
Expression and characterization of thermostable glycogen branching enzyme from Geobacillus mahadia Geo-05
- Published
- Accepted
- Received
- Academic Editor
- Christopher Cooper
- Subject Areas
- Biotechnology, Molecular Biology
- Keywords
- 1-4-alpha-glucan branching enzyme, His-patch thioredoxin, Geobacillus sp, Glycogen branching enzyme, Genome mining
- Copyright
- © 2016 Mohtar et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2016. Expression and characterization of thermostable glycogen branching enzyme from Geobacillus mahadia Geo-05. PeerJ 4:e2714 https://doi.org/10.7717/peerj.2714
Abstract
The glycogen branching enzyme (EC 2.4.1.18), which catalyses the formation of α-1,6-glycosidic branch points in glycogen structure, is often used to enhance the nutritional value and quality of food and beverages. In order to be applicable in industries, enzymes that are stable and active at high temperature are much desired. Using genome mining, the nucleotide sequence of the branching enzyme gene (glgB) was extracted from the Geobacillus mahadia Geo-05 genome sequence provided by the Malaysia Genome Institute. The size of the gene is 2013 bp, and the theoretical molecular weight of the protein is 78.43 kDa. The gene sequence was then used to predict the thermostability, function and the three dimensional structure of the enzyme. The gene was cloned and overexpressed in E. coli to verify the predicted result experimentally. The purified enzyme was used to study the effect of temperature and pH on enzyme activity and stability, and the inhibitory effect by metal ion on enzyme activity. This thermostable glycogen branching enzyme was found to be most active at 55 °C, and the half-life at 60 °C and 70 °C was 24 h and 5 h, respectively. From this research, a thermostable glycogen branching enzyme was successfully isolated from Geobacillus mahadia Geo-05 by genome mining together with molecular biology technique.
Introduction
The branching enzyme (EC 2.4.1.18) is a type of transferase that carries out the transglycosylation reaction of starch and glycogen making the structures branched out (Abad et al., 2002). Glycogen branching enzymes (GBE) are commercialised for applications in the beverage, food processing and nutraceutical industries. Studies have been done to utilize this enzyme either in vivo or in vitro in order to boost the quality of starchy food by increasing the branches in starch molecules (Kortstee et al., 1996; Kawabata et al., 2002; Kim et al., 2005; Lee et al., 2008). The branching enzyme has been used to produce cyclodextrin, a compound that is used as an ingredient in sports drinks, to enhance the taste of food and also as a spray-drying aid (Takata et al., 2010). Other than that, the branching enzyme also used in bread as an anti-staling agent, produce low viscosity and high molecular weight starch, use for paper coating and even warp sizing textile fibers to make the fibers stronger (Van der Maarel et al., 2002). Studies of GBE are also emerging with therapeutic applications; for example, against tuberculosis and glycogen branching enzyme deficiency disease (Pal et al., 2010; Garg et al., 2007; Bruno et al., 1993). The thermostable GBE is very practical in industries, but the production of this enzyme in its thermophilic host is very low. Therefore, recombinant DNA technologies, such as Escherichia coli cloning and expression systems, were often utilized in order to maximize enzyme production. The E. coli system is often preferred, as this system is easy to manipulate, capable of producing enzyme rapidly and reasonably cheap.
‘Genome mining’ is a term given to a technique that uses basic bioinformatics tools and databases to search for genes with a specific function, such as enzymes, natural products and metabolites, from genome sequences of numerous kinds of organisms (Van der Maarel et al., 2002; Ferrer, Martínez-Abarca & Golyshin, 2005; Challis, 2008). This technique exploits the readily accessible public databases that store gene and genome sequences; for example, GenBank at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov), the UCSC Genome Browser (http://genome.ucsc.edu) and the Ensembl Genome Browser (http://www.ensembl.org) (Corre & Challis, 2007; Schattner, 2009).
For this research, a thermophilic bacterium, Geobacillus mahadia Geo-05, was sampled from Sungai Klah Hot Springs, Sungkai, Perak, Malaysia at 90 °C and therefore it was postulated that this bacterium species would produce thermostable glycogen branching enzyme that is active at high temperature. The objectives of this research are to isolate and characterize glycogen branching enzyme gene (glgB) from Geobacillus mahadia Geo-05.
Materials and Methods
Genome mining
The genome sequence of Geobacillus mahadia Geo-05 used in this research was contributed by Malaysia Genome Institute. Known glgB nucleotide sequences from other Geobacillussp. were obtained from GenBank and were used in sequence alignment softwares, local BLAST and ClustalW, to locate the position of the open reading frame (ORF) of glgB in the G. mahadia Geo-05 genome (Hall, 2010; EMBL-EBI, 2010; NCBI, 2010). glgB sequences of Geobacillus sp. obtained from GenBank that were used are Bacillus sp. NBRC 15315 (AB294568), Geobacillus stearothermophilus(M35089), Geobacillus sp. Y412MC10, Geobacillus sp. Y412MC61 (CP001794) and Geobacillus thermodenitrificans NG80-2. The similarity of amino acid sequence of GBE from Geobacillus mahadia Geo-05 compared to GBE from the other Geobacillus sp. are 97%, 81%, 51%, 99% and 91%, respectively.
Microorganisms and media
The Geobacillus mahadia Geo-05 used in this research was contributed by the Malaysia Genome Institute (DSMZ accession number: DSM 29729). G. mahadia Geo-05 was grown in nutrient broth and nutrient agar (Merck). The bacteria were cultivated at 60 °C for 18 h. The genomic DNA was purified using Qiagen DNeasy® Blood and Tissue Kit.
