
Loss of CclA, required for histone 3 lysine 4 methylation, decreases growth but increases secondary metabolite production in Aspergillus fumigatus
- DOI
- 10.7717/peerj.4
- Published
- Accepted
- Received
- Academic Editor
- Dee Carter
- Subject Areas
- Microbiology, Molecular Biology, Mycology
- Keywords
- COMPASS complex, Gliotoxin, Histone methylation, Secondary metabolism, Growth and development, 6-azauracil sensitivity, Aspergillus fumigatus
- Copyright
- © 2013 Palmer et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
- Cite this article
- 2013. Loss of CclA, required for histone 3 lysine 4 methylation, decreases growth but increases secondary metabolite production in Aspergillus fumigatus. PeerJ 1:e4 https://doi.org/10.7717/peerj.4
Abstract
Secondary metabolite (SM) production in filamentous fungi is mechanistically associated with chromatin remodeling of specific SM clusters. One locus recently shown to be involved in SM suppression in Aspergillus nidulans was CclA, a member of the histone 3 lysine 4 methylating COMPASS complex. Here we examine loss of CclA and a putative H3K4 demethylase, HdmA, in the human pathogen Aspergillus fumigatus. Although deletion of hdmA showed no phenotype under the conditions tested, the cclA deletant was deficient in tri- and di-methylation of H3K4 and yielded a slowly growing strain that was rich in the production of several SMs, including gliotoxin. Similar to deletion of other chromatin modifying enzymes, ΔcclA was sensitive to 6-azauracil indicating a defect in transcriptional elongation. Despite the poor growth, the ΔcclA mutant had wild-type pathogenicity in a murine model and the Toll-deficient Drosophila model of invasive aspergillosis. These data indicate that tri- and di-methylation of H3K4 is involved in the regulation of several secondary metabolites in A. fumigatus, however does not contribute to pathogenicity under the conditions tested.
Introduction
Aspergillus fumigatus is a saprophytic filamentous fungus commonly found in diverse environments throughout the world. Whereas this fungus appears to have always been associated with aspergillomas since such structures were recognized by medical science in the 1800s, the incidence of invasive aspergillosis (IA) is a more recent phenomenon directly correlated to the increase in immune-compromised patients originally treated for other diseases. Why A. fumigatus predominates as the causative agent of IA instead of the 200 or so other Aspergilli is a question of considerable interest as the answers should help in devising strategies to control this often fatal disease.
A. fumigatus possesses several attributes that are thought to contribute to its predominance in IA and other Aspergillus-associated diseases; this topic has been the subject of several recent reviews (Dagenais & Keller, 2009; Abad et al., 2010; Mccormick, Loeffler & Ebel, 2010). The reviews indicate that this species may not possess unique classical virulence factors, but rather orchestrates the expression of conserved fungal characteristics in such a way as to evade or overcome host defenses more so than other Aspergilli. There are, however, at least two attributes that have been associated with A. fumigatus virulence that vary considerably among Aspergillus species: its remarkable thermo-tolerance and its unique repertoire of toxic secondary metabolites (SM) (Dagenais & Keller, 2009; Abad et al., 2010).
Many studies dating back almost 30 years have implicated several toxins as important in host – Aspergillus interactions (Mullbacher, Waring & Eichner, 1985; Eichner et al., 1986; Sutton et al., 1994; Amitani et al., 1995; Khoufache et al., 2007; Fallon, Reeves & Kavanagh, 2011). In recent years, individual genes required for SM production have been removed from the A. fumigatus genome and the resultant deletants tested for virulence in various animal models (D’enfert et al., 1996; Reeves et al., 2006; Ben-Ami et al., 2009). Through these studies it was discovered that a global regulator of SM production, LaeA, was shown to be important in IA development (Bok et al., 2005; Sugui et al., 2007a). Furthermore, the LaeA regulated SM gliotoxin contributes to A. fumigatus virulence in several animal models of IA (Reeves et al., 2004; Stanzani et al., 2005; Cramer et al., 2006; Coméra et al., 2007; Sugui et al., 2007b; Ben-Ami et al., 2009; Lionakis & Kontoyiannis, 2010). Since then, global regulation of SM gene clusters has been found to be regulated in part through global changes in histone modifications, particularly acetylation and methylation of histone 3 tail residues (Bok et al., 2005) and reviewed in Strauss & Reyes-Domínguez (2011). These modifications have been linked to LaeA activity (Reyes-Dominguez et al., 2010). A recent report in A. nidulans showed that CclA, a member of the conserved eukaryotic COMPASS complex that methylates histone 3 lysine 4 (H3K4), was also critical in the regulation of several SMs (Bok et al., 2009). H3K4 can have up to five different modifications: acetylated, unmodified, mono-methylated, di-methylated, and tri-methylated. Histone tail modifications were once thought to be permanent, however more recent data has indicated that these modifications are dynamic, i.e. they are reversible. For example, while the COMPASS complex methylates H3K4 there is a reciprocal enzyme that demethylates H3K4. In mammalian systems, a flavin containing amine oxidase named LSD1 (lysine specific demethylase 1) is responsible for demethylation of H3K4 (Shi et al., 2004).
Based on our previous research in the model fungus A. nidulans, we identified the A. fumigatus cclA and LSD1 homologs from the genome sequence. To investigate a possible role of H3K4 methylation in SM cluster regulation and pathogenicity in A. fumigatus, we deleted both cclA and the putative LSD1 homolog, which we named hdmA (histone demethylase A). Whereas the hdmA deletant was indistinguishable from wild type, the cclA deletant, similar to the phenotype of the A. nidulans ΔcclA mutant, was deficient in tri- and di-methylation of H3K4 and exhibited slow growth but increased activation and expression of several A. fumigatus SMs, including gliotoxin. The mutant showed wild type pathogenicity in both a neutropenic murine model and Toll-deficient Drosophila model of invasive aspergillosis. These data suggest that tri- and di-methylation of H3K4 is not required for pathogenicity under the conditions tested and raises the possibility that pathogenicity of a slow growing mutant strain may be compensated by enhanced gliotoxin synthesis.
Materials and Methods
Strains, growth conditions, and DNA manipulations
All A. fumigatus strains used in this study are derivatives of the clinical isolate AF293 (Xue et al., 2004) and are listed in Table 1. Strains were maintained on glucose minimal media (GMM) (Shimizu & Keller, 2001) and when appropriate the media was supplemented with 5 mM uracil and 5 mM uridine for pyrG1 strains, 5 mM arginine for argB1 strains, and hygromycin was used at a final concentration of 100 ug/ml. Radial growth experiments and 6-azauracil sensitivity assays were carried out essentially as in Palmer et al. (2008). All DNA manipulations were conducted according to standard protocols (Sambrook & Russell, 2001) and fungal transformation was done as previously described (Miller, Miller & Timberlake, 1985) with a minor modification of protoplasts being plated in 0.75% molten top agar. Primers used in this study are listed in Table 2.
