and biological relevance. These proteins were classified by their gene functions and biological pathways using the freely available gene ontology (GO) database provided by the GO consortium. The retrieved sequences were locally searched against NCBI nr using the NCBI BLAST+ client software (ncbi-blast-2.2.28+−win32.ext) to find homologous proteins, from which functional annotations were transferred to the target proteins. The top 10 BLAST hits with an E-value of less than 1e−3 for each query protein were retrieved and loaded into Blast2GO (Version 2.7.2) for GO mapping and annotation. In addition, differentially expressed proteins, proteins participating in the immune system, and proteins involved in the reproductive process were evaluated using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING; http://string.embl.de/) to build a functional protein association network.
Results
Percent females of crosses and wasp longevity with different infection statuses
Among the four crosses (♀ W+ × ♂ W+, ♀ W+ × ♂ W−, ♀ W− × ♂ W−, and ♀ W− × ♂ W+), the percent females of uninfected females and infected males (♀ W− × ♂ W+, 0.11) was significantly lower than that of the other three hybridization groups (P < 0.001; Fig. 1A), confirming that the reproductive regulation of Wolbachia on N. vitripennis was CI. The average offspring numbers of Wolbachia-infected wasps with one (W+*1), three (W+*3), and five pupa (W+*5) per day were 215.17 ± 19.30, 391.83 ± 12.95, and 444.17 ± 20.32 respectively, whereas the offspring numbers of Wolbachia-free wasps with one (W−*1), three (W−*3), and five pupa (W−*5) per day were 176.17 ± 13.53, 304.83 ± 24.86, and 407.17 ± 11.18, respectively (Fig. 1B). With the increase in pupae number, the progeny size of Wolbachia-infected N. vitripennis increased faster, implying that Wolbachia significantly enhanced host fecundity (one pupae: df = 1, F = 1.572, P = 0.002; three pupa: df = 1, F = 1.208, P < 0.001; five pupa: df = 1, F = 2.965, P = 0.003).
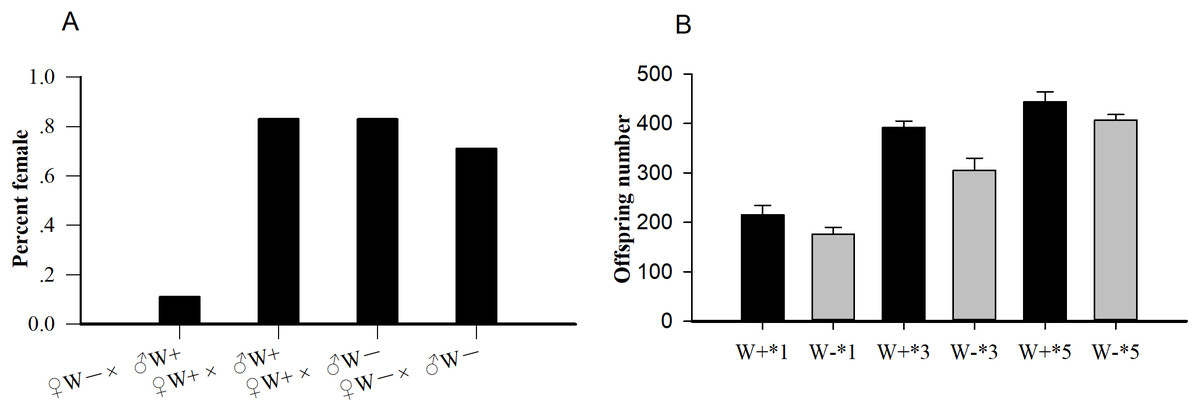
Figure 1: The statistics of percent females and progeny size.
(A) Percent females of four cross types (♀ W− × ♂ W+, ♀ W+ × ♂ W+, ♀ W+ × ♂ W−, ♀ W− × ♂ W−) based on different Wolbachia infection statuses. (B) Comparisons of progeny sizes with different Wolbachia infection statuses and pupae numbers. W+, Wolbachia-infected group; W−, Wolbachia-free group. ♀, female, and ♂, male. The number after “*” is the number of fly pupae replaced every day.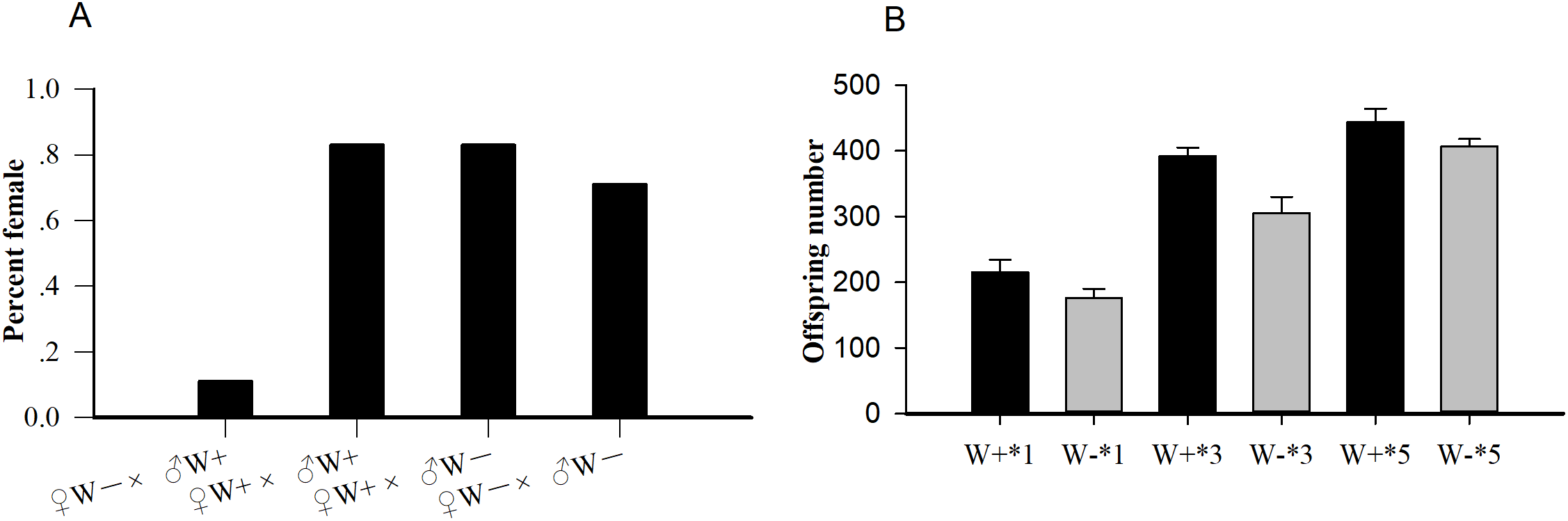{kind=link}
The average longevities of Wolbachia-infected and Wolbachia-free females were 18 and 16 days (F1,288 = 0.19, P = 0.07), respectively, whereas those of Wolbachia-infected and Wolbachia-free males were nine and eight days (F1,347 = 0.36, P = 0.12), respectively (Fig. 2). The existence of Wolbachia did not significantly alter the life span of N. vitripennis.