Cloning and expression
The glgB from G. mahadia Geo-05 were amplified using polymerase chain reaction (PCR). The forward primer has additional four bases at the 5′ end to prepare the insert for cloning reaction into pET102/D-TOPO® vector (Invitrogen). Forward primer: 5′–CACCATG CGA TCC AGC TTG ATT GC–3′; Reverse primer: 5′–TCA ATG ATC CGG TAC TTC CC–3′. Amplification process was carried out in a reaction mixture containing 20–50 ng DNA template, 0.2 µM forward and reverse primers, 0.2 mM dNTP mix, 1.2 U Pfu DNA polymerase and 1×Pfu Buffer with MgSO4.The genes were amplified using a thermocycler (MyCycler™, BioRad) with the temperature program of predenaturation at 95 °C for 5 min; 35 cycles of 30 s denaturation at 95 °C, 30 s annealing at 57 °C and 4 min extension at 72 °C; followed by final elongation step at 72 °C for 7 min and hold at 10 °C. Fresh PCR products were cloned into pET102/D-TOPO® vector from Champion™ pET Directional TOPO® Expression Kit expressed in E. coli BL21 Star™ (DE3).
Expression was done in 200 mL LB broth containing 100 µg/mL ampicillin in 1 L shake flask, incubated at 37 °C with 250 rpm shaking in INFORS HP (Ecotron) incubator shaker. The expression was induced with 0.75 mM IPTG when optical density A600nm reached 0.5 for 8 h. After induction, cell culture was centrifuged at 12,000× g for 20 min at 4 °C.
Protein purification
The cell pellet was resuspended in 10 mL of 50 mM sodium phosphate buffer (pH 7.0), sonicated (Branson Digital Sonifier; 2 min with 30 s lapse; amplitude: 30%) and protein aggregates was separated from soluble protein by centrifugation (12,000× g, 20 min, 4 °C). Recombinant GBE (GBE-05) (soluble protein) was purified by affinity chromatography technique using Äkta Explorer (GE Healthcare). The cleared cell lysate was loaded into 1 mL HisTrap HP column (GE Healthcare) at flow rate of 1 mL/min. The column was then washed with 20 column volume of binding buffer (20 mM sodium phosphate, 0.5 M NaCl, 30 mM imidazole, pH 7.4) and the bound enzyme was eluted with elution buffer (20 mM sodium phosphate, 0.5 M NaCl, 0.5 M imidazole, pH 7.4) by a linear gradient. Eluted protein fractions were pooled and subjected to buffer exchange using 30,000 mwco spin column (Millipore) to the buffer that was used for the assay and analysed using SDS-PAGE. SDS-PAGE (12% running gel, 6% stacking gel) was done using Laemmli’s method (Laemmli, 1970). The sample (10 µL) was loaded into the gel and run at 180 volts for 1 h. The gel was then stained with Coomassie Brilliant Blue R-250 solution. The protein content was determined by Quick StartTM Bradford protein assay (Biorad).
Iodine stain assay
Enzyme solution in 50 mM sodium phosphate buffer, pH 7.0 (50 µl) was incubated with 50 µl of substrate at 50 °C for 30 min. The substrate was 0.1% amylose from potato (Sigma) dissolved in 50 mM sodium phosphate buffer (pH 7.0) and 10% (v/v) of DMSO. The reaction was terminated by the addition of 1 mL of iodine reagent. Iodine reagent was prepared fresh from 0.5 mL of stock solution (0.26 g of I2 and 2.6 g of KI in 10 mL of distilled water), 0.5 mL of 1 M HCl and diluted to 130 mL in distilled water. One unit (U) of enzyme activity was defined as the decreased of A660nm reading by 1% per minute. The decreased of A660nm reading represents the amylose-iodine complex (Shinohara et al., 2001).
Enzyme characterization
The effect of temperature on GBE-05 activity was studied at temperatures from 30 °C to 80 °C with 5 °C intervals. The enzyme thermostability test was done by incubating the enzymes at 40 °C–80 °C for 24 h with 4 h intervals. After the incubation, the enzyme was immediately cooled in an ice bath prior to assay. GBE activity was assayed at 50 °C, pH 7.0. The effect of pH on GBE-05 activity was studied at pH 4–pH 10. GBE-05 activity was assayed in 50 mM acetate buffer for pH 4–6, 50 mM potassium phosphate buffer for pH 6–8, 50 mM Tris-Cl buffer for pH 8–9 and50 mM glycine-NaOH for pH 9–10. The effect of pH on GBE-05 stability was studied by incubating the enzyme in the buffers mentioned at 25 °C for 1 h. GBE activity was assayed at 50 °C, pH 7.0. To study the effect of metal ions on GBE-05 activity, GBE-05 was treated with 1 mM and 5 mM of metal ions (Mg2+, Ca2+, Fe2+, Mn2+, Zn2+ and Cu2+) for 30 min at 25 °C and immediately assayed after the treatment at 50 °C, pH 7.0.
Nucleotide sequence accession number
The nucleotide sequence data reported in this paper are registered with the GenBank nucleotide sequence databases under accession number KC951870.
| Conserved region | ||||
|---|---|---|---|---|
| I | II | III | IV | |
| Geobacillus mahadia Geo-05 | HQAGLGVIIDWVPGHFCK | HVDGFRVDAVAN | VLMIAEDSTDW | FILPFSHDEVV |
| Geobacillus sp. Y412MC10 | HQAGIGVLLDWVPAHFAK | HIDGLRVDAVTS | ALMMAEESSAW | FTLPLSHDEVV |
| Geobacillus sp. Y412MC61 | HQAGLGVIIDWVPGHFCK | HVDGFRVDAVAN | VLMIAEDSTDW | FILPFSHDEVV |
| Geobacillus sp. NBRC 15315 | HQAGIGVILDWVPGHFCK | HVDGFRVDAVAN | VLMIAEDSTDW | FILPFSHDEVV |
| Bacillus stearothermophilus | HQQGIGVILDWVPGHFCK | HVDGFRVDAVAN | ILMIAEDSTDW | FILPFSHDEVV |
| Geobacillus thermodenitrificans NG80-2 | HQAGIGVIMDWVPGHFCK | HIDGFRVDAVAN | VLMIAEDSTDW | FILPFSHDEVV |
| Escherichia coli | HAAGLNVIMDWVPGHFPT | GIDALRVDAVAS | AVTMAEESTDF | FILPFSHDEVV |
| Mycobacterium tuberculosis | HQAGIGVIVDWVPAHFPK | HIDGLRVDAVAS | IVTIAEESTPW | YVLPLSHDEVV |
Notes:
The conserved amino acids are in bold.