| Strain | Genotype | Source |
|---|---|---|
| AF293 | Wild type | (Xue et al., 2004) |
| AF293.1 | pyrG1 | (Xue et al., 2004) |
| AF293.6 | pyrG1, argB1 | (Xue et al., 2004) |
| TJW84.1 | pyrG1, ΔcclA::A. parasiticus pyrG | This study |
| TJW88.21 | pyrG1, ΔcclA::A. parsasiticus pyrG, cclA::hygromycin | This study |
| TJMP2.36 | pyrG1, argB1, ΔhdmA::A. parasiticus pyrG | This study |
| TJMP3.52 | pyrG1, argB1, ΔhdmA::A. parasiiticus pyrG, A. fumigatus argB | This study |
| TJMP29.1 | pyrG1, argB1, ΔhdmA::A. parasiticus pyrG, hdmA:A. fumigatus argB | This study |
| Name | Sequence (5′ to 3′): Restriction sites underlined | Purpose |
|---|---|---|
| Fbre5F | ATATCAGTTGTTGCTCCTAGGGC | 5′ Flank ΔcclA |
| Fbre5R-EcoRI | CATGTGGAATTCAATAACGGTTCACGAGTAAATTG | 5′ Flank ΔcclA |
| Fbre3F-HindIII | TGGGGTAAGCTTGTTCCGAGCCATATCTGTC | 3′ Flank ΔcclA |
| Fbre3R | ACAACAAAACTAGCTCTCTCGGC | 3′ Flank ΔcclA |
| FbreCOMF-NotI | TTGCTCCAGCGGCCGCATCCTCAAACCTCGCCAAGTATG | cclA complementation |
| FbreCOMR-SpeI | GGAAATACTAGTGAAGCTGAAAGTGACGTCTAGAA | cclA complementation |
| JP1 | TATAGGGACGAATTCACAGACAATG | 5′ Flank ΔhdmA |
| JP2 | CGCTGAATTCGGGTTTGATGGACATTGGAC | 5′ Flank ΔhdmA |
| JP3 | CCGGTCTAGAGGTAATGCTTAGACTCCCGTA | 3′ Flank ΔhdmA |
| JP4 | GTCATGCGGCCGCCACTGCCCTCGTTAAGG | 3′ Flank ΔhdmA |
| JP lsdA comp For | GCTTGGATCCGACGTAGCAGGTGAAC | hdmA complementation |
| JP lsdA comp Rev | CATTGGATCCTCCTCGCCTTTCTCC | hdmA complementation |
| JP Afumi argB For | GAACGCGGTCTGCATCCAAG | Af-argB cloning |
| JP Afumi argB Rev | GAAGGAGAGACCCATACATCC | Af-argB cloning |
| FumlaeAF | ATGTTTCTCAACGGGCAGGGC | laeA probe |
| FumlaeAR | ATTGGCGAGAGGTTTTCGAGCC | laeA probe |
| FbreIF | CTTTCCACACATCAAGTACCGGC | cclA probe |
| FbreIR | ATTGAAGCGTTCGCCAATGCC | cclA probe |
| JP LSD1 | TCTCACGCAACTACATACGTCAAC | hdmA probe |
| JP LSD2 | TTTTTCCTCTTCGCTGGCTTGCC | hdmA probe |
| GZINTF | AAGGGCCGGTAGTCTACCTCTTC | gliZ probe |
| GZINTF | CGATCTGGTAGCTGCCCAGCTGGAAG | gliZ probe |
Construction of mutant A. fumigatus strains
An A. fumigatus cclA disruption cassette was constructed using double-joint PCR consisting of the following: 1-kb DNA fragment upstream of the cclA start codon (primers Fbre5F and Fbre5R-EcoRI), a 3-kb EcoRI-HindIII DNA fragment of A. parasisitus pyrG via pJW24 (Calvo et al., 2004), and a 1-kb DNA fragment downstream of the cclA stop codon (primers Fbre3F and Fbre3R-HindIII). The fusion PCR product was then used to transform AF293.1 to prototrophy creating TJW84.1. Homologous single-gene replacement of cclA was confirmed by Southern analysis (Figs. 1A and 1B). In order to complement the ΔcclA mutant, a complementation plasmid (pJW109.5) was constructed. The plasmid contained a 3.8-kb wild-type cclA gene including a 1-kb native promoter and a 1-kb native termination cassette, which was amplified by primers FbreCOMF-NotI and FbreCOMR-SpeI. The PCR product was cloned into a NotI-SpeI site of pUCH2-8 (Alexander, Hohn & Mccormick, 1998), which contains the selectable marker hygromycin B phosphotransferase. TJW84.1 was transformed with pJW109.5 to hygromycin resistance and Southern blot confirmed single copy integration for TJW88.21 (Fig. 1B).
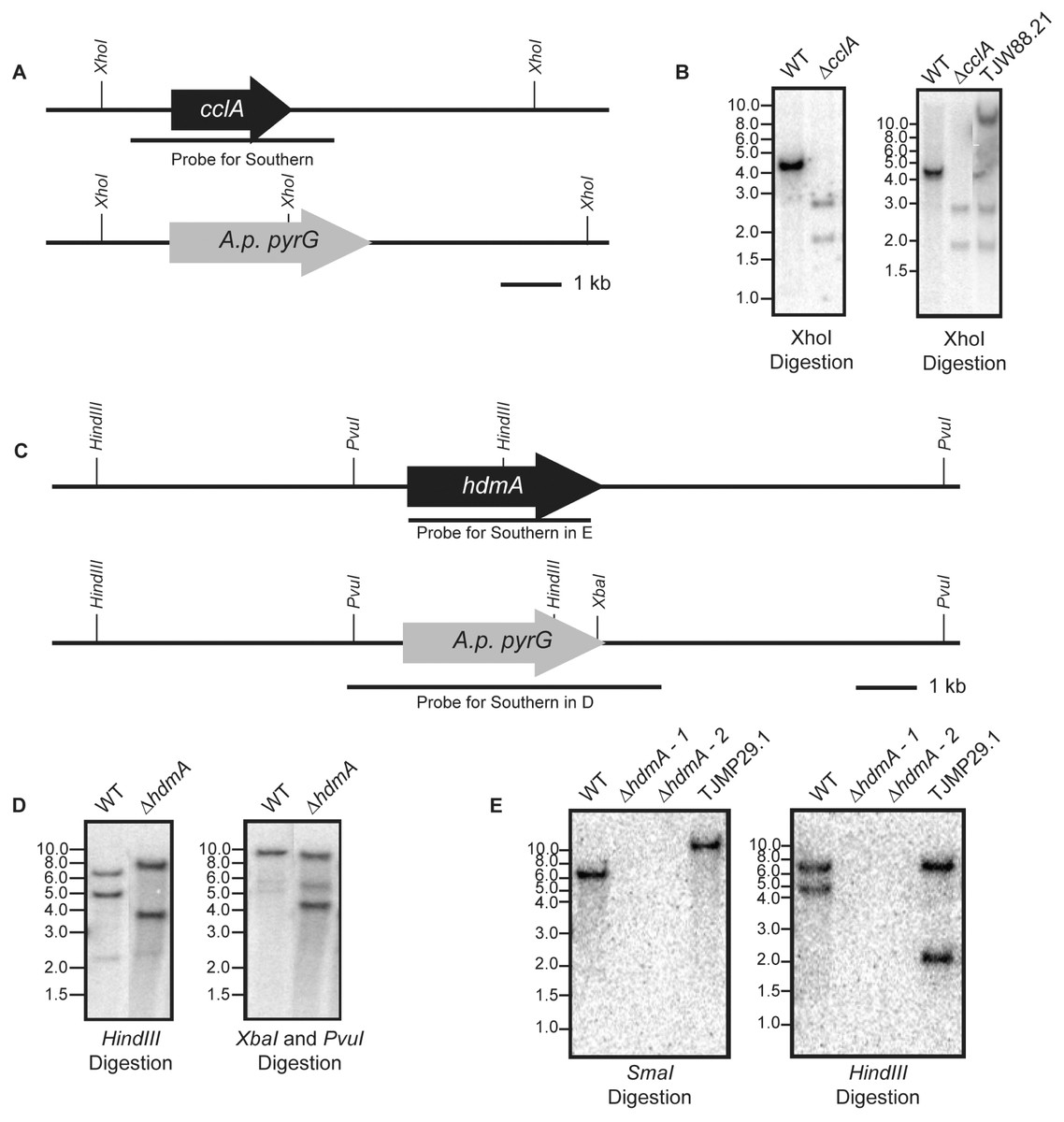
Figure 1: Construction of deletion and subsequent complementation strains of cclA and hdmA in A. fumigatus.