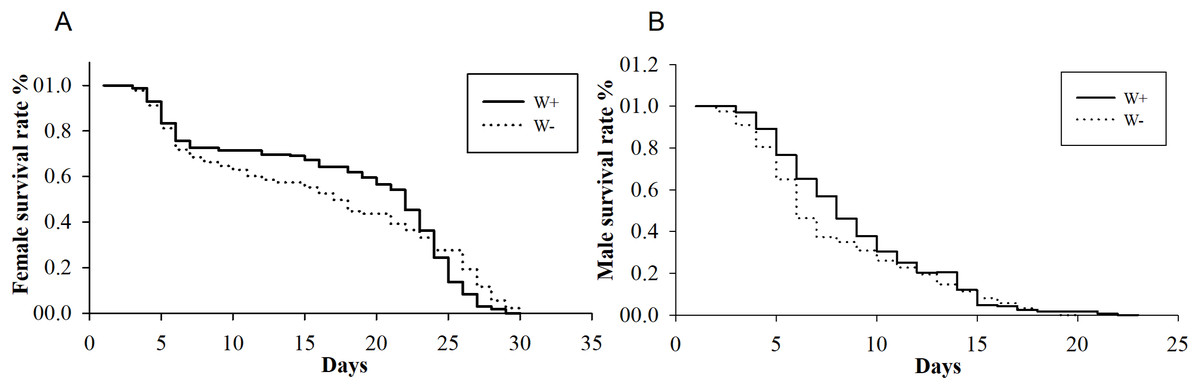
Figure 2: Longevity of female and male N. vitripennis with different Wolbachia infection statuses.
(A) Female longevity. (B) Male longevity. “——” indicates Wolbachia-infected N. vitripennis longevity, and “-–-” indicates Wolbachia-free N. vitripennis longevity.{kind=link}
Identification, GO analysis, and KEGG analysis of Nasonia proteomics by iTRAQ
A total of 3,109 proteins were identified from 22,946 unique peptides based on the N. vitripennis database with a peptide FDR of less than or equal to 0.01. From these, 3,096 proteins were quantified in three Wolbachia-infected and three Wolbachia-free N. vitripennis libraries (Table S2). Statistical analysis showed that 40.11% of the identified proteins had coverage greater than 20, whereas only 4.54% of proteins had coverage below 2. In addition, 2,520 proteins (81.03%) were identified with at least two peptides. Proteins with masses of more than 10 kDa had broader coverage in protein mass distribution (Fig. S1).
All 3,096 proteins were annotated using Blast2GO (Table S3) and analyzed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (Table S4). In total, 2,578 proteins were annotated by 9,297 GO terms, covering a wide range of BPs (33.47%), MFs (41.47%), and CCs (25.26%; Fig. 3). Notably, 13 proteins participated in the immune system process, and 15 proteins were involved in both the reproductive process and reproduction (Table 1). In particular, these immune proteins were involved in regulation of the immune system process, immune response, innate immune response, and immune response-regulating cell surface receptor signaling pathways. Reproduction proteins participated in the regulation of reproductive processes, sexual reproduction, developmental processes involved in reproduction, reproductive system development, and other pathways.
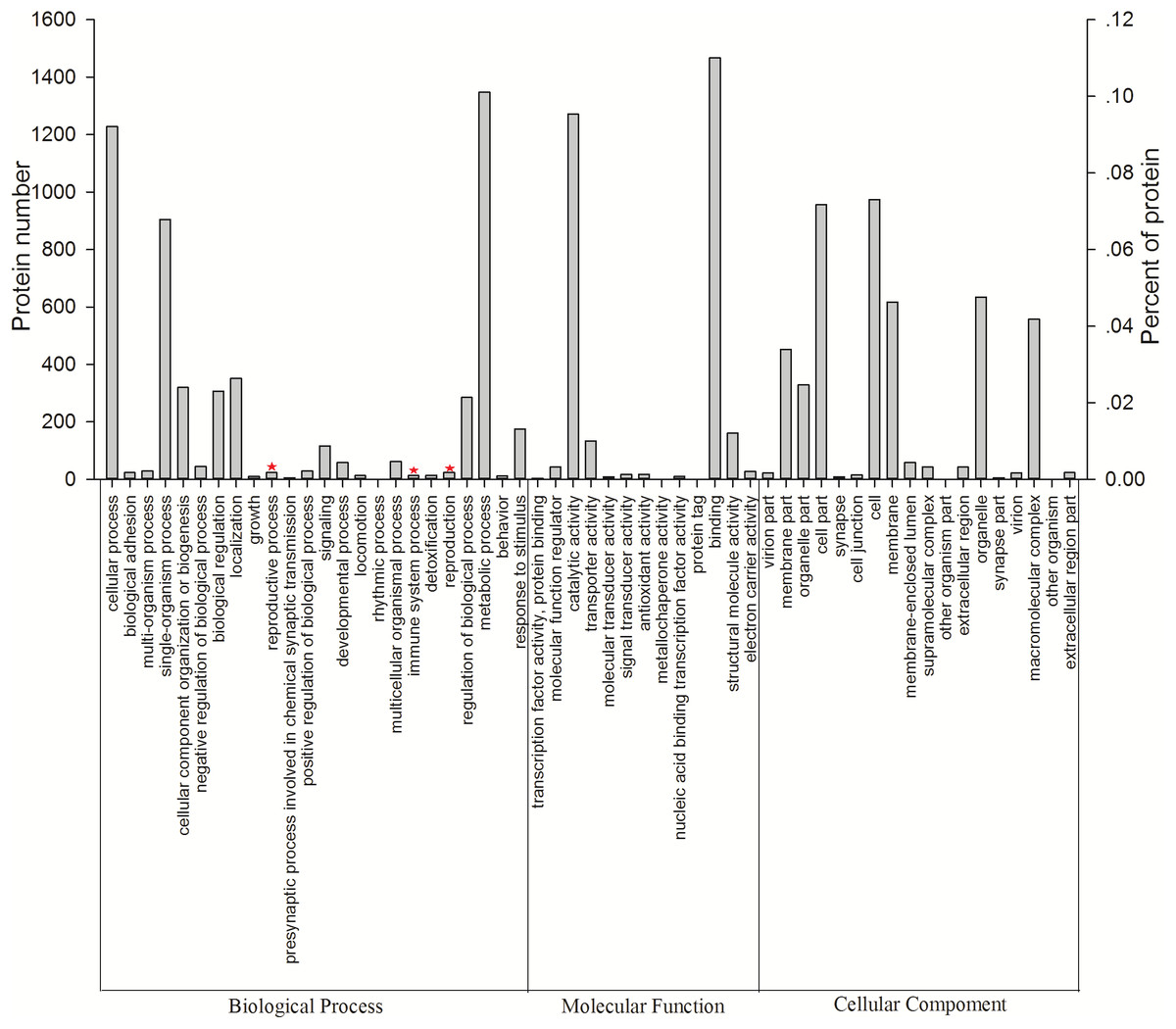
Figure 3: Vertical graph of gene ontology classifications for proteins identified from Wolbachia-infected and Wolbachia-free N. vitripennis proteomics.