Results and discussion
Genome mining
glgB of G. mahadia Geo-05 has the size of 2013 bp that codes for 670 amino acids. The theoretical molecular weight is 78.43 kDa, predicted using the “Compute pI/Mw tool” from ExPASy Bioinformatics Resource Portal (http://web.expasy.org/compute_pi/). The four conserved regions of α-amylase family enzymes were determined (Table 1). Within the four conserved regions, there are seven highly conserved amino acids that have important roles in the catalysis and substrate binding. Three of the conserved residues are the catalytic residues; Asp313 in region II, Glu356 in region III and Asp424 in region IV. Four other conserved residues; Asp243 and His248 in region I, Arg311 in region II and His423 in region IV are responsible for substrate binding (Abad et al., 2002; Van der Maarel et al., 2003).
| Sample | Total protein (mg) | Total activity (u) | Specific activity (u/mg) | Purification fold | Recovery (%) |
|---|---|---|---|---|---|
| Cell extract | 4.86 | 1314.50 | 270 | 1 | 100 |
| Purified GBE | 0.43 | 1105.28 | 2,598 | 10 | 84 |
Protein purification
GBE-05 produced by pET102/D-TOPO® expression vector has His-Patch thioredoxin fused to the protein. His-Patch thioredoxin is a mutated thioredoxin that has a metal binding domain, which has been shown to have high affinity for divalent cations and therefore, the fusion protein can be purified using metal chelating resins like nickel sepharose (Lu et al., 1996). The recovery of protein obtained after the purification process was high with the enzyme activity increased by ten fold (Table 2). The SDS-PAGE result shows a single band for the purified enzyme (pooled eluted fractions) in lane 3, which means that the enzyme was successfully purified (Fig. 1). The theoretical molecular weight of GBE was 78 kDa and with the addition of His-Patch thioredoxin (13 kDa), the expected size of the recombinant protein would be 91 kDa.

Figure 1: SDS-PAGE of purified enzyme.
M: Broad Range Prestained Protein Marker (Nacalai). Lane 1: Crude enzyme. Lane 2: Protein in flowthrough fractions. Lane 3: Purified enzyme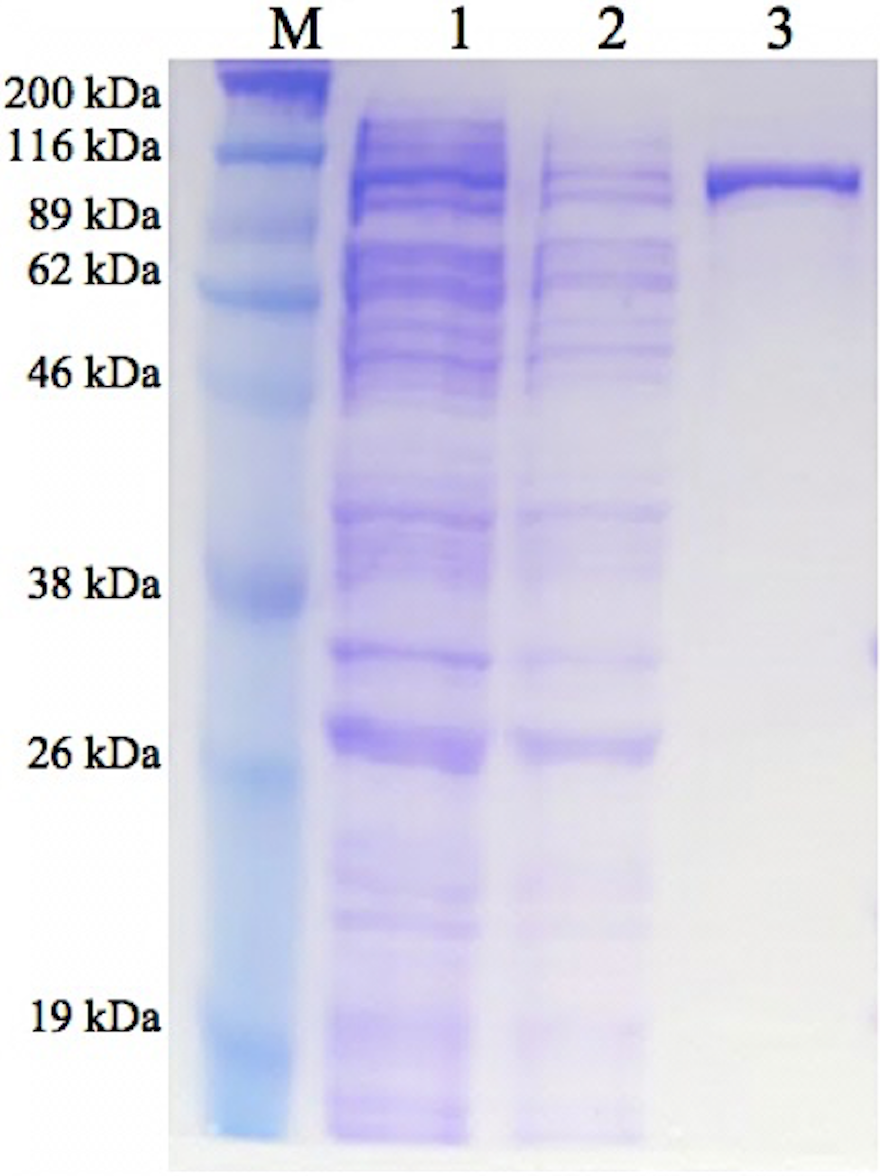{kind=link}
Enzyme characterization
GBE-05 was generally active at 45 °C–60 °C and enzyme activity was highest when assayed at 55 °C (Fig. 2). This optimum temperature of GBE-05 was higher than GBEs isolated from G. stearothermophilus and A. gottschalkii, which has the optimum temperature of 50 °C (Takata et al., 1994; Thiemann et al., 2006). However, GBEs isolated from extreme thermophilic bacteria, Rhodothermus obamensis, R. marinus and A. aeolicus showed higher optimum temperature, that is between 65 °C–80 °C (Shinohara et al., 2001; Van der Maarel et al., 2003; Yoon et al., 2008). These bacteria produce enzymes that are active at higher temperature comparatively to their optimal growth temperatures.