(A) A schematic drawn to scale illustrates the replacement of the cclA ORF with the A. parasiticus pyrG gene and the location of XhoI restriction enzyme sites. (B) A radiolabeled probe consisting of the cclA ORF and ∼1 kb on either side was used to probe a Southern blot of XhoI digested genomic DNA. The expected banding pattern was observed. Confirmation of a complemented cclA strain (TJW88.21) was achieved via Southern blot using the described conditions above. An additional band is shown that is greater than 10 kb that is attributed to the random integration of the cclA complementing plasmid. (C) Schematic drawn to scale of the restriction enzyme cut sites in the wild type and ΔhdmA backgrounds. (D) Southern blot using a HindIII digestion and a XbaI–PvuI double digestion using a radiolabeled probe consisting of the gene deletion construct shows the expected size differences between wild type and the ΔhdmA strains. (E) Complementation of the ΔhdmA mutant was confirmed using a SmaI and subsequent HindIII digestion to determine single integration of the complementing plasmid (TJMP29.1).{kind=link}
An hdmA disruption cassette was constructed by PCR amplifying a 1.1-kb DNA fragment upstream of the hdmA ORF using the primer pair JP1 and JP2 containing EcoRI restriction sites. The subsequent 1.1-kb EcoRI-EcoRI fragment was cloned into pJW24 to create pJMP1. A downstream 1.0-kb fragment was amplified using the primer pair JP3 and JP4 containing XbaI and NotI restriction sites respectively. The 1.0-kb XbaI–NotI fragment was cloned into pJMP1 to create the hdmA disruption cassette named pJMP2. The hdmA ORF was disrupted by transformation of linearized pJMP2 into AF293.6 creating the ΔhdmA argB1 auxotrophic strain (TJMP2.36), which was confirmed by Southern blot (Figs. 1C and 1D). In order to generate a prototrophic deletion strain, an argB complementation plasmid was constructed by amplifying a 2.6-kb fragment corresponding to A. fumigatus argB using the primer pair ‘JP Afumi argB For’ and ‘JP Afumi argB Rev’, which was subsequently cloned into pCR2.1 TOPO (Invitrogen) to create pJMP4. This plasmid was transformed into TJMP2.36 to generate a prototrophic ΔhdmA strain (TJMP3.52). For complementation of ΔhdmA, a 4.6-kb fragment corresponding to ∼0.9-kb up stream and ∼0.5-kb downstream of the hdmA ORF was amplified with the primer pair ‘JP lsdA comp For’ and ‘JP lsdA comp Rev’ each containing a BamHI site. The subsequent BamHI-BamHI fragment was cloned into pJMP4 to create pJMP13. TJMP2.36 was transformed with pJMP13 to create a complemented control strain TJMP29.1 (ΔhdmA + hdmA) and single integration was confirmed by Southern blot (Fig. 1E).
Physiology experiments
Radial growth measurements and sensitivity to 6-azauracil (6AU) was conducted as described previously (Palmer et al., 2008). Four replicates were used for each assay and statistical significance was calculated with ANOVA analysis using Prism 5 software (Graph Pad).
Alterations in H3K4 methylation
Nuclear extracts were isolated as previously described (Palmer et al., 2008). Approximately 50 µg of nuclear protein extract was electrophoresed on a 10% Tricine-SDS-PAGE gel (Schägger, 2006) and subsequently electroblotted to nitrocellulose membranes. Detection of H3K4 modifications was conducted using the following primary antibodies and dilutions: 1:1,000 anti- histone 3 (Upstate, #07-690), 1:1,000 anti-H3K4 mono-methylation (Upstate, #07-436), 1:2,000 anti-H3K4 di-methylation (Upstate, #07-030), 1:2,000 anti-H3K4 tri-methylation (Upstate, #07-473). Chemiluminescent detection was employed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and a secondary goat anti-rabbit-horseradish peroxidase conjugate antibody (Pierce, #31460) diluted 1:15,0000 (Fig. 2).
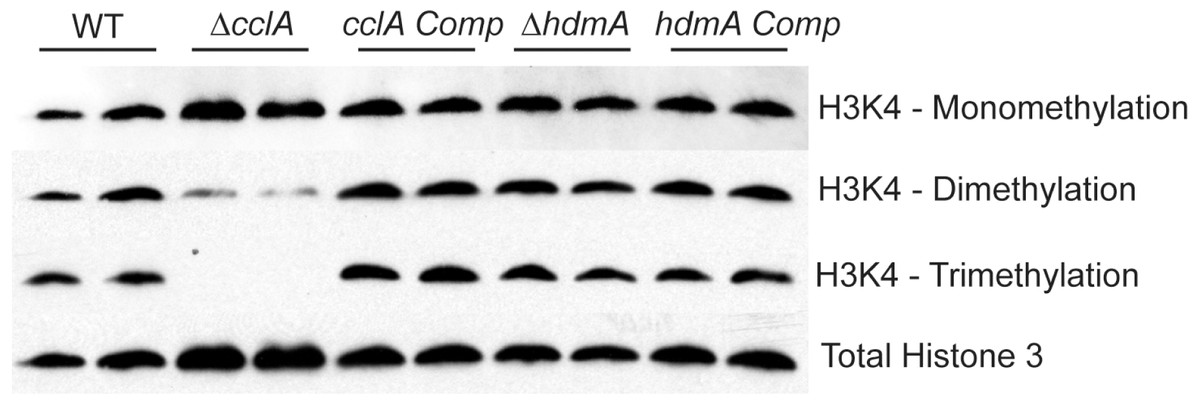
Figure 2: Deletion of cclA results in a reduction in di-methylation and loss of tri-methylation of lysine 4 of histone 3 (H3K4).