The proteins were found to be associated with a wide range of biological process (BP), molecular function (MF), and cellular component (CC). Immune system processes, reproductive processes, and reproduction are indicated by red stars.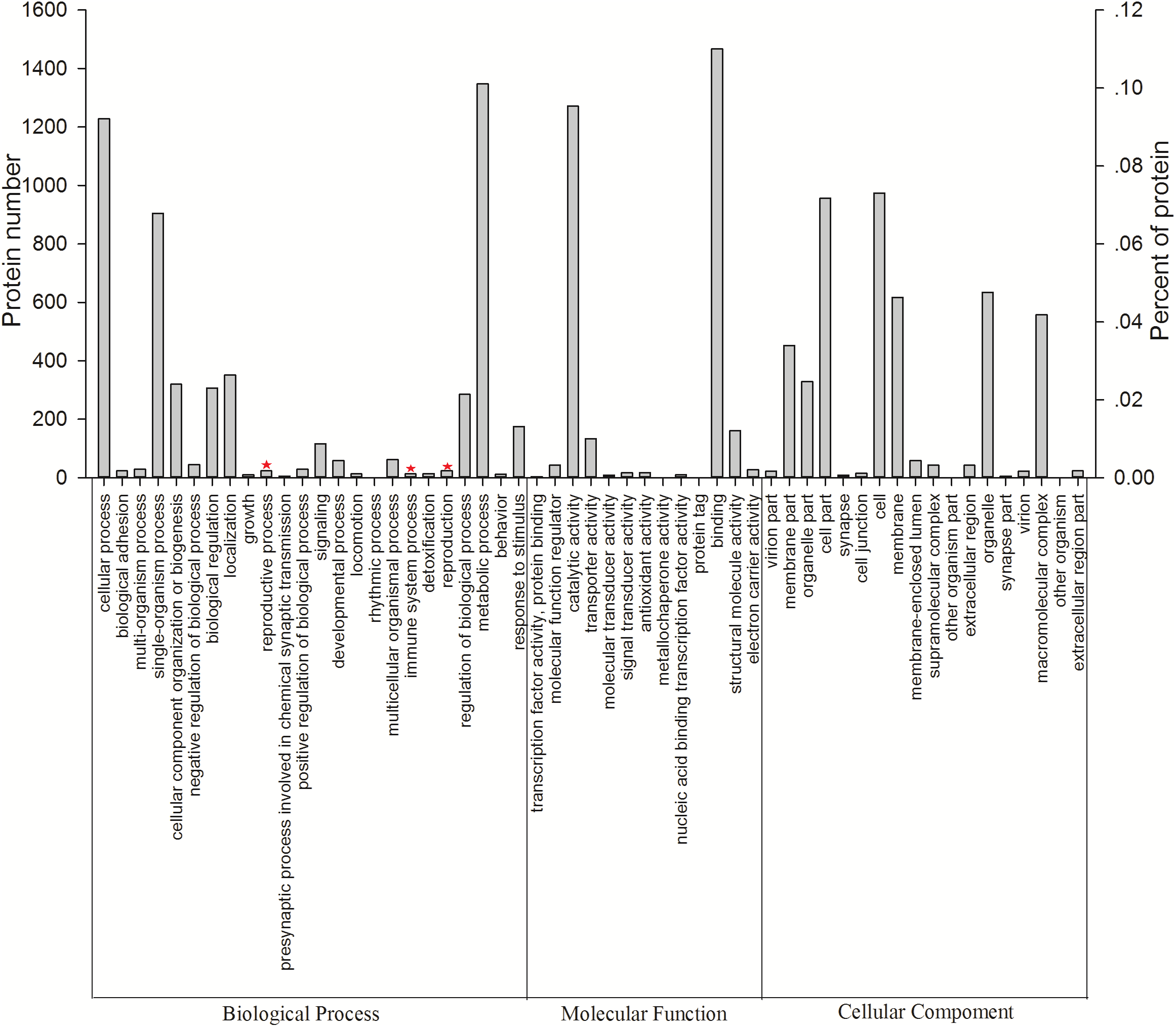{kind=link}
| Accession | NV database | Description | Protein functions | W+/W− |
|---|---|---|---|---|
| 156540814 | NV18390 | ADP-ribosylation factor 1 | Immune | 0.9413223 |
| 156547980 | NV13565 | Peptidoglycan recognition protein 1-like isoform X2 | Immune | 1.14620672 |
| 156551611 | NV15422 | Histone H4-like | Immune | 0.96703548 |
| 283436140 | NV17164 | Peptidoglycan-recognition protein S2-like protein precursor | Immune | 0.99915634 |
| 345486704 | NV50172 | Histone H3 | Immune | 0.93876689 |
| 345491753 | NV10830 | Interleukin enhancer-binding factor 2 isoform X2 | Immune | 0.98109544 |
| 645034592 | NV13687 | Guanine nucleotide-binding protein G(q) subunit alpha isoform X4 | Immune | 0.93153917 |
| 645037639 | —— | Flotillin-2 | Immune | 0.98390399 |
| 645038121 | NV18120 | Microtubule-associated protein RP/EB family member 1 isoform X1 | Immune | 1.01723944 |
| 156537767 | NV10430 | Ras-related protein Rac1, partial | Immune/reproduction | 0.97298107 |
| 645015241 | NV15633 | Ras-related protein Ral-a isoform X5 | Immune/reproduction | 0.97905013 |
| 645034685 | NV13706 | Filamin-A isoform X1 | Immune/reproduction | 1.0087272 |
| 645035835 | —— | Superoxide dismutase [Cu–Zn]-like isoform X2 | Immune/reproduction | 0.92936684 |
| 156550189 | NV14758 | Ubiquitin-conjugating enzyme E2-17 kDa isoform X2 | Reproduction | 1.00353964 |
| 254910945 | NV16613 | Actin related protein 1 | Reproduction | 0.96004749 |
| 299782477 | NV13256 | Dynein light chain A | Reproduction | 0.98722231 |
| 345494735 | NV16743 | Ras-related protein Rab6 isoform X2 | Reproduction | 0.97798355 |
| 644992919 | NV14154 | Histone H3.3 | Reproduction | 0.91800771 |
| 645001627 | NV11130 | Nuclear factor of activated T-cells 5 | Reproduction | 1.03309889 |
| 645005129 | NV13395 | Serine/threonine-protein phosphatase 2B catalytic subunit alpha isoform-like isoform X2 | Reproduction | 0.9490586 |
| 645012252 | NV14708 | Heterogeneous nuclear ribonucleoprotein R-like | Reproduction | 1.00903299 |
| 645031721 | NV12583 | Tropomyosin 1 isoform X14 | Reproduction | 0.97277993 |
| 645034137 | NV13603 | Calmodulin isoform X2 | Reproduction | 0.97721229 |
| 645041050 | NV18956 | Moesin/ezrin/radixin homolog 1-like isoform X6 | Reproduction | 0.97626768 |
Notes:
Protein functions were determined based on GO annotations. W+/W−, the protein expression ratio of Wolbachia-infected and Wolbachia-free N. vitripennis. “——” indicates no corresponding proteins in the N. vitripennis (NV) database.