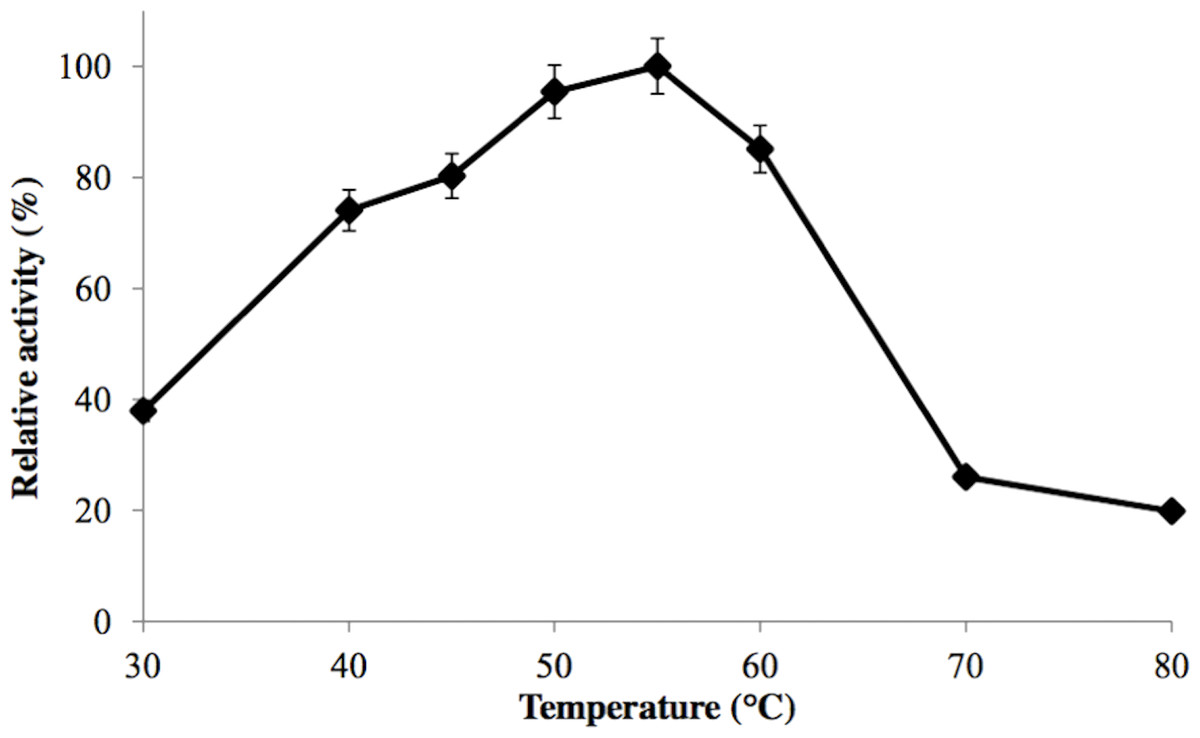
Figure 2: Effect of temperature on enzyme activity.
GBE activity was assayed at temperature between 30 °C–80 °C. 100% of activity is 476 U/mg using iodine stain assay. Note: error bars represent means ±5% for triplicate determinations.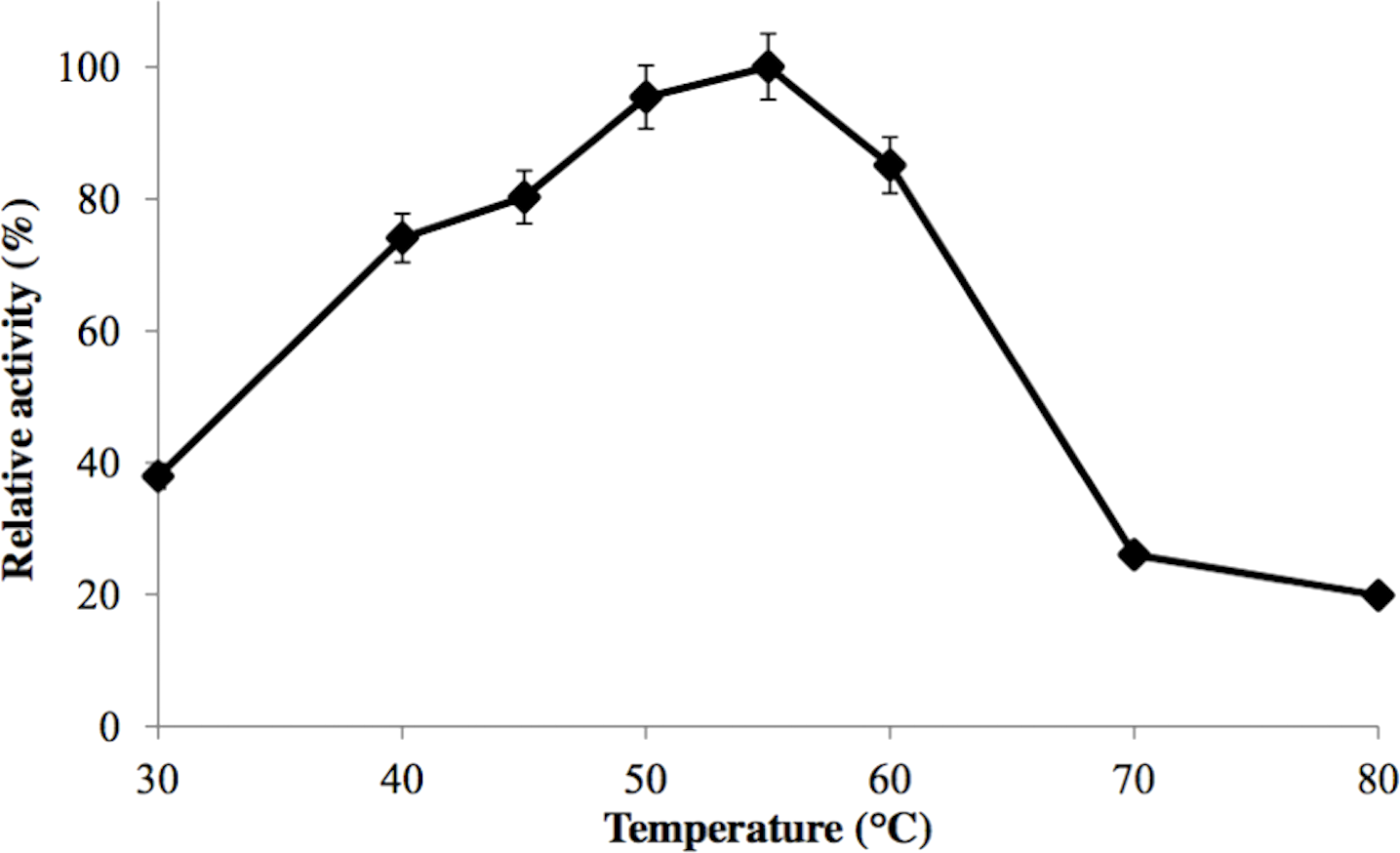{kind=link}
The half-life of the enzyme at 60 °C was 24 h while at 70 °C, 5 h (Fig. 3). GBE-05 is more stable compared to GBE from G. stearothermophilus that has lost 20% of enzyme activity at 60 °C in just 30 min and A. gottschalkii that has a half-life of only 55 min at 55 °C (Takata et al., 1994; Thiemann et al., 2006). Since GBE-05 does not have any disulphide bonds predicted, therefore the stability of this enzyme is possibly due to the high composition of aromatic amino acid residues. The thermostability of an enzyme can be presumed