Western blot of nuclear protein extracts clearly indicate that cclA is required for proper methylation of H3K4. While H3K4 mono-methylation is unaffected by loss of cclA, di-methylation is reduced and tri-methylation was absent. These data are consistent with S. cerevisiae BRE2 null mutants. No difference in global methylation patterns of H3K4 was detected in the ΔhdmA strain compared to wild type.{kind=link}
Northern analysis and secondary metabolite extraction
Fifty milliliter cultures of liquid GMM + 0.1% yeast extract were inoculated with 1 × 107 spores per ml and incubated at 250 rpm 29 °C for 24 h, followed by a reduction in temperature to 25 °C for an additional 48 h. Mycelia were harvested, lyophilized overnight, and total RNA was extracted using Isol-RNA Lysis Reagent (5 Prime) according to manufacturers recommendations. Subsequent northern analysis was done using radiolabeled probes for the corresponding transcript (Fig. 4C) (primers are listed in Table 2). Secondary metabolites were extracted from 20 ml of culture filtrate with an equal volume of chloroform. The air-dried chloroform layer was resuspended in 75 µl chloroform and 5 µl was separated on a thin layer chromatography plate (Whatman, #4410 222) using chloroform: acetone (7:3) as a solvent. The TLC plate was then dried and imaged under 254 nm and 366 nm ultraviolet light (Fig. 4A). Organic extracts from solid minimal medium cultures was accomplished by taking equal sized cores from the center of point-inoculated cultures, homogenized in 3 ml of water, and extracted with an equal volume of chloroform. Quantification of TLC spots was achieved through densitometry analysis using Image J and normalized to wild type levels.
Pathogenicity assays
The neutropenic murine model of aspergillosis was conducted as described previously (Bok et al., 2005) with approval of the William S. Middleton VA and the University of Wisconsin Animal Care Committees in accordance with the MV2344 animal use protocol. Toll-deficient flies were generated by crossing flies carrying a thermosensitive allele of Toll (Tl r632) with flies carrying a null allele of Toll (Tl I-RXA) (Lionakis et al., 2005). Two- to four day old adult female Toll-deficient flies were used in all of the experiments. Twenty flies were infected with each A. fumigatus strain used in this study. All of the experiments were performed in triplicate. To minimize circadian rhythm variability, all experiments were performed at 9 AM. A. fumigatus isolates were grown on yeast extract agar glucose (YAG) at 37 °C. Conidia were collected in sterile 0.9% saline from 2 days old cultures. The conidial concentration suspension was determined by using a hemacytometer and adjusted to 1 × 107 per ml. The dorsal side of the thorax of twenty CO2 anesthetized flies was punctured with a thin (10 µm) sterile needle that had been dipped in a concentrated solution of A. fumigatus conidia (107 cells/ml). In our previous work, this method was shown to deliver a reproducible inoculum of 700 to 800 conidia (Lionakis et al., 2005). As a negative control group, Toll-deficient flies were punctured with a 10 µm sterile needle and monitored daily for survival. Flies that died within 3 h of the injection were considered to have died as a result of the puncture procedure and were not included in the survival rate analysis. The flies were housed in a 29 °C incubator to maximize expression of the Tl r632 phenotype (Lemaitre et al., 1996). The Toll-deficient flies were transferred into fresh vials every 3 days. Fly survival was assessed daily over 8 days.
Results
Deletion of cclA but not hdmA affects global histone 3 lysine 4 methylation patterns
Aspergillus fumigatus cclA was identified through BLASTp analysis of the A. fumigatus genome with the A. nidulans CclA amino acid sequence (Bok et al., 2009), which yielded one homolog Afu3g04120. The human LSD1 protein sequence (Genbank, #O60341) was used to BLASTp the A. fumigatus genome sequence, which provided one potential ortholog, Afu4g13000. Afu4g13000 is 29% identical to human LSD1 and contains the conserved SWIRM domain, a flavin amine oxidase domain, as well as an HMG Box domain (Shi et al., 2004) and thus was named hdmA for histone demethylase A.
The cclA and hdmA alleles were independently disrupted in the A. fumigatus AF293 genetic background. Southern analysis of transformants for each gene confirmed simple gene replacements (Fig. 1). One representative deletant, TJW84.1 for ΔcclA and TJMP3.52 for ΔhdmA, was chosen for each gene replacement for the studies presented below. Both deletants were complemented with their respective genes to generate a single copy cclA + ΔcclA strain and a single copy hdmA + ΔhdmA strain as shown in Table 1.
Our first experiment was to assess if loss of either gene affected histone 3 lysine 4 methylation (H3K4). The ΔcclA strain showed a clear loss of whole genome H3K4 tri-methylation and a considerable decrease in di-methylation (Fig. 2); this tallied with earlier studies in our lab assessing H3K4 methylation in an A. nidulans ΔcclA mutant (Bok et al., 2009). These data are also consistent with BRE2 mutants in Saccharomyces cerevisiae, where loss of BRE2 results in strains with diminished di-methylation and no tri-methylation of H3K4 (Schneider et al., 2005). The complemented strain was restored in H3K4 methylation to wild type levels. However, loss of hdmA did not exhibit a detectable impact on whole genome H3K4 methylation (Fig. 2).
CclA mutants are crippled in growth and sensitive to 6-Azauracil
As reported for the A. nidulans ΔcclA mutant (Giles et al., 2011), the A. fumigatus ΔcclA strain exhibited poor growth as exhibited by decreased radial growth on several different media, including rich media (YPD and Champs) as well as our standard minimal medium (GMM) (Fig. 3A). Additionally, the ΔcclA mutant had a measurable growth defect at all temperatures tested (Fig. 3B) and decreased mass when grown in liquid shaking culture (data not shown). This defect was rescued in the complemented strains. Neither the hdmA deletant nor its complement exhibited any growth phenotype.

Figure 3: Null mutants of cclA result in reduced growth and increased sensitivity to 6-azauracil.