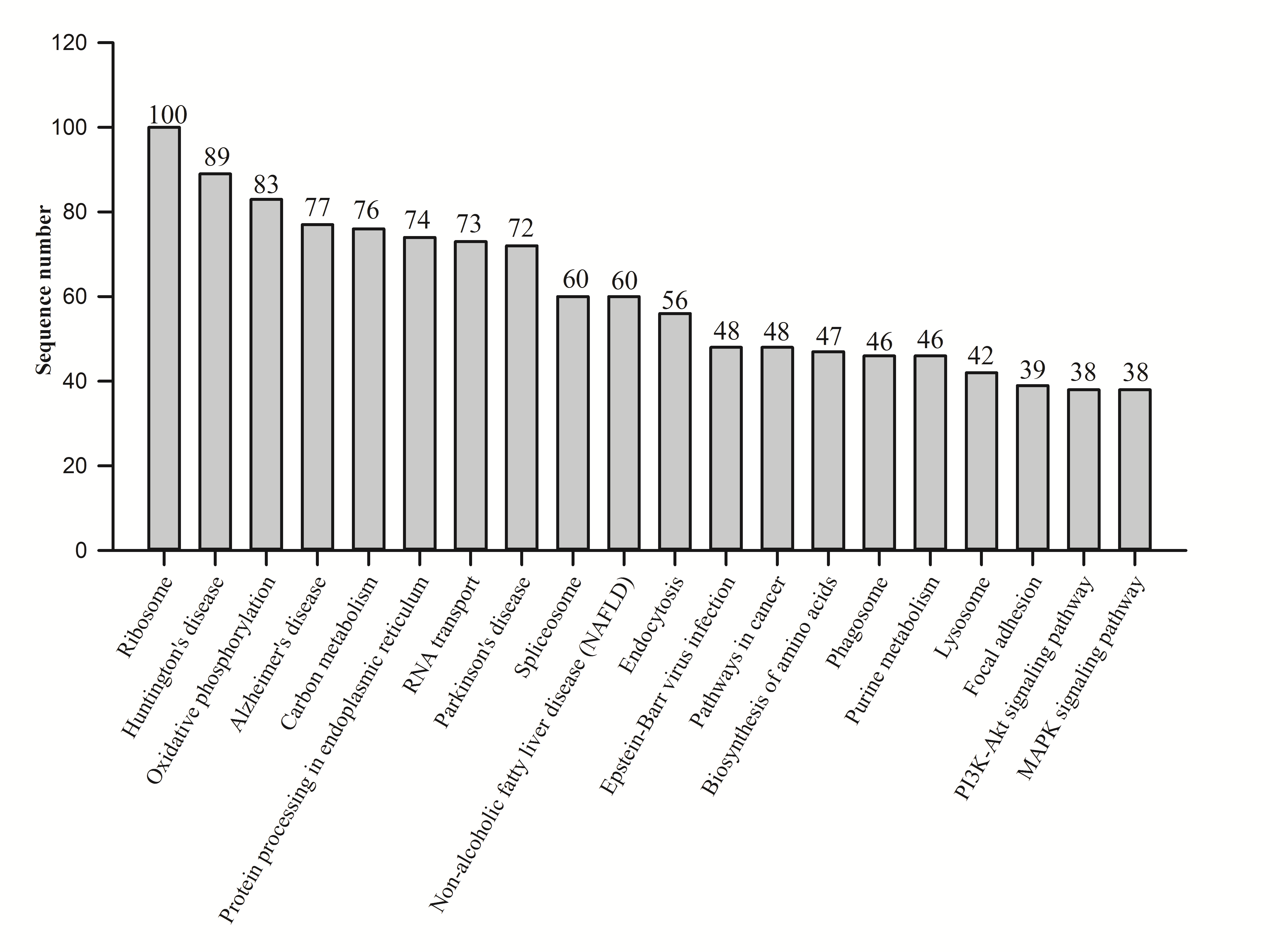
KEGG analysis showed that 1,975 proteins were assigned to 1,962 KEGG orthologies and were involved in 337 maps. The top 20 maps were analyzed (Fig. S2), and the ribosome contained the most proteins.
Significantly differentially expressed proteins
Based on the iTRAQ-LC-MS/MS proteomic analysis, proteins with more than 1.2-fold differences and P values of less than 0.05 were regarded as significantly differentially expressed. There were only 23 proteins that were significantly differentially expressed between Wolbachia-infected and uninfected N. vitripennis based on the proteomics analysis, including 15 upregulated and eight downregulated proteins (Table 2). Fifteen proteins among the 23 proteins were involved in catalytic activity and binding in the MF category and in metabolic processes in the BP category. These differentially expressed proteins belonged to the biosynthesis, metabolism, and degradation processes, including Ras signaling, peroxisomes, vascular smooth muscle contraction, inflammatory mediator regulation of TRP channels, and chemical carcinogenesis. Interestingly, neither immune-related proteins nor reproductive proteins were significantly differentially expressed. Some immune genes, particularly those involved in the Toll or IMD pathways, as well as some antimicrobial peptides, were not found in either Wolbachia-infected or Wolbachia-free organisms. Protein LOC 100121395 (NV15328), which was uncharacterized in the research database, was found as putative odorant binding protein 69. Uncharacterized protein LOC1003315494 and LOC100678792 may be venom proteins by BLAST. NV11606, which was described as hydroxyacid oxidase 1, may participate not only in the carbon metabolism, but also in redox signaling and lipid homeostasis. NV16308, alcohol dehydrogenase class-3, also participated in multiple metabolic pathways, including glycolysis/gluconeogenesis, carbon metabolism, metabolism of xenobiotics by cytochrome P450, retinol metabolism, tyrosine metabolism, and fatty acid degradation.