from its primary sequence information as there are correlations between the number of aromatic amino acids (phenylalanine, tryptophan and tyrosine), glutamine and asparagine with the thermostability (Burley & Petsko, 1985; Serrano, Bycroft & Fersht, 1991; Vieille et al., 2001; Van der Maarel et al., 2002). Enzymes with a high number of aromatic residues in combination with low number of glutamine and asparagine would show higher temperature stability. The reason behind this is that the hydrophobic interactions between the aromatic groups are responsible for the stability of a thermophilic protein, while the deamination of thermolabile amino acids (asparagine and glutamine) resulted in the inactivation of enzymes at elevated temperature (Vieille et al., 2001).
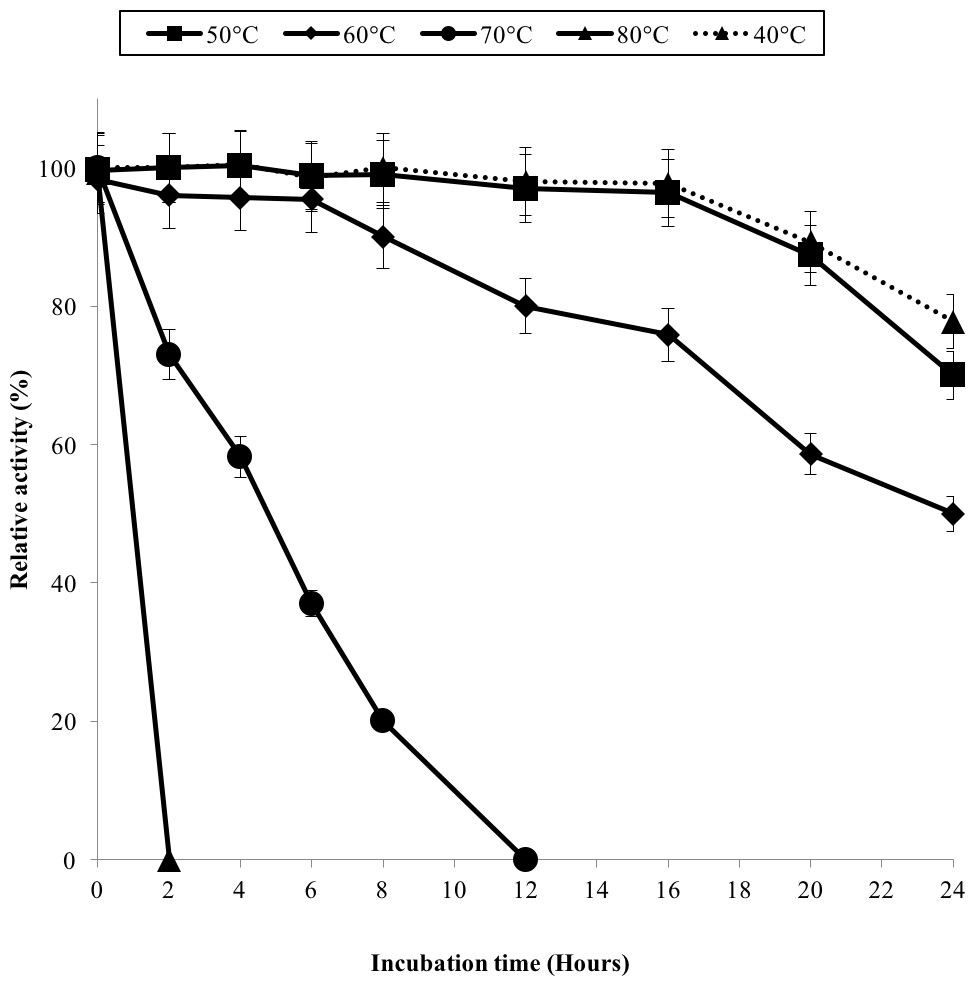
Figure 3: Effect of temperature on enzyme stability.