(A) The ΔcclA strain is defective in radial growth on rich media (YPD, Champs) as well as minimal media (GMM) using both nitrate and ammonium as a nitrogen source. The complemented control strain restores wild-type levels of growth. (B) Quantification of radial growth on solid media at three different temperatures (25 °C, 29 °C, and 37 °C) illustrates a significant growth reduction in the ΔcclA strain in all conditions tested. No phenotype was observed for a mutant lacking hdmA. (C) Radial growth assays conducted on GMM amended with 100 ug/ml of 6-azauracil indicated that null mutants of cclA are more sensitive to 6-azauracil than wild type. The cclA complemented control strain restores sensitivity to 6-azauracil to wild type levels. Asterisk indicates p < 0.001 using an ANOVA test for statistical significance with Prism 5 software.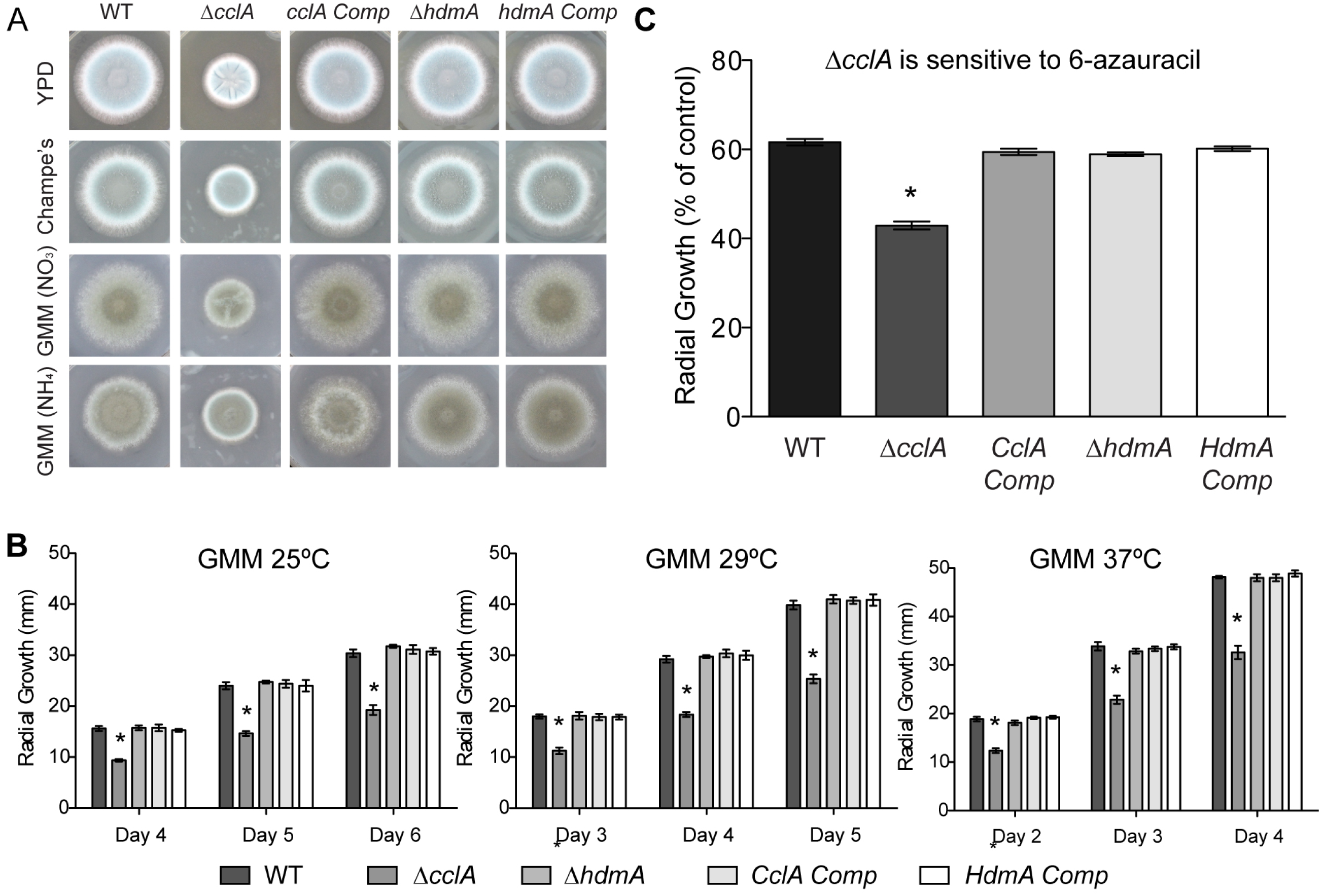{kind=link}
The phenotype of the ΔcclA strain shared some similarities to that of a previously described A. fumigatus chromatin mutant, ΔclrD (Palmer et al., 2008), impaired in methylation of H3K9. As histone methyltransferase mutants are often found to be sensitive to chemicals that target various cellular processes, the ΔclrD mutant had been assessed for sensitivity to various chemical agents and found to be sensitive to 6-azauracil (6AU); an inhibitor of guanine nucleotide synthesis and indicator of transcriptional defects (Riles et al., 2004; Zhang et al., 2005; Palmer et al., 2008). Following that protocol, the ΔcclA and ΔhdmA strains were similarly screened and the former but not the latter found to be also sensitive to 6AU (Fig. 3C). Resistance to 6AU was rescued in the complement strain. Moreover, we did not find differences in sensitivity of ΔhdmA or ΔcclA strains compared to wild type when exposed to several chemical agents that included hydroxyurea (DNA synthesis inhibitor), thiabendazole (microtubule destabilizer), 1.2 M Sorbitol (osmotic stress), 0.6 M KCl (osmotic stress), Congo red (cell wall inhibitor), or Calcofluor white (cell wall inhibitor) (data not shown).
CclA loss results in increased secondary metabolism
Mutations in chromatin remodeling genes have frequently been associated with alterations in secondary metabolite production, both in A. nidulans (Shwab et al., 2007; Bok et al., 2009) and A. fumigatus (Lee et al., 2009). In particular the A. nidulans ΔcclA strain was found to produce novel metabolites with antimicrobial properties (Bok et al., 2009; Giles et al., 2011). An examination of extracts from three different conditions of the two mutants showed that the A. fumigatus ΔcclA strain but not the ΔhdmA strain was increased in the production of several metabolites (Fig. 4). In particular, this strain produced over 4 times as much gliotoxin compared to wild type when grown in liquid shaking culture (Figs. 4A and 4B). The high level of gliotoxin was associated with a large increase in transcription of gliZ, the C6 transcription factor required for gli expression (Fig. 4C). Under these growth conditions, loss of cclA also resulted in several fold increases of numerous other metabolites (Figs. 4A and 4B). The ΔcclA mutant was also analyzed under alternative growth conditions and produced similar amounts of metabolites on solid medium at 29 °C compared to wild type (Fig. 4D), however ΔcclA mutants produce more of a few unidentified metabolites when grown 37 °C on solid medium (Fig. 4D).