| Accession | NV database | Description | W+/W− |
|---|---|---|---|
| 645042578 | NV19267 | Chitotriosidase-1-like, partial | 0.6936338 |
| 345492571 | —— | Uncharacterized protein LOC100679225 | 0.7228549 |
| 156546540 | NV12994 | Cytochrome b5-like | 0.8075551 |
| 156544032 | NV11606 | Hydroxyacid oxidase 1 | 0.8137581 |
| 645035666 | NV12106 | Acyl-coenzyme A thioesterase 13 | 0.825083 |
| 645005765 | NV14562 | Alcohol dehydrogenase-like | 0.8277345 |
| 156551475 | NV15328 | Uncharacterized protein LOC100121395 | 0.8285397 |
| 645026174 | NV18416 | Protein henna isoform X2 | 0.8316617 |
| 156544652 | NV11923 | 85/88 kDa calcium-independent phospholipase A2-like | 1.2050817 |
| 345488667 | SP6 | Trypsin-1-like | 1.2074925 |
| 345487828 | —— | Polyamine-modulated factor 1-binding protein 1-like | 1.2602131 |
| 645009869 | NV11878 | Uncharacterized protein LOC100119601 | 1.2892213 |
| 289177071 | NV17138 | Carboxylesterase clade A, member 4 | 1.3667084 |
| 239048037 | —— | Venom protein V precursor | 1.386467 |
| 156540800 | SP31 | Trypsin beta | 1.390753 |
| 239735550 | CCE-B2 | Carboxylesterase clade B, member 2 precursor | 1.3934924 |
| 238859625 | NV18383 | Serine protease 33 precursor | 1.4075765 |
| 645027109 | NV16308 | Alcohol dehydrogenase class-3 | 1.4157821 |
| 156552724 | NV18294 | Endothelin-converting enzyme 1-like | 1.4191134 |
| 645038835 | NV12901-PA | Glutamic acid-rich protein-like | 1.4199756 |
| 644992473 | —— | Uncharacterized protein LOC103315494 | 2.0601506 |
| 345485039 | NV21224-PA | Uncharacterized protein LOC100678792 | 2.1550859 |
| 156554004 | NV15950 | Uncharacterized protein LOC100119759 | 4.0840236 |
Notes:
W+/W−, ratio of protein expression for Wolbachia-infected N. vitripennis relative to that of Wolbachi-free N. vitripennis. Downregulated proteins had a W+/W− ratio of less than or equal to 0.83, whereas upregulated proteins had a W+/W− ratio of greater than or equal to 1.2. “——” indicates no corresponding proteins in the N. vitripennis (NV) database.
Quantitative real-time reverse transcription PCR
Seven genes encoding significantly differentially expressed proteins (NV11606, NV11923, NV11878, SP6, NV18383, NV16308, and NV21224-PA; Table 2) were selected for quantitative real-time reverse transcription PCR (RT-qPCR) validation; these genes encoded calcium-independent phospholipase, serine protease, alcohol dehydrogenase, hydroxyacid oxidase, trypsin, and two uncharacterized proteins. Moreover, seven genes encoding putative immune-related proteins or reproductive proteins (NV16743, NV10430, NV15633, NV13603, NV12583, NV18120, and NV10830; Table 1) were chosen for RT-qPCR. With the exception of NV11606 and SP6, all RT-qPCR results showed similar changes compared with the proteomic analyses (Fig. 4), confirming the reliability of our proteomic analysis.
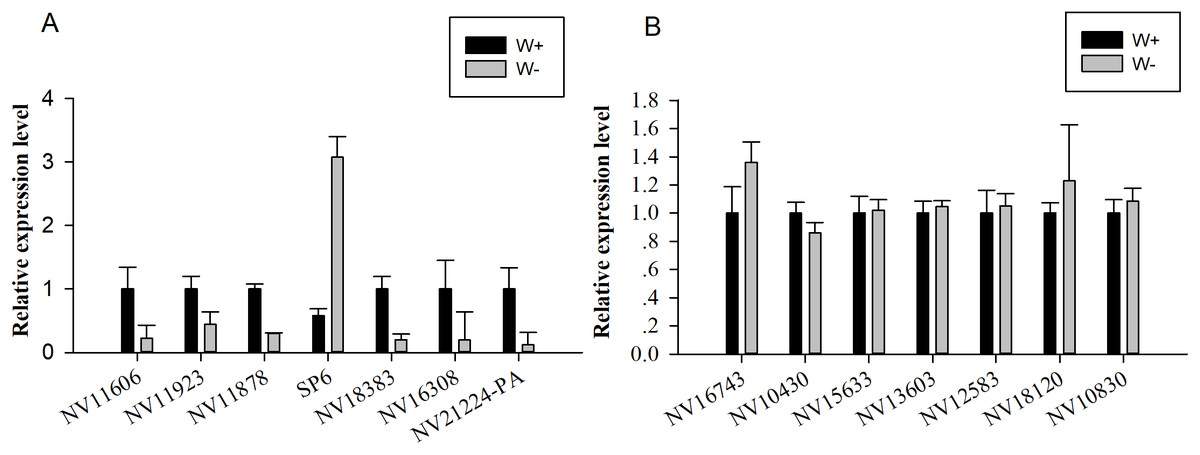
Figure 4: Quantitative real-time reverse transcription PCR was used for validation of selected genes.
(A) Relative expression levels of genes encoding differentially expressed proteins (NV11606, NV11923, NV11878, SP6, NV18383, NV16308, and NV21224-PA). (B) Relative expression levels of genes encoding immune-related or reproductive proteins (NV16743, NV10430, NV15633, NV13603, NV12583, NV18120, and NV10830).{kind=link}
Interactions between putative immune-related proteins, reproductive proteins, and significantly differentially expressed proteins
The online database STRING 10.0 (Search Tool for Retrieval of Interacting Genes/Proteins database) is a system that searches for interactions between known and predicted proteins based on both direct physical and functional correlations among proteins. To explore whether the significantly differentially expressed proteins influenced the immune or reproductive processes in N. vitripennis indirectly, the relationships among 23 significantly differentially expressed proteins, 13 proteins with putative roles in the immune system process, and 15 proteins involved in the reproductive process were studied.
The results showed that all significantly differentially expressed proteins located on the edge of the network map were not associated with any immune-related proteins or reproductive proteins, and there were no direct interactions between significantly differentially expressed proteins (Fig. 5). Some immune-related proteins and reproductive proteins formed a network with complex interactions. For example, four Ras family proteins formed a complex interaction network, with functions in the cell cycle, cell adhesion force, cell phagocytic activity, and other processes.