GBE was incubated at 40 °C–80 °C prior to enzyme assay. Enzyme assay was done at 50 °C. 100% of activity is 793 U/mg using iodine stain assay. Note: Error bars represent means ±5% for triplicate determinations.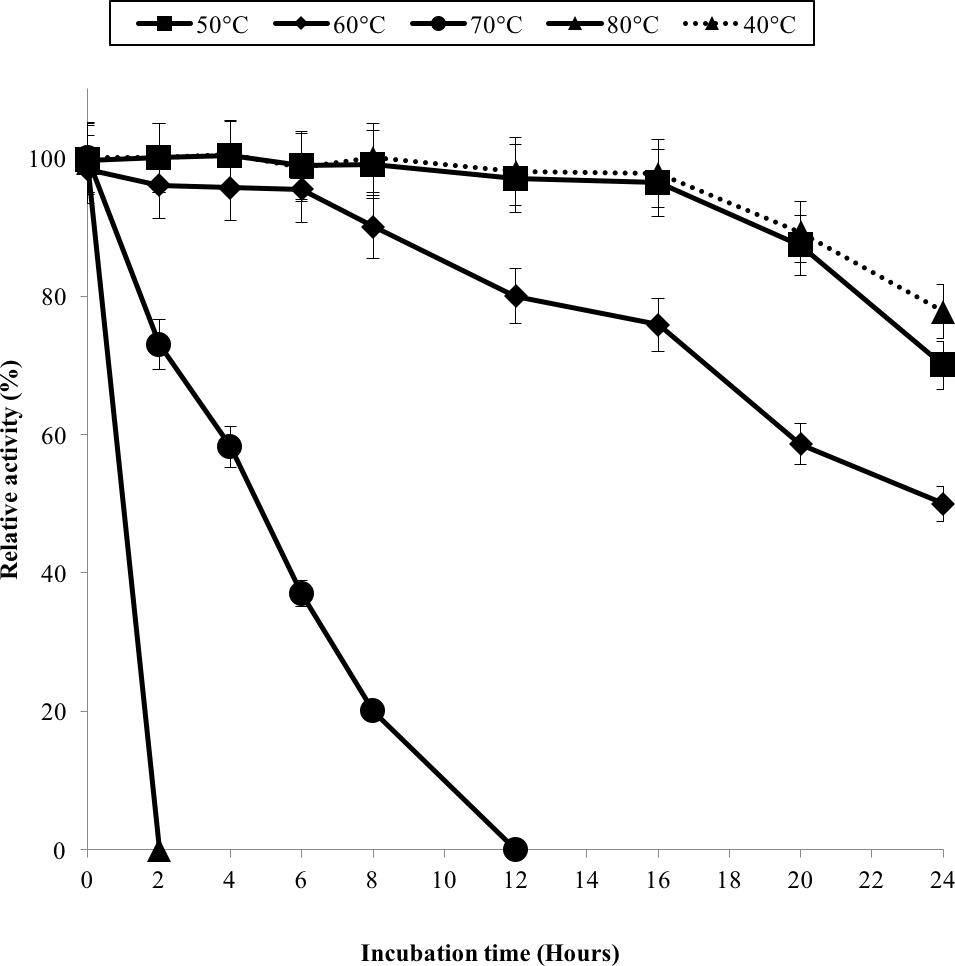{kind=link}
GBE-05 displayed relatively high activity in broad pH range, where more than 60% of enzyme activity remained when assayed at pH 5–pH 9 (Fig. 4A), and was found to be most active at pH 6. The stability test shown that the enzyme was stable between pH 5–pH 9 where more than 50% of enzyme activity remained after the 30 min of pH treatment (Fig. 4A). It is important for GBE-05 to be active and stable in wide range of pH if this enzyme were to be applied industries.
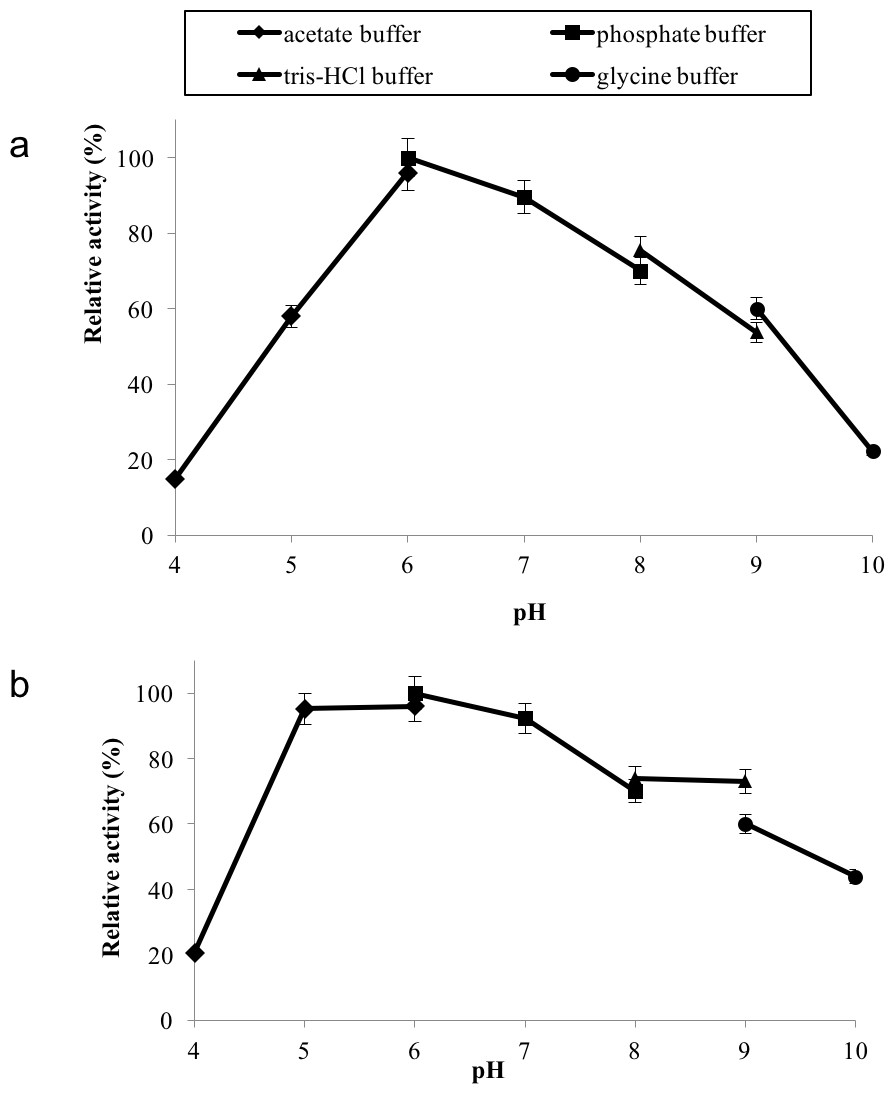
Figure 4: (A) Effect of pH on enzyme activity. (B) Effect of pH on enzyme stability.