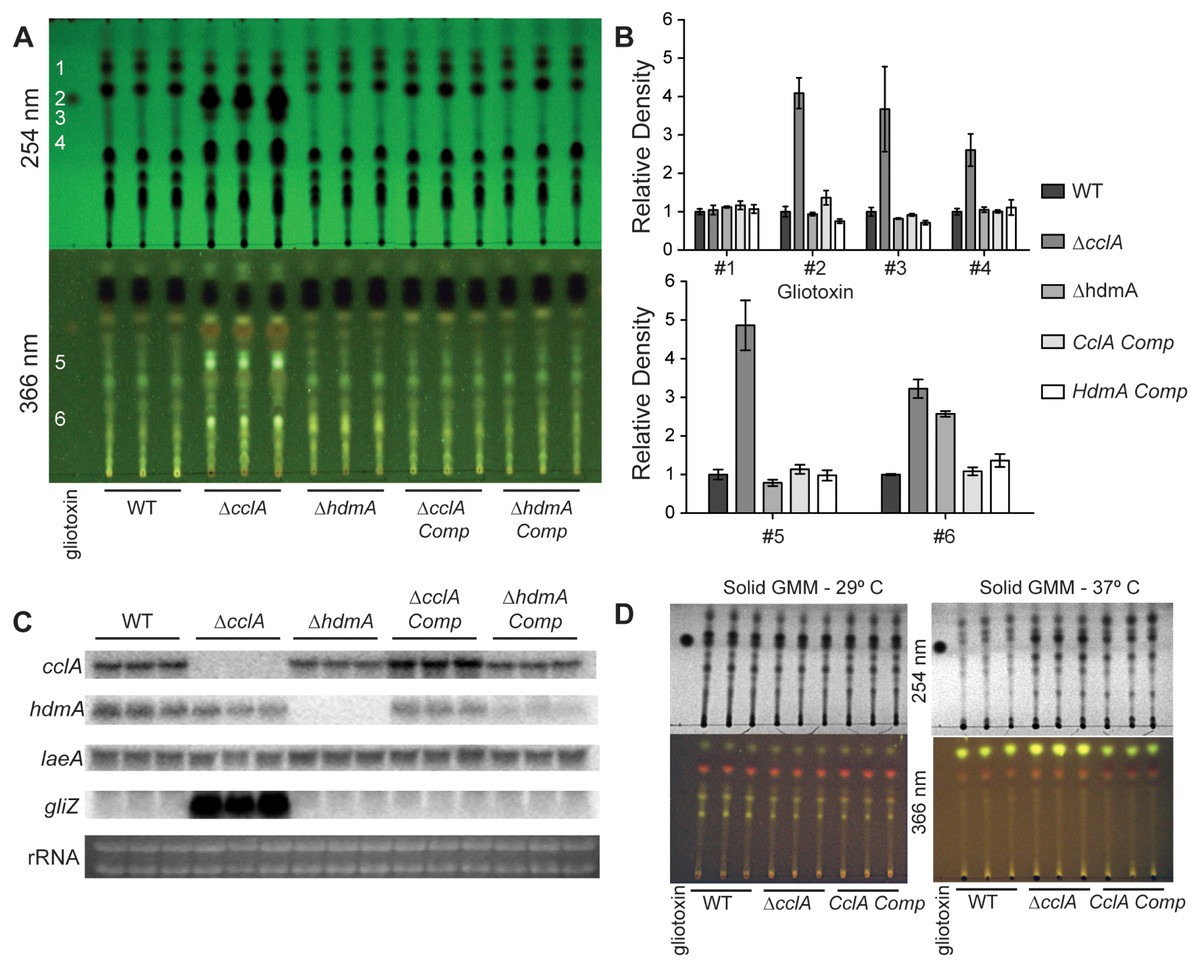
Figure 4: CclA, but not HdmA, controls production of several secondary metabolites, including gliotoxin.
(A) Strains were grown in liquid minimal media shaking culture and analyzed for production of secondary metabolites by thin layer chromatography (TLC) using gliotoxin as a standard. UV infused TLC plates were photographed under 254 nm and 366 nm light. (B) Quantification of TLC spots was achieved by densitometry with Image J software and relative density was normalized to wild type extracts, which reveals that null mutants of cclA produced more than 4 times more gliotoxin as well as several fold more production of several unidentified metabolites. (C) Northern analysis from the same growth conditions confirms the strains as well as identifies a large increase in gliZ transcript in the ΔcclA mutant background compared to wild type. There are no differences in transcription of the regulator of secondary metabolism laeA. (D) Organic extracts were prepared from cultures grow on solid media and subsequently analyzed via TLC analysis using chloroform:acetone (7:3) as a solvent. Growth at 29 °C results in no detectable difference between ΔcclA and wild type, however at 37 °C a few metabolites are slightly increased in ΔcclA compared to wild type.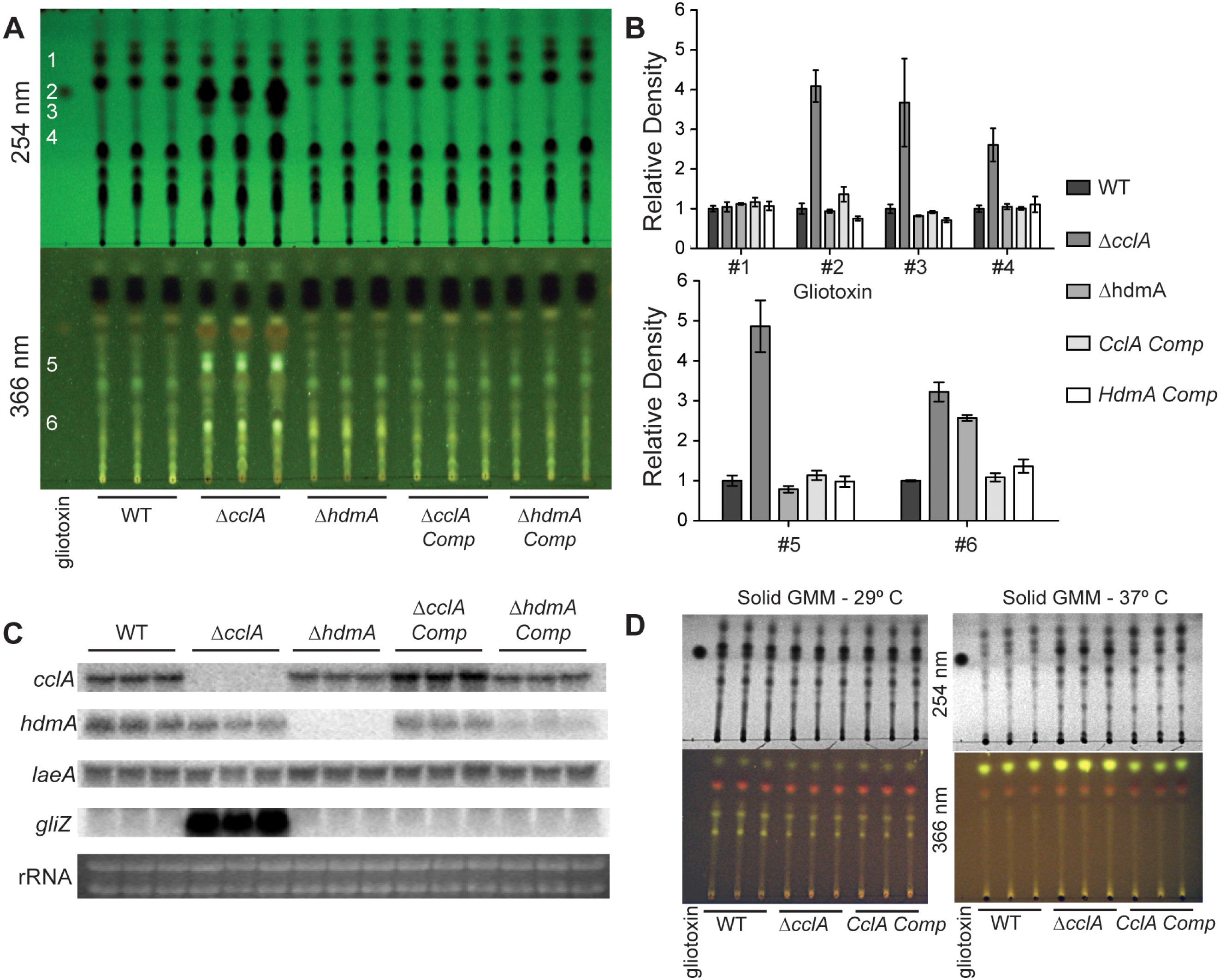{kind=link}
Virulence of ΔcclA is unaltered in a mammalian and Drosophila model
Although poorly growing A. fumigatus mutants have often been found to exhibit a decrease in virulence (D’enfert et al., 1996; Brown et al., 2000), the increased toxin production in the ΔcclA strain coupled with the poor growth presented a unique phenotype that disallowed an obvious prediction on its pathogenicity attributes. Therefore, to assess cclA or hdmA loss on virulence, we examined virulence in two animal models. Assessment of the mutants in a neutropenic murine model showed no significant difference between the wild type and mutant strains (Fig. 5A). This was also true for the Toll-deficient Drosophila model (where only ΔcclA was assessed, Fig. 5B).