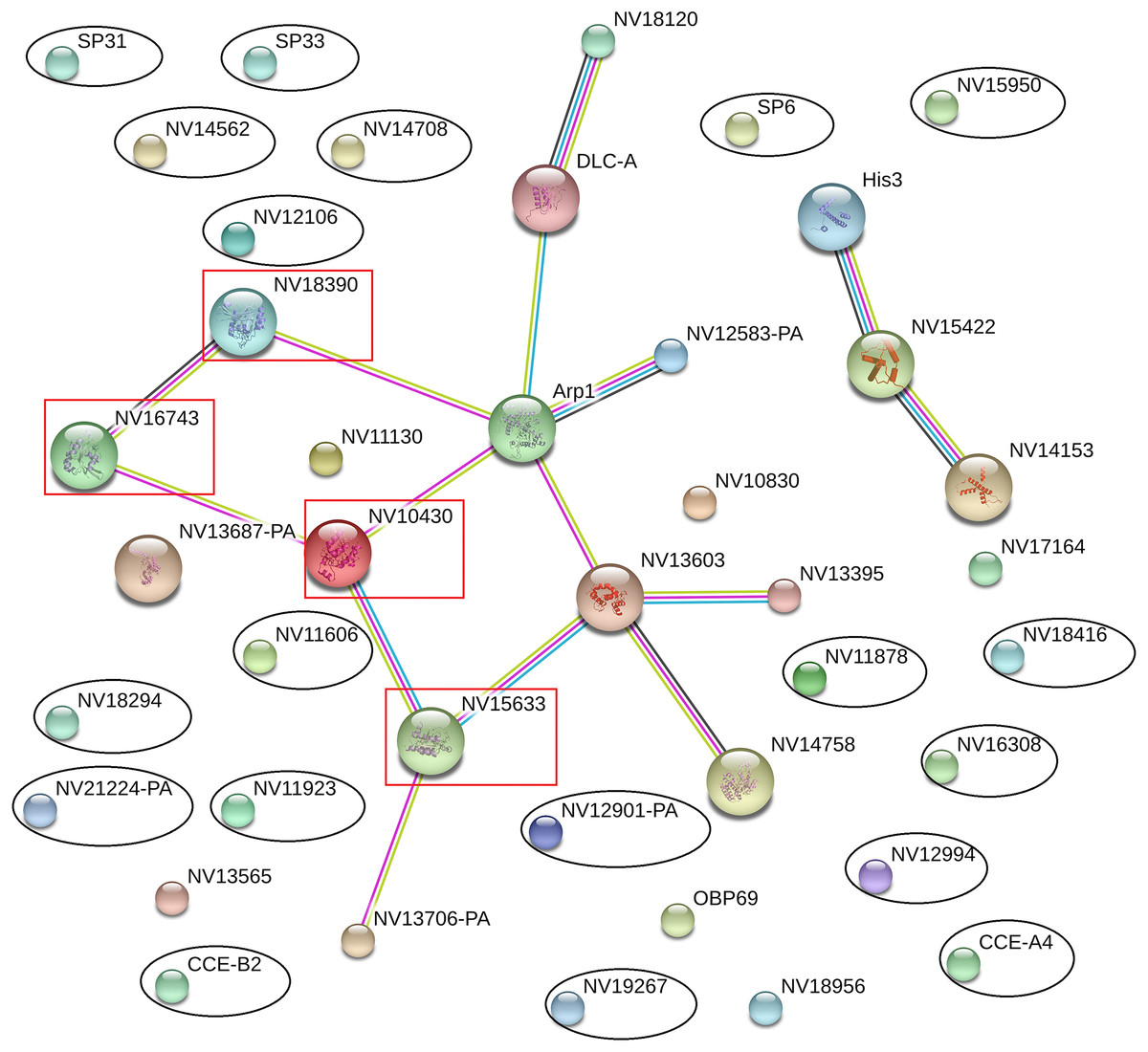
Figure 5: Interaction network among differentially expressed proteins, immune-related proteins, and reproductive proteins, as revealed by the STRING database.
Proteins with black ovals are differentially expressed proteins, whereas proteins with red squares are Ras family proteins.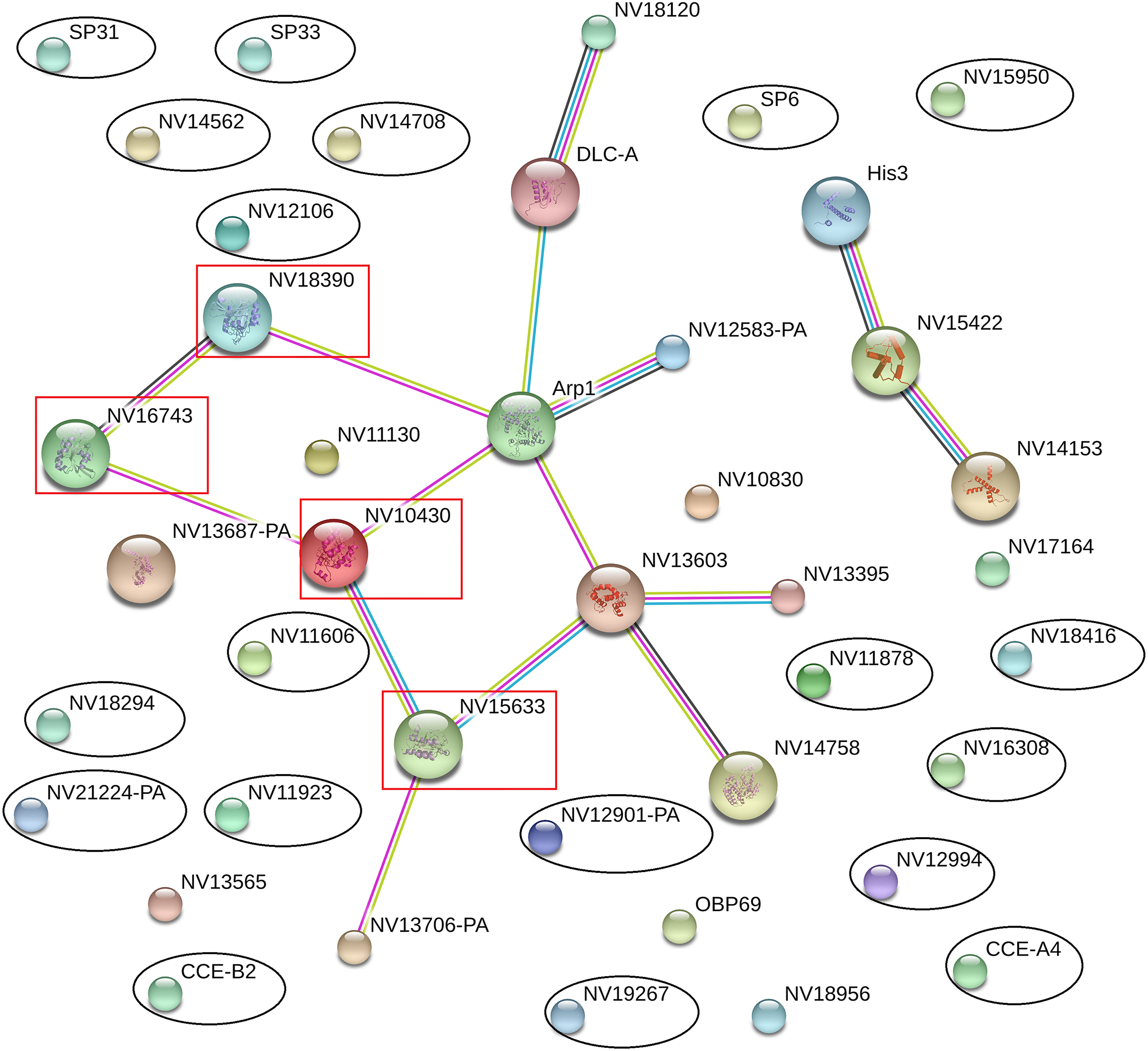{kind=link}
Lipid metabolic pathway
Next, we analyzed the pathways involved for all proteins from Drosophila melanogaster, Aedes aegypti, Solenopsis invicta, Apis mellifera, and N. vitripennis using the KEGG database and filtered out fatty acid metabolism pathways (Table 3). Fatty acid metabolism involves a total of 16 pathways. The proteins involved in the glycerophospholipid metabolism pathway, glycerolipid metabolism, and fatty acid degradation were relatively highly expressed. In N. vitripennis, proteins involved in primary bile acid biosynthesis were more than D. melanogaster, Aedes aegypti, Solenopsis invicta and Apis mellifera, approximately 6–13-fold.