Note: data represents mean ± SE (n = 3).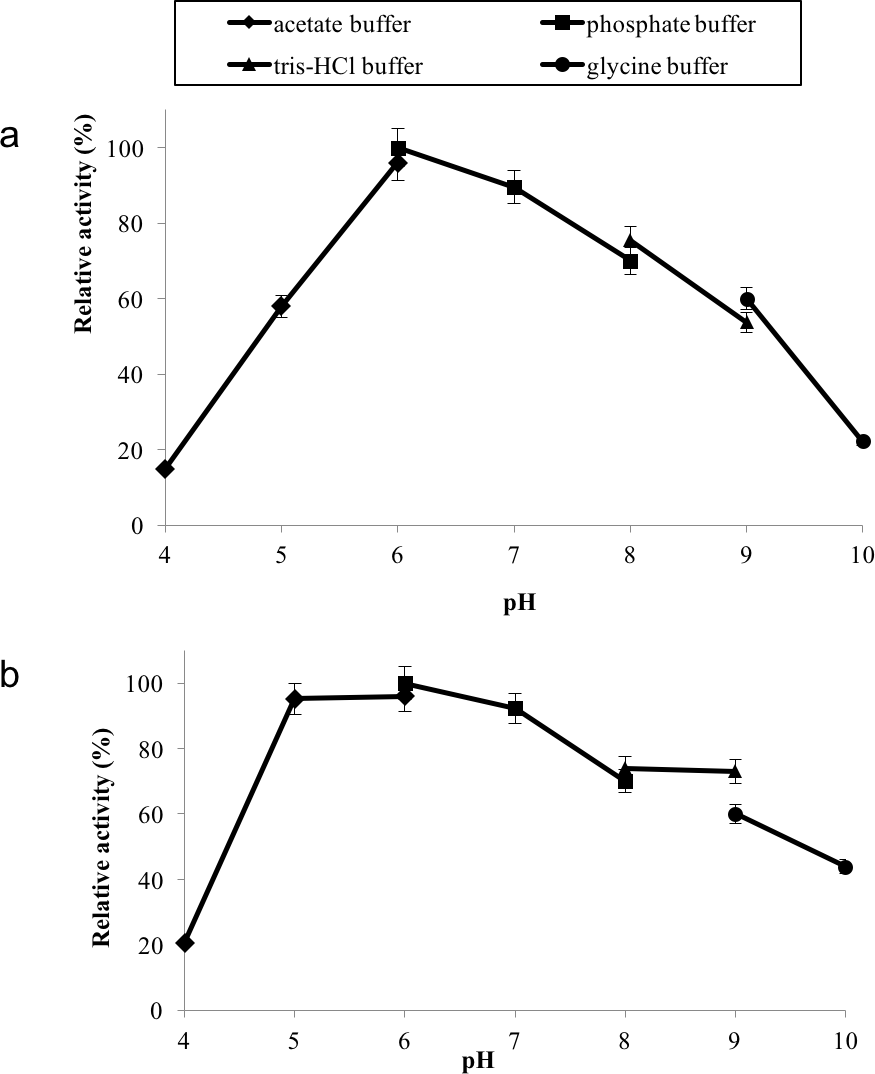{kind=link}
Metal ions had different effects on GBE-05 activity but none of the metal ions experimented upon enhanced the enzyme activity (Fig. 5). Two alkaline earth metals of group 2 elements (Mg2+ and Ca2+) were tested to have no effect on enzyme activity. However, GBE activity was slightly lowered to 73% when the concentration of Ca2+ increased to 5 mM. Similar results are also observed in GBE from M. tuberculosis but Mg2+ seems to enhance the activity of GBE by 15% for R. marinus (Garg et al., 2007; Yoon et al., 2008). Four transition metals (Mn2+, Fe2+, Cu2+and Zn2+) were also tested out. 1 mm Mn2+ did not affect enzyme activity but the activity was decreased by 14% in 5 mM Mn2+. Mn2+ also showed slight inhibition on GBE activity isolated from Anaerobranca gottschalkii and R. marinus (Thiemann et al., 2006; Yoon et al., 2008). Zn2+ and Cu2+ repressed the enzyme activity as only 40% and less remained. These metal ions also appear to restrain GBE activity from other bacteria, A. gottschalkii, R. marinus and M. tuberculosis(Thiemann et al., 2006; Garg et al., 2007; Yoon et al., 2008). 5 mM of Fe2+ inhibits the enzyme by 60%, same as R. marinus (Yoon et al., 2008).