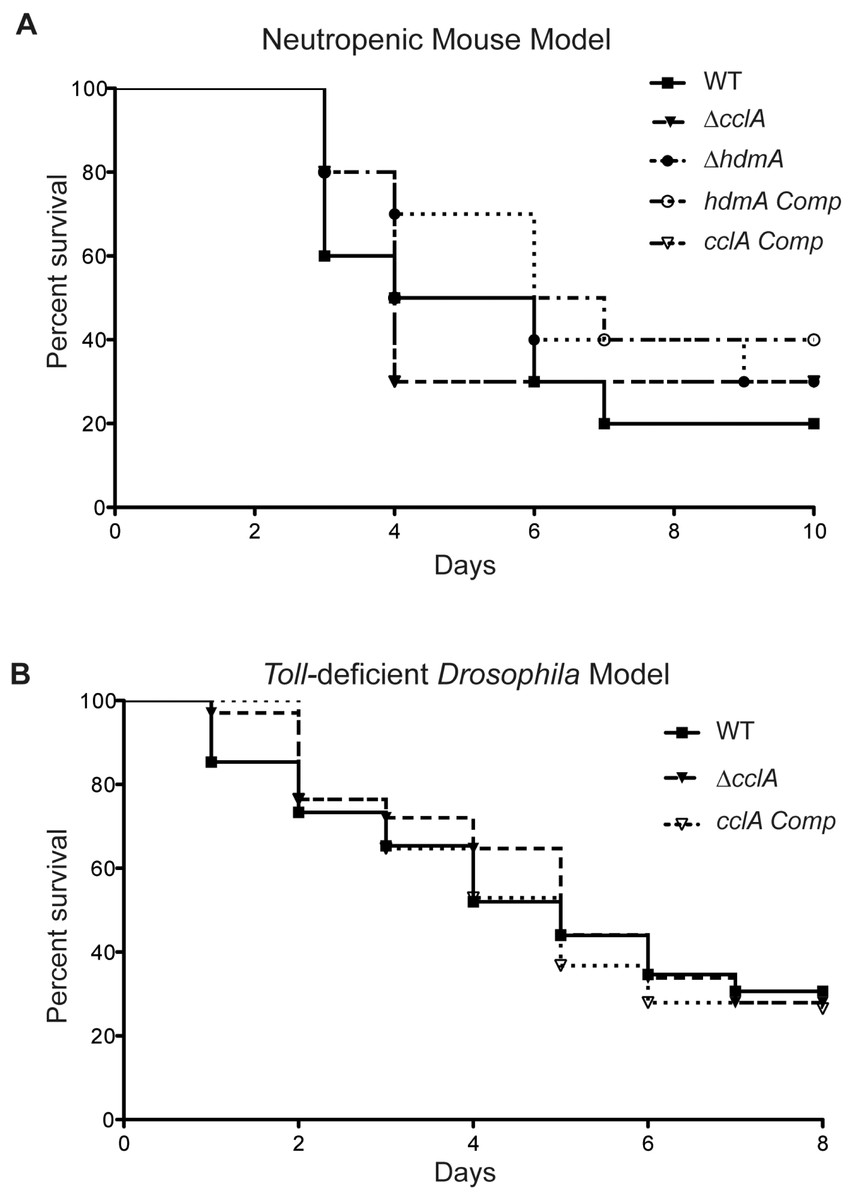
Figure 5: Pathogenicity of ΔcclA and ΔhdmA strains are wild type in murine model of IA and ΔcclA strains are wild type in Toll-deficient Drosophila model.
(A) Pathogenicity of WT, ΔcclA, cclA complement, ΔhdmA, and hdmA complement strains was assessed using the neutropenic murine model of aspergillosis. No differences were measured in any of the strains tested in comparison to wild type (AF293). (B) An alternative pathogenicity assay using Toll-deficient fruit flies illustrates that the ΔcclA mutant is no different than wild type. The Mantel–Cox statistical test was used to assess differences in survival curves.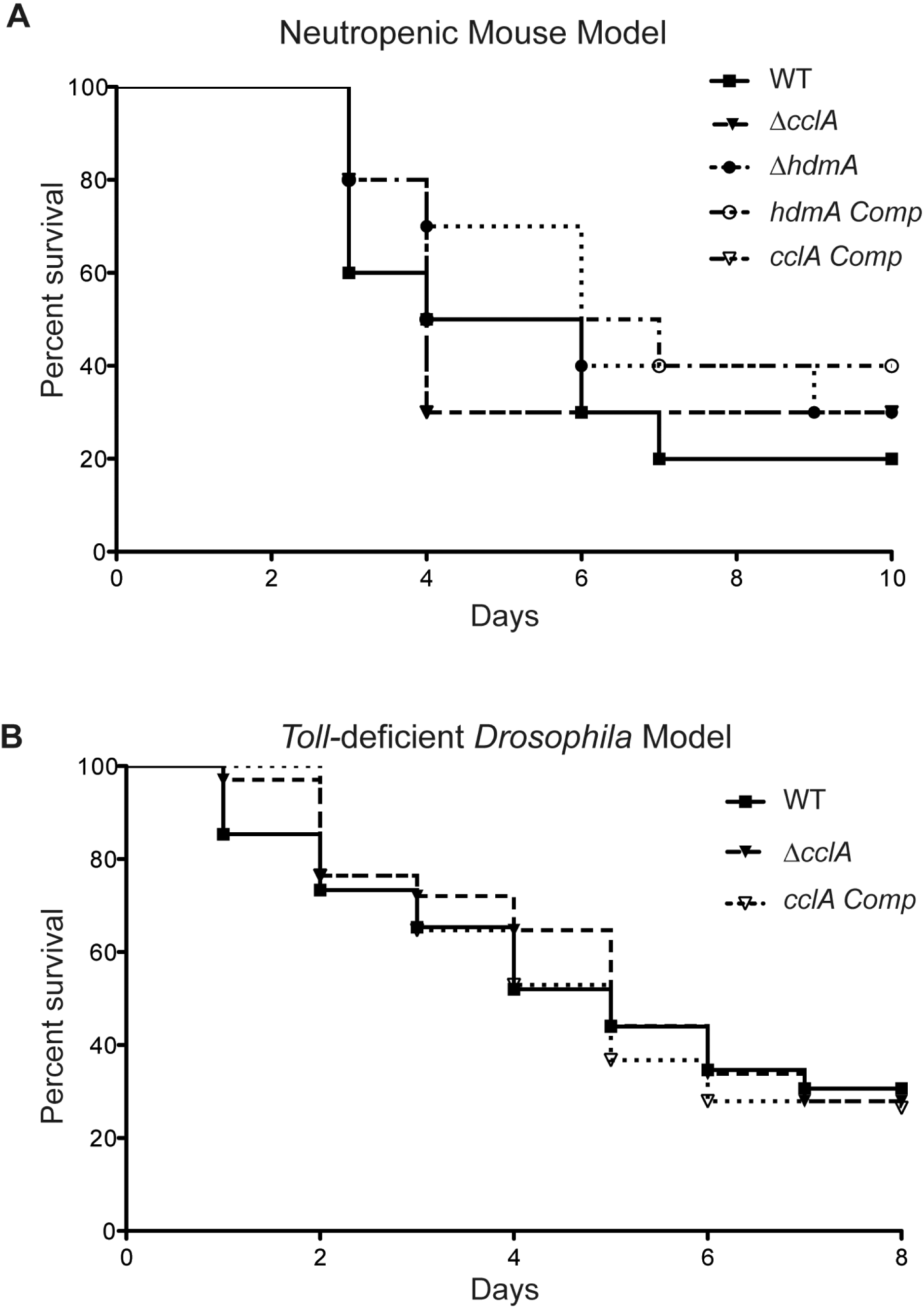{kind=link}
Discussion
Genes involved in secondary metabolism in fungi are arranged in a clustered format that is easily impacted by chromatin level alterations (Palmer & Keller, 2010). This characteristic has been illustrated primarily by two ways: either through manipulation (deletion or overexpression) of chromatin modifying genes or SM induction via chemical epigenetics (Bok et al., 2009; Cichewicz, 2010). LaeA, a global regulator of SM (Reyes-Dominguez et al., 2010), is a conserved virulence factor in filamentous fungi including A. fumigatus, A. flavus and Gibberella zeae (Bok et al., 2005; Sugui et al., 2007a; Amaike & Keller, 2009). Although the mechanism of LaeA action is not known, the results of its activity, such as decrease in heterochromatin marks (e.g. H3K9 tri-methylation, (Reyes-Dominguez et al., 2010)) correlated with increased SM cluster expression, are thought to contribute to its role as a virulence factor in IA. Extrapolation of LaeA impact on chromatin led to the hypothesis that other chromatin remodeling genes, such as histone methyltransferases, acetylases and their cognate demethylases and deacetylases would also impact SM production and, possibly, virulence attributes in pathogenic fungi such as A. fumigatus.