| Metabolic pathway name | Nasonia vitripennis (%) | Drosophila melanogaster | Apis mellifera (%) | Solenopsis invicta (%) | Aedes aegypti |
|---|---|---|---|---|---|
| 00061 Fatty acid biosynthesis | 12(3.34) | 20(3.57) | 8(2.89) | 45(15.85) | 13(2.91) |
| 00062 Fatty acid elongation | 12(3.34) | 22(3.92) | 17(6.14) | 11(3.87) | 29(6.50) |
| 00071 Fatty acid degradation | 26(7.24) | 47(8.38) | 24(8.66) | 24(8.45) | 41(9.19) |
| 00072 Synthesis and degradation of ketone bodies | 5(1.39) | 6(1.07) | 5(1.81) | 6(2.11) | 5(1.12) |
| 00073 Cutin, suberine and wax biosynthesis | 23(6.41) | 23(4.10) | 11(3.97) | 21(7.39) | 31(6.95) |
| 00100 Steroid biosynthesis | 22(6.13) | 18(3.21) | 4(1.44) | 13(4.58) | 30(6.73) |
| 00120 Primary bile acid biosynthesis | 33(9.19) | 5(0.89) | 4(1.44) | 3(1.06) | 3(0.67) |
| 00140 Steroid hormone biosynthesis | 23(6.41) | 37(6.60) | 13(4.69) | 16(5.63) | 41(9.19) |
| 00561 Glycerolipid metabolism | 38(10.58) | 97(17.29) | 38(13.72) | 28(9.86) | 51(11.43) |
| 00564 Glycerophospholipid metabolism | 51(14.21) | 139(24.78) | 59(21.30) | 43(15.14) | 68(15.25) |
| 00565 Ether lipid metabolism | 18(5.01) | 32(5.70) | 17(6.14) | 9(3.17) | 23(5.16) |
| 00600 Sphingolipid metabolism | 24(6.69) | 41(7.31) | 23(8.30) | 19(6.69) | 40(8.97) |
| 00590 Arachidonic acid metabolism | 28(7.80) | 28(4.99) | 16(5.78) | 13(4.58) | 24(5.38) |
| 00591 Linoleic acid metabolism | 10(2.79) | 9(1.60) | 8(2.89) | 6(2.11) | 9(2.02) |
| 00592 alpha-Linolenic acid metabolism | 12(3.34) | 16(2.85) | 10(3.61) | 9(3.17) | 13(2.91) |
| 01040 Biosynthesis of unsaturated fatty acids | 22(6.13) | 21(3.74) | 20(7.22) | 18(6.34) | 25(5.61) |
Notes:
The metabolic pathways were analyzed using the KEGG pathway database. The entire proteomes of D. melanogaster, Solenopsis invicta, and Aedes aegypti were downloaded from UniProtKB, and proteomes of N. vitripennis and Apis mellifera were downloaded from the Hymenoptera Genome Database. The number and the percentage represent the protein number and proportion of the proteins involved in lipid metabolism pathway.
Discussion
In this study, we used a combination of ecological experiments, quantitative proteomics, and bioinformatics data mining to explore the effects of native Wolbachia infection on N. vitripennis. Ecological experiments confirmed the effects of native Wolbachia on the reproduction of N. vitripennis via CI and showed that Wolbachia enhanced the fecundity of infected females. However, there were no significant differences in life span between Wolbchia-infected and Wolbachia-free N. vitripennis. In total, 3,096 proteins from N. vitripennis were quantified by iTRAQ; these proteins were involved in various processes, including binding activity, metabolic processes, cellular processes, and other life processes. Interestingly, the proteomes of native Wolbachia-infected and Wolbachia-free N. vitripennis showed few expression differences in reproduction and immune processes.
Differently expressed proteins involved in some metabolic pathways, for example, alpha-Linolenic acid metabolism, ether lipid metabolism, and carbon metabolism. It should be noted that NV18383 was known as SP33, belonging to serine protease with SP6 and SP31 (Table 2). Serine protease is a type of venom proteins, the venom glands of parasitoid wasps build an environment conducive to survival and development by influencing the arthropod host’s physiology (de Graaf et al., 2010). SP6 and SP31 were trypsin, which yielded the enzymes responsible for vital processes such as digestion, blood coagulation, fibrinolysis, development, fertilization, apoptosis, and immunity (Page & Di Cera, 2008). NV12994 was known as cytochrome b5-like, and Takamiya et al. (2016) confirmed the hypothesis that cytochrome b5 played vital roles in parasitic adaptation, together with oxygen-avid haemoglobin. Using BLAST, we found uncharacterized protein LOC1003315494 and LOC100678792 may be venom proteins as the homologous genes code venom proteins in Trichomalopsis sarcophagae (Martinson et al., 2017).