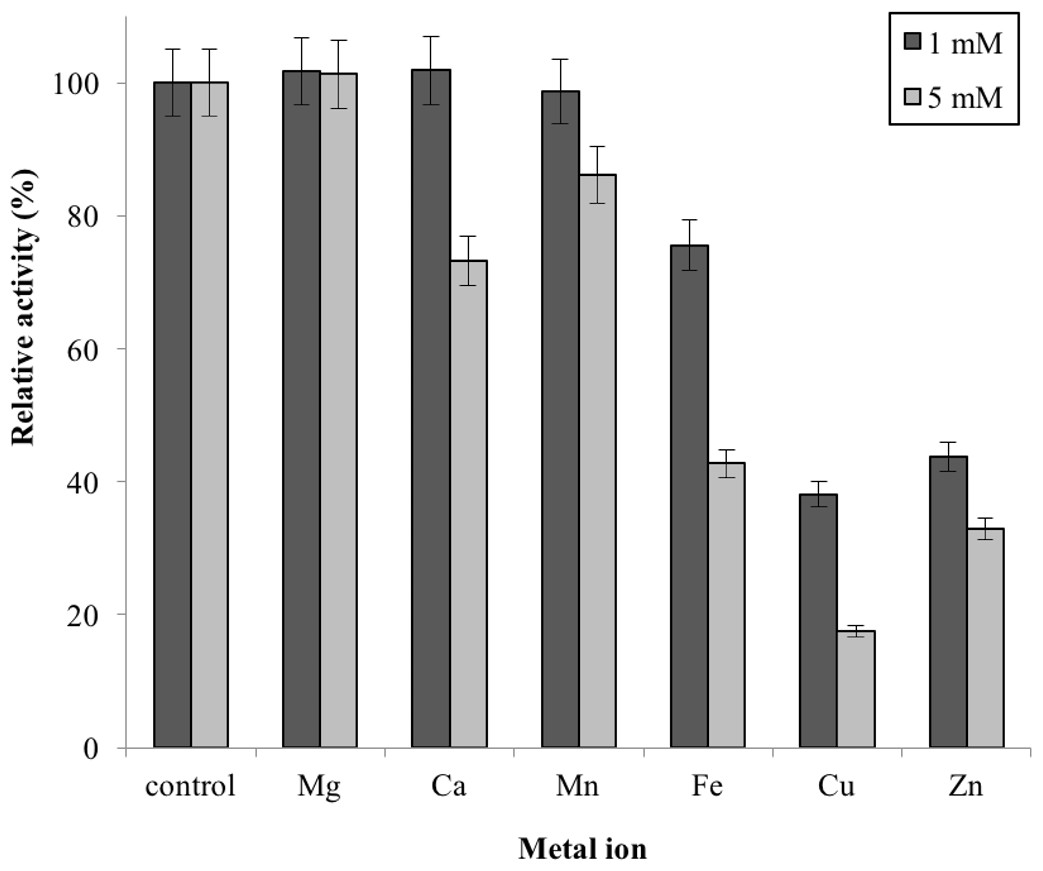
Figure 5: Effect of metal ion on enzyme activity.
Enzyme activity was assayed with two concentrations of metal ions, 1mM and 5 mM. 100% of activity is 641 U/mg using iodine stain assay. Note: error bars represent means ±5% for triplicate determinations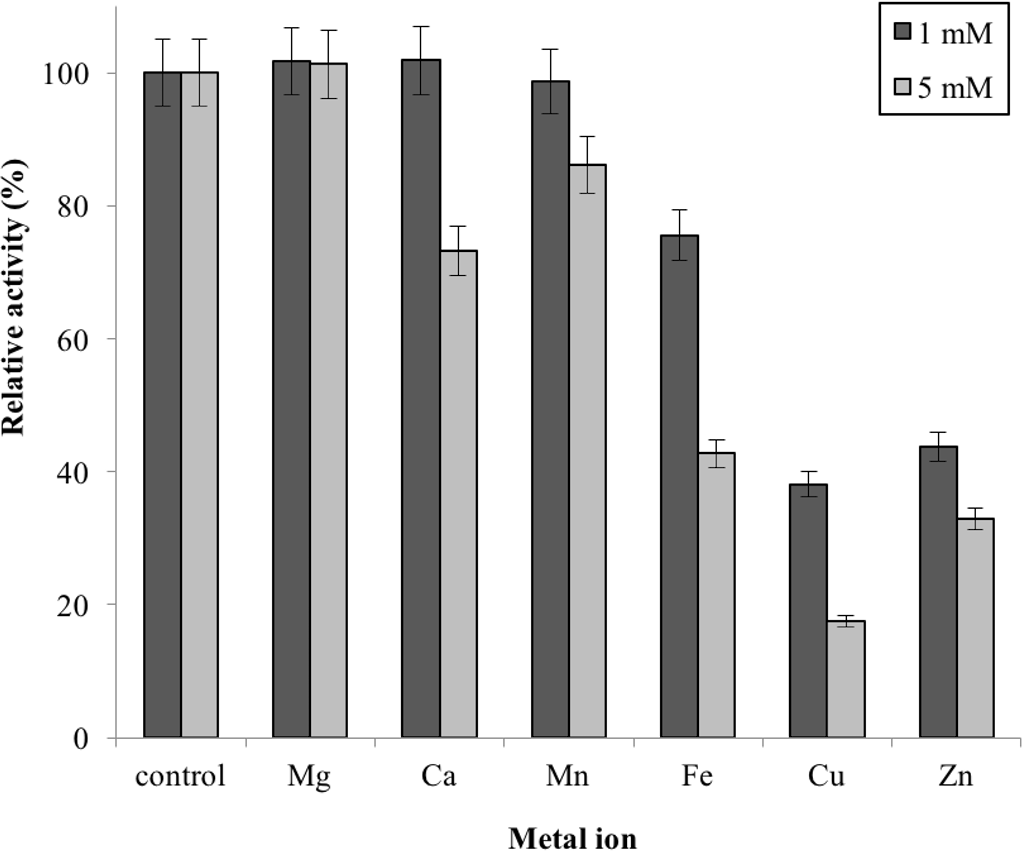{kind=link}
Conclusions
In conclusion, GBE-05 is stable and active at high temperature and therefore is very applicable in industries. The results of genome mining and computational prediction complement the results obtained from wet laboratory experiments. The vast information on genome sequence together with latest development in structural prediction software and algorithms enables scientists to compute data from genes to protein structure and function accurately.