Here our efforts focused on two conserved genes that in other systems are involved in methylation and demethylation of histone 3 on lysine 4. The methylation state of H3K4 is associated with both gene activation and repression in eukaryotes (Krogan et al., 2002; Mueller, Canze & Bryk, 2006). The conserved eukaryotic COMPASS complex is required for methylation of H3K4 with one of its key members a SPRY domain protein termed Bre2p in Saccharomyces cerevisiae (Krogan et al., 2002), Ash2p in Schizosaccharomyces pombe (Roguev et al., 2001) and CclA in A. nidulans (Bok et al., 2009). Demethylation of H3K4 is achieved through activity of an amine oxidase called LSD1 in higher eukaryotes (Shi et al., 2004; Shi et al., 2005). Aspergillus species contain one putative LSD1 homolog, which we termed hdmA in this work due to the existence of a previously named lsdA gene – containing no similarity to hdmA – involved in late sexual development in A. nidulans (Lee et al., 2001).
Our previous results in A. nidulans demonstrated a critical role for CclA in both SM production as well as normal growth (Bok et al., 2009; Giles et al., 2011). This phenotype was replicated in the A. fumigatus ΔcclA mutant. On the other hand, the hdmA deletant in A. nidulans had a more subtle effect, and similar to what we describe here for the A. fumigatus ΔhdmA mutant, small impact on fungal morphology (data not shown). Notably, deletion of cclA in either species was accompanied by an easily detectable decrease in H3K4 methylation, however under the conditions used in this study, no difference in H3K4 methylation was observed in the ΔhdmA strain. Deletion or down regulation of this protein in other organisms can be detected by increased H3K4 methylation (Shi et al., 2004); possibly this increased H3K4 methylation is not observable in bulk histone assessment as measured in this study or, alternatively, HdmA is not the (major) H3K4 demethylase in A. fumigatus. An additional class of Jumonji C domain (JmjC) containing proteins has been shown to demethylate lysine residues in histone tails. While chromatin structure in the aspergilli is thought to be more similar to fission yeast and higher eukaryotes, in S. cerevisiae a JmjC domain containing protein, Jhd2p, is the major demethylase of H3K4 (Liang et al., 2007; Seward et al., 2007). Moreover there is a Jhd2 homolog (Afu5g03430) present in the A. fumigatus genome and thus perhaps Afu5g03430 is the major demethylase of H3K4 in the aspergilli.
Stress assays suggested that the ΔcclA strain was more sensitive to 6AU, a chemical used to identify strains impaired in transcriptional processes, than wild type or the complemented strain. Use of this chemical has identified several S. cerevisiae histone methyltransferase mutants defective in transcriptional elongation (Exinger & Lacroute, 1992; Li, Moazed & Gygi, 2002; Zhang et al., 2005), therefore it is perhaps not surprising that two A. fumigatus histone methyltransferase mutants – ΔcclA described in this study and ΔclrD previously described – are also sensitive to this chemical. Whereas the mechanism of ΔcclA sensitivity to 6AU is unknown, this sensitivity could reflect a defect in the fungal transcriptional machinery in this strain, which highlights the pleiotropic phenotypes of these mutants.
The increased SM output from the ΔcclA strain (Fig. 4) likely arises from aberrancies in transcriptional activity, in this case primarily enhancing transcription of a subset of SM clusters as previously demonstrated in the A. nidulans ΔcclA mutant (Bok et al., 2009). The increased expression of gliZ correlated well with the significant increase in gliotoxin synthesis in this strain. Although we have not characterized the other up regulated metabolites in this strain, we speculate that some of them may be a result of ‘turning on’ formerly silent SM clusters and we will be investigating this hypothesis in future studies.
In A. fumigatus, mutants that have been shown to display poor growth in vitro have also been shown to be less virulent in a model of IA (D’enfert et al., 1996; Brown et al., 2000). However, it was recently shown in Candida albicans that mutants that grow poorly in vitro do not always show a decrease in virulence (Noble et al., 2010). Similarly, disruption of ClrD in A. fumigatus results in a mutant that is defective in growth and asexual sporulation (Palmer et al., 2008) however did not result in a decrease in pathogenicity in the mouse model of IA (T. Dagenais, D. Andes & N. Keller, unpublished data). Thus, the pleiotropic phenotype of poor growth in the laboratory with increased secondary metabolite synthesis of the ΔcclA mutant presented an interesting and potentially opposing coupling of putative A. fumigatus virulence attributes, as enhanced gliotoxin production would be expected to increase virulence (Bok et al., 2006; Cramer et al., 2006; Sugui et al., 2007b). The results from our studies suggest that loss of the CclA gene had no significant effect on pathogenicity in either a neutropenic murine or Drosophila model of IA. Although we did not test growth rate in the host organisms, our data could suggest that pathogenicity can be restored in poor growing strains by increased secondary metabolite (gliotoxin) production, or alternatively, could indicate that genetic factors regulating in vitro growth may differ from those controlling in vivo growth. Moreover, this study supports the view that a composition of many A. fumigatus characteristics contributes to the pathogenicity of this species.