The immune system of insects is sensitive to the presence of symbionts (Gross et al., 2009) and plays vital roles in maintaining the host balance. Innate immunity has been extensively studied in D. melanogaster using expression-based methodologies, such as RNA-sequencing. One of the important biological consequences of pathogenic infection is the rapid induction of several classes of effector proteins, along with upregulation of a number of other pathway components (Apidianakis et al., 2005; De Gregorio et al., 2001, 2002; Pal, Wu & Wu, 2008; Vodovar et al., 2005). Analysis of RNA transcripts in Nasonia showed that infection of Serratia marsecens and Enterococcus faecalis induced upregulation of 5.6% of immune genes, compared with 1.2% of nonimmune genes, and these effects were particularly obvious for some antimicrobial peptides and recognition genes (Sackton, Werren & Clark, 2013). The homology-based Nasonia immune catalogue was updated by characterization of many new immune-inducible genes, some of which were taxonomically restricted to either the wasp lineage or to Hymenoptera as a whole (Sackton, Werren & Clark, 2013). In contrast, our proteomics analysis showed that natural infection by Wolbachia did not induce significant up- or downregulation of any immune-related proteins. We only found 13 proteins with immune-related functions in our data. Compared with the transcriptome of the homology-based Nasonia immune catalogue, three immune-related proteins (encoded by NV10430, NV14758, and NV17164) were found in the catalogue, but were not significantly regulated by short-term infection with Serratia marsecens or E. faecalis.
Transfection of Wolbachia has caused concerns in mosquitos because this endosymbiont may be used for control of Dengue virus (Ruang-Areerate & Kittayapong, 2006; Schnettler et al., 2016; Walker et al., 2011; Zhang, Hussain & Asgari, 2014). Short-term infection with Wolbachia reduces reproductive regulation in insect hosts, such as CI, male-killing, and sperm modification (Blagrove et al., 2013; Jeffries & Walker, 2016; McGraw et al., 2001). Studies have shown that short-term infection with Wolbachia significantly affects the expression of immune genes, including those involved in the Toll and IMD immune pathways, as well as a number of antimicrobial genes (Bian et al., 2013; Pan et al., 2012; Rances et al., 2012). In contrast, in long-term native Wolbachia infection, induction of Toll-like receptors or IMD immune-related genes is not observed in Aedes fluviatilis mosquitos (Caragata et al., 2017). Our proteomics analysis of N. vitripennis also showed that no immune proteins were significantly differentially expressed following Wolbachia infection. We speculate that insect hosts may have adapted to infection by endosymbiotic Wolbachia through long-term intergrowth by adjusting its physiological and biochemical activities in vivo. As proposed by Zug, a recently acquired infection is likely to trigger immune responses as a key resistance mechanism, whereas in co-evolved associations, resistance may no longer be the best response to infection (Zug & Hammerstein, 2015b). The absence of systemic immune activation is not rare among native Wolbachia infections and may be symptomatic of increased tolerance on the part of the host and reduced pathogenicity on the part of the symbiont (Bourtzis, Pettigrew & O’Neill, 2000; Caragata et al., 2017). This could explain our proteomic results to some extent. Notably, stable infection with Wolbachia in D. melanogaster has been shown to yield different results. Gene expression analysis of the testes dissected from third instar larvae of both Wolbachia-infected and Wolbachia-free D. melanogaster showed that a total of 296 genes were significantly identified by microarray analysis. Differential expression of genes related to metabolism, immunity, reproduction, and other functions was observed (Zheng et al., 2011). Moreover, proteomics analysis showed that 83 proteins, including 14 and five proteins involved in reproduction and immunity, respectively, were significantly differentially expressed between one-day-old Wolbachia-infected and one-day-old Wolbachia-free D. melanogaster (Yuan et al., 2015). We used local BLASTp to search those 83 significantly differentially expressed proteins in our N. vitripennis proteomics data and found 10 proteins with more than 70% identity (E score < 10−5), as shown in Table S5. In particular, three homologous proteins (NCBI ID: 299782477, 156537767, 645012252) were involved in reproduction, and protein 156537767 was also involved in immunity. However, these three proteins did not show significantly differential expression in our proteomics analysis of native Wolbachia-infected and Wolbachia-free N. vitripennis.
As a parasitoid, N. vitripennis is a carnivore, feeding on an amino acid-rich diet both as larva and adults (Werren & Loehlin, 2009a). The Nasonia genome revealed loss or rearrangement of some amino acid metabolic pathways, including tryptophan, and aminosugar metabolism, which may reflect its specialized carnivorous diet (Werren et al., 2010). We hypothesized that Nasonia may have more lipid metabolism proteins; thus, we compared the numbers of lipid metabolism proteins from D. melanogaster, Aedes aegypti, Solenopsis invicta, Apis mellifera, and N. vitripennis (Table 3). To our surprise, with the exception of more proteins involved in primary bile acid biosynthesis, Nasonia did not exhibit changes in lipid metabolism genes compared with four other insect species. Our proteomics analysis of Nasonia, as a model insect, provided important insights that will facilitate the elucidation of various metabolic and immune-related mechanisms.
The interactions between insect hosts and symbionts are being extensively studied, particularly with regard to unknown molecular mechanisms. Our current findings provided novel insights into the complex interactions between Nasonia and native Wolbachia infection. Based on our findings, we propose the gradual adaptation of Wolbachia in insects, although some phenotype is obvious. With long-term coexistence, both insect hosts and symbionts may adapt to each other. More studies are needed to verify this hypothesis.
Conclusions
In this study, we evaluated the proteomics of N. vitripennis and the effects of native infection by the widespread endosymbiont Wolbachia using iTRAQ-LC-MS/MS. In total, 3,096 proteins were identified from native Wolbachia-infected and antibiotic-treated Wolbachia-free N. vitripennis samples, including a wide range of proteins involved in BPs, MFs, and CCs. Interestingly, although the phenotype was obvious, there were few significant changes in immune or reproductive proteins between samples with different Wolbachia infection statuses, and most differentially expressed proteins belonged to categories of binding processes, catalytic activity, and metabolic processes. Furthermore, there were no direct correlations in differential expression between immune and reproductive proteins. Our findings provided valuable insights into the mechanisms underlying the interactions between insect hosts and endosymbionts.
Supplemental Information
{kind=link}
Fig. S2. Enriched KEGG pathway analysis of proteins quantified from N. vitripennis proteomics by iTRAQ.
The top 20 pathways are shown.
{kind=link}
Table S3. Details of gene ontology classifications for proteins identified from Wolbachia-infected and Wolbachia-free N. vitripennis proteomics.
Table S5. Proteins with more than 70% identity analysed by local BLASTp.
W+/W-, the protein abundance ratio of Wolbachia-infected N. vitripennis compared to Wolbachia-free N. vitripennis. Identities were analysed by local BLASTp (E-value < 10